- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The metal-support interfaces between metals and oxide supports have long been studied in catalytic applications, thanks to their significance in structural stability and efficient
catalytic activity. The metal-rare earth oxide interface is particularly interesting because these early transition cations have high electrophilicity, and therefore good binding strength
with Lewis basic molecules, such as H2O. Based on this feature, here we design a highly efficient composite Ni-Y2O3 catalyst, which forms abundant active Ni-NiO_x_-Y2O3 interfaces under the
water-gas shift (WGS) reaction condition, achieving 140.6 μmolCO gcat−1 s−1 rate at 300 °C, which is the highest activity for Ni-based catalysts. A combination of theory and ex/in situ
experimental study suggests that Y2O3 helps H2O dissociation at the Ni-NiO_x_-Y2O3 interfaces, promoting this rate limiting step in the WGS reaction. Construction of such new interfacial
structure for molecules activation holds great promise in many catalytic systems. SIMILAR CONTENT BEING VIEWED BY OTHERS REVERSING SINTERING EFFECT OF NI PARTICLES ON Γ-MO2N VIA STRONG METAL
SUPPORT INTERACTION Article Open access 30 November 2021 TACKLING ACTIVITY-STABILITY PARADOX OF RECONSTRUCTED NIIROX ELECTROCATALYSTS BY BRIDGED W-O MOIETY Article Open access 04 December
2024 BOOSTING CO HYDROGENATION TOWARDS C2+ HYDROCARBONS OVER INTERFACIAL TIO2−_X_/NI CATALYSTS Article Open access 07 November 2022 INTRODUCTION Physicochemical interfaces between the metal
and support are important in heterogeneous catalysis1. Construction of catalytically active interfaces between metals and supports has attracted extensive attentions. The metal support
interaction leads to high dispersion2,3 or even over-encapsulation of active metals4. Therefore, it is crucial to design and control suitable metal-support interaction and thus create
abundant and effective interfacial active sites for molecule transformation. In particular, the metal interaction with third row (MgO5, Al2O36 and SiO27) oxides and fourth row 3_d_ metal
oxides (TiO21,4,8,9,10,11,12, FeO_x_13,14,15, Co3O416,17) have been widely explored and discussed in terms of electron transfer process18,19, oxygen vacancies1,11,12,20 and surface wetting4.
In comparison, fifth or sixth row oxides, such as rare earth oxides, are seldom used as catalysts support except for CeO22,3,20,21,22,23,24,25. These early transition rare earth cations are
strong Lewis acid and have strong binding to Lewis base such as H2O and NH3. The metal-rare earth oxide interface should have very different properties to those third and fourth row metals.
It is a complete surprise that they have been rarely investigated in the H2O or NH3 based reactions, such as water-gas shift (WGS) reaction11,21−23. WGS is an irreplaceable reaction to
produce H2 from CO and H2O, which is responsible for ammonia synthesis, hydrogenation reactions and hydrogen fuel cells26. On one hand, WGS is exothermic and limited by thermodynamic
equilibrium at high-temperature (HT). On the other hand, H2O is a very weak oxidant for CO and has very sluggish kinetics at low-temperature (LT). In comparison, medium-temperature (MT) WGS
can achieve rapid kinetics with high CO conversion equilibrium27,28. Therefore, developing effective and stable catalytic interfaces in the MT region is a practical route to replace the
existing cascade HT-LT WGS process. This is usually achieved with Ni-based catalysts11,12,29,30,31,32,33,34,35,36, which is mostly active between 300 and 400 °C. Existing research focus on
CeO2 based support30,31,32,33,34,35 mostly due to its abundant oxygen vacancy. However, at MT region, such oxygen vacancy may not be necessary, whereas the surface adsorption and activation
of H2O is the key. This is an area that rare earth support, such as Y2O3, has good competitivity. In addition, Y2O3 also offers good thermal stability, high chemical durability and high
mechanical strength, promoting the stability of the metal oxide interface. Here we reported an in situ formed Ni-NiO_x_-Y2O3 interface during MT-WGS reaction, achieving 140.6 μmolCO gcat−1
s−1 of CO conversion at 300 °C, which was 4 times to the existing literature reports. Such unique interface was only formed with a 9:1 molar ratio of Ni to Y. The addition of only 10% Y2O3
played a key role in catalytic activity improvement and prevented the catalyst from sintering. In addition, through theoretical simulation we have demonstrated the electron transfer at the
interface, which indicated the existence of the interaction intrinsically. The Ni-NiO_x_-Y2O3 interface was clearly characterized with aberration corrected electron microscopy and in situ
Raman spectra. Combined with DFT calculation, H2O molecules dissociated effectively on the Ni-NiO_x_-Y2O3 interfacial site was proved. And the calculation of the whole reaction process also
proved that the Ni-NiO_x_-Y2O3 interface was very efficient for WGS reaction. The excellent catalytic performance of Ni-NiO_x_-Y2O3 interface suggested the potential of studying and applying
rare earth elements in catalysis, creating new metal-support interface with unconventional surface behaviors. RESULTS CATALYTIC PERFORMANCE OF THE NI9Y1O_X_ CATALYST IN WGS REACTION Series
of Ni_a_Y_b_O_x_ (_a_: _b_ was the molar ratio of Ni to Y) samples were synthesized via the ultrasonic spray method21,37,38. The simultaneous decomposition of Ni and Y precursors in the same
sprayed droplet ensured the formation of maximized interface between the two metals (Supplementary Fig. 1). The droplets were sent into a high temperature tube furnace under the purging of
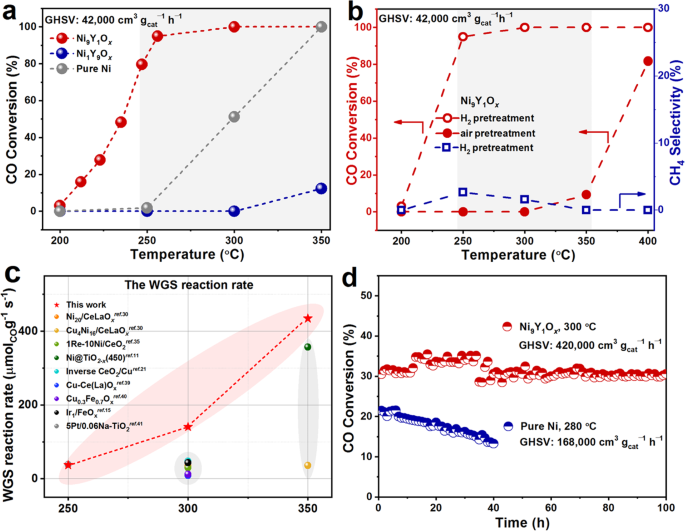
N2 to sufficiently undergo pyrolysis reaction, forming the Ni_a_Y_b_O_x_ catalyst. Supplementary Figs. 2, 3a and Fig. 1a showed the activities of Ni9M1O_x_ (M = Y, Ce, Sm and Al) catalysts
and the Ni_a_Y_b_O_x_ catalysts with different Ni/Y ratios for the WGS reaction. The CO conversion of the Ni9Y1O_x_ catalyst was much higher than other catalysts, reaching more than 90% at
250 °C (pretreatment in the H2 atmosphere, the gas hourly space velocity (GHSV) was 42,000 cm3 gcat−1 h−1). This catalytic activity transferred the HT to MT WGS reaction for Ni-based
catalyst. The different catalytic performance of Ni_a_Y_b_O_x_ catalysts correlated well with their reducing ability (Supplementary Fig. 4). Catalyst with low reduction temperature and large
H2 consumption in the H2 temperature-programmed reduction (H2-TPR) profile showed high CO conversion. The Ni9Y1O_x_ has a small reaction peak at 220 °C and a major one at 400 °C. In
comparison, both NiO and Y2O3 have only one feature at 350 and 575 °C, respectively (Supplementary Fig. 5). The peak at 220 °C was therefore related to the Ni-Y2O3 interface. As shown in
Fig. 1b, pretreatment in the H2 atmosphere shifted the CO conversion to lower temperature range for almost 150 °C, compared to the air-pretreated one. This result suggested that the
oxidation state of the Ni played an important role. The formation of CH4 was only detected at 250 and 300 °C with less than 3% selectivity. Compared to other catalyst system in the
literature11,15,21,30,35,39,40,41, the Ni9Y1O_x_ catalyst achieved the highest reaction rate and turnover frequency (TOF) of Ni-based catalysts, even higher than some Cu-based and noble
metal catalysts between 250 and 350 °C (Fig. 1c, Supplementary Table 1). It also maintained solid time-on-stream stability at high temperature and with an ultra-high GHSV, in which the
conversion decayed by only 2% in 100 h (Fig. 1d). Even in the stream also contained CO2 and H2 (5% CO, 23.3% H2O, 10% H2, 5% CO2, N2), Ni9Y1O_x_ could still maintain similar catalytic
performance comparable to that of commercial Cu-Zn-Al (Supplementary Fig. 3b). The addition of Y2O3 significantly improved the activity and stability of the catalyst, compared to pure Ni
sample (with NiO as precursor). STRUCTURAL CHANGE OF THE NI9Y1O_X_ CATALYST The specific surface areas (_S__BET_) increased from 12.9 to 55.5 m2 g−1 when the Ni content was increased from
5.4% to 67.3% (Supplementary Table 2). Combined with the transmission electron microscope (TEM) and scanning electron microscope (SEM) characterizations (Supplementary Fig. 6), it could be
observed that the Ni9Y1O_x_ catalyst presented spherical morphology (250–1500 nm). Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM)
images showed that the sample composed of uniformly dispersed nano-particles (Fig. 2). The inter-planar spacing of the fresh Ni9Y1O_x_ (Fig. 2d) sample was the same to the lattice fringe of
NiO (111), which was consistent with the X-ray diffraction (XRD) results (Fig. 3a). In addition, no lattice fringe of Y2O3 was observed, indicating that Y2O3 was poorly crystallized due to
the low calcination temperature. In comparison, Ni9Y1O_x_-used (Fig. 2h, i) mainly showed the lattice fringe of metallic Ni, indicating the reduction of Ni2+ to Ni0 during the H2
pretreatment. In addition, lattice fringes of Y2O3 were found between Ni particles (Fig. 2h) indicated the partial crystallization of Y2O3. The structural changes of the catalyst were
observed more intuitively based on the results of elemental mapping. As shown in Fig. 2e, j, the initial uniform distribution of Ni, Y and O in the fresh Ni9Y1O_x_ catalyst segregated after
reaction. For Ni9Y1O_x_-used, the Ni particles grew larger to about 8 nm, and the Y and O were mainly distributed in the surroundings of Ni particles. This structure prevented the further
growth of Ni particles and provided abundant interfaces between Ni and Y2O3. Comparing with TEM images of pure Ni sample (Supplementary Fig. 7), it was clear that Y2O3 played the crucial
role in forming interfaces and stabilizing the catalyst structure. We further explored the chemical state of the catalyst surface by X-ray photoelectron spectroscopy (XPS). The fitting
results of XPS data were integrated in Supplementary Table 3. From Ni 2_p_ XPS spectra (Fig. 3b, d), it was seen that the peak of Ni9Y1O_x_-fresh mainly corresponded to Ni2+ species, while
that of Ni9Y1O_x_-used sample mainly corresponded to Ni0 species12,30,31,32,33. This observation was consistent well with the observed lattice fringes (Fig. 2h). Trace amount of Ni2+ species
were detected for Ni9Y1O_x_-used, which was caused by the partial oxidation when exposed in air. From the result of quasi in situ XPS, only Ni0 species were detected on the surface (Fig.
3d). The Y 3_d_ (Fig. 3c, e) spectra showed the Y3+ species for both samples42,43,44. The main difference was that Y3+ in Y2O3 was mainly detected from the fresh sample; but for the used
samples, it was mainly Y carbonate. The catalyst surface change might result from the WGS reaction containing CO2, which was easily absorbed by Y2O3 on the surface. The bulk Y3+ was still in
the form of Y2O3 as confirmed in XRD results (Fig. 3a). The local structure of Y3+ was further examined in the Y _K_ edge X-ray absorption fine structure (XAFS). At the near edge, pure Y2O3
crystal showed peaks after the edge (Fig. 3f). Literature suggested that there were two Y3+ sites inside Y2O3 crystal45. Site 1 at 17054.4 eV had the same Y-O bonding length, whereas site 2
at 17063.0 eV had different Y-O lengths. Both fresh and used Ni9Y1O_x_ only had the site 1 feature, suggesting weak crystallinity and uniform Y-O bonding length. The near edge spectra also
confirmed the Y3+ states for both fresh and used catalysts (Fig. 3f). The Y-O coordination numbers (C.N.) decreased from 7.671 ± 2.033 Å to 5.155 ± 0.392 Å after catalysis (Fig. 3g and
Supplementary Table 4). This was associated with the formation of Y-Y coordination at 3.613 ± 0.098 Å. The change in the coordination situation was mainly related to the better
crystallization of Y2O3 after the catalytic reaction, which was consistent with the XRD results (Fig. 3a). The combination of XRD, TEM, XPS and XAFS results suggested that Y3+ doped into the
NiO lattice at the fresh state. During the reduction with H2, the NiO was reduced to Ni, leaving no space for Y3+. Therefore, Y3+ was partially converted into crystalline Y2O3 and formed
the Ni-Y2O3 interface. Then we explored the nature of the interfacial interactions. Firstly, the theoretical and actual hydrogen consumption of the catalysts was quantitatively calculated
according to the H2-TPR result (Supplementary Fig. 4, Supplementary Table 2). The actual H2 consumption was higher than the theoretical value, which indicated that the Y2O3 species in the
catalyst were also reduced due to the formation of the interface. Besides, DFT calculations demonstrated the nature of the interaction was an electrostatic attraction (Supplementary Fig. 8).
The charge density calculations suggested that the combination Y3O4/NiO_x_/Ni{111} from Y3O4, NiO_x_ and Ni{111} appeared noticeable electronic migration (NiO_x_ gained 1.643 electrons from
Y3O4 and 0.187 electron from Ni{111}). THE DETERMINATION OF NI-NIO_X_-Y2O3 INTERFACES The phase change of Ni9Y1O_x_ catalyst was further examined with in situ XRD under 5% H2/Ar atmosphere
(Fig. 4a). It was found that NiO phase dominated from room temperature to 300 °C. The formation of metallic Ni appeared at 350 °C, which was in good agreement with the ex situ XRD results
(Fig. 3a). This also explained the low activity of the air-activated catalyst below 350 °C because NiO was not reduced below this temperature under reaction condition. In addition, the
crystalline dominate size of metallic Ni increased gradually with the temperature rise (Supplementary Table 5), 5.5 nm at 400 °C and 9.6 nm at 500 °C, according to the calculation using
Scherrer formula. We further verified microscopic structure change of the interface during the WGS reaction by in situ Raman spectra. The vibration peak around ~505 cm−1 which corresponds to
NiO46,47,48,49,50 was observed in the Ni9Y1O_x_ and pure NiO sample before reaction (Fig. 4b, d, Supplementary Fig. 9). The change of temperature and particle size would cause the shift of
NiO vibration peak50. In addition, the change of peak position could also be considered as the formation of other Ni-O coordination forms, resulting in the formation of NiO_x_ (0 < _x_
< 1) species. Pretreatment at 400 °C under 5%H2/Ar led to disappearance of NiO vibration peak (Fig. 4b), indicating that NiO was fully reduced to metallic Ni. This observation was
consistent with the XRD results (Fig. 4a). Very interestingly, the vibration peak of NiO appeared again in the WGS reaction atmosphere, suggesting the formation of Ni-O bond on the surface.
Furthermore, by switching the feeding gas between CO and H2O (Fig. 4c), it was found that the vibration peak of NiO disappeared when CO was introduced at 400 °C, and then it appeared again
after feeding H2O vapor. This indicated that the O atoms around Ni were almost fully consumed when the catalyst was treated at high temperature under CO, which was evidenced by the in situ
XRD results (Supplementary Fig. 10). The formation of NiO_x_ was not observed with pure Ni sample obtained from ultrasonic spray method (Fig. 4d), regardless of oxidation or reduction
conditions. Therefore, without the Ni-Y2O3 interface, Ni alone could not directly react with H2O to formed NiO_x_. The in situ Raman and XRD experiments suggested that when exposed to H2O
vapor, active oxygen species were formed over the Ni-Y2O3 interface and subsequently converted into Ni-NiO-Y2O3 interface as the active states for WGS reaction. Therefore, the roles of Y2O3
were: (I) restricting the growth of the Ni particles under the reduction conditions; (II) forming the Ni-NiO_x_-Y2O3 interfacial sites under the WGS condition. REACTION PATHWAY WITH
NI-NIO_X_-Y2O3 INTERFACE PARTICIPATION At present, there were mainly two recognized mechanisms for the WGS reaction: redox mechanism and associative mechanism51,52,53,54. For the redox
mechanism, CO reacted with the surface oxygen to form CO2 and vacancies, where H2O dissociated to form H2. Following this mechanism, the produced H2 could be detected during the H2O
dissociated process. For the associative mechanism, CO and H2O adsorbed on the surface of the catalyst to form intermediate, and then the intermediate further decomposed to produce CO2 and
H2 simultaneously. In that case, H2O tended to form surface hydroxyl species, which could react with CO20,21. Here, the H2O dissociation experiment was conducted (Supplementary Fig. 11). No
H2 was produced in either the thermostatic test or the temperature-programmed heating test, ruling out the existence of redox mechanism. Furthermore, the temperature programmed surface
reaction (TPSR) experiment was performed. The results showed a wide range of desorption signals of CO2 (Supplementary Fig. 12), which was caused by the oxidation of CO with surface O. The
strong adsorption of CO was also found in the temperature-programmed desorption (TPD) using Ar as the carrier gas (Supplementary Fig. 13) and CO kinetic order (Supplementary Fig. 14c). From
the results of transient isothermal surface reaction (250 °C, Fig. 5a) after WGS reaction, it could be observed that the concentration ratio of CO2/H2 was ~2 when the reaction reached a
stable state. It suggested that H2O might be dissociated to OH* first, and then OH* reacted with CO to generate CO2 and H*, following the associative mechanism20,21 according to 2 OH + 2 CO
→ H2 + 2 CO2. In addition, the cyclic CO temperature-programmed reduction (CO-TPR) was used to verify the production of hydroxyl36. As shown in Fig. 5b, the catalyst was treated in the WGS
reaction atmosphere at room temperature between two CO-TPR experiments. For the CO-TPR-1, there were mainly three reduction peaks, among which the one at lower temperature (182 °C) was the
reduction of small-size NiO, and the one at higher temperature (413 °C) should be the reduction of large-size NiO. The reduction peak at 253 °C was accompanied by the generation of H2, which
resulted from the reaction between CO and hydroxyl species20,21,36. The formation of hydroxyl was caused by the dissociation of H2O in the WGS reaction before CO-TPR-1. For CO-TPR-2, two
main reduction peaks could be observed, among which the higher temperature reduction peak was considered as the reduction of NiO_x_ species formed in the reaction atmosphere. This reduction
was similar to the H2-TPR result of the used sample (Supplementary Fig. 15a). Accordingly, the reduction peak at 252 °C was also accompanied by the generation of H2 signal, indicating the
generation of surface hydroxyl was recyclable. The same result was also observed for the catalyst treated in the H2O/Ar atmosphere between CO-TPR-1 and CO-TPR-2 (Supplementary Fig. 15b). In
order to ensure that pure Y2O3 did not contribute to the dissociation of H2O, we demonstrated this by activity test, in situ Raman and H2O dissociation experiments of pure Y2O3
(Supplementary Fig. 16). It was believed that the interface played an important role in the dissociation of H2O. We further calculated the activation of the H2O (300 °C) at different surface
locations to verify the hypothesis of interfacial active sites. H2O adsorbed weakly on metallic Ni surface (Ni{111}). When the partial pressure of H2O was equal to 10 kPa, the _∆G_ of
adsorbing on the top position and the bridge position were 0.516 eV and 0.758 eV, respectively. (Supplementary Fig. 17). However, the adsorption of H2O molecules in the Y3O4/NiO_x_/Ni{111}
model was completely different. The _∆G_ of H2O adsorption at the three Y sites were preponderant (−0.428, −0.468 and 0.219 eV) because Y2O3 was a stronger Lewis acid than Ni (Fig. 5c). The
coordination numbers of the Y atom at these three sites were 5, 4, and 4 in turn, and the Bader charges value were 1.84, 2.01 and 2.01, respectively. This was agreement with the EXAFS result
of the used sample (the C.N. for Y-O was 5.155 ± 0.392). This result indicated that the lower coordination number (the C.N. of Y atom in Y2O3 was 6) and higher positive charge of Y atoms
led to its stable binding with H2O molecules. Besides, two models (the Ni{111} and Y3O4/NiO_x_/Ni{111}) for H2O dissociation were also different. For the Ni{111}, although the adsorbed H2O
molecules dissociated to *OH and *H were the process of decreased _∆G_, the whole process of H2O adsorption and dissociation was still a non-spontaneous process (∆_G_ > 0) (Supplementary
Fig. 17). For the Y3O4/NiO_x_/Ni{111} model, although the H2O adsorption at site I (−0.428 eV), site II (−0.468 eV) and site III (0.219 eV) were various, the Gibbs free energies of the
decomposition products (IMI-2, IMII-2 and IMIII-2) were significantly lower than those of the Y3O4/NiO_x_/Ni{111} plus vapor, i.e., the ∆_G_ were down around 0.6 eV (Fig. 5c). In general,
the Ni-NiO_x_-Y2O3 interface was much more favorable for the activation of H2O than pure Ni. The results of H2O reaction order measurements also supported the same conclusion (Supplementary
Fig. 14a, b). The H2O reaction order of the pure Ni sample (0.57 at 270 °C) was obviously larger than that of the Ni9Y1O_x_ sample (0.32 at 270 °C), indicating that the latter was easier to
adsorb and dissociate H2O. The whole reaction path of the WGS reaction was simulated and presented in Fig. 5d and Supplementary Table 7. H2O was adsorbed on the atom Y at the first step and
CO was adsorbed on the Ni atom at third step. The rate-determining step was the fourth elementary reaction, i.e., *OH migration from atom Y to atom C, and the most probable cause was the
bonding force of CO were too strong. As a whole, the WGS reaction was spontaneous because the sign of ∆_G_ was negative (−1.476 eV at 300 °C, the partial pressure of each gas species was set
to the value at the outlet of reactor, shown as Supplementary Table 6). In addition, the ∆_G_ at the inlet of reactor were also provided and the larger spontaneous tendency was presented
for the lower product concentrations (shown as Supplementary Fig. 18 and Supplementary Table 8). From the conclusion of the reaction pathway study, we proposed the associative reaction
mechanism involving the NiO_x_ species at the interface (Supplementary Fig. 19). The adsorbed H2O molecules first dissociated at the interface NiO_x_ site to generate two OH* molecules, and
then the active hydroxyl species further reacted with the adsorbed CO to generate CO2 and H2. Therefore, the reactive Ni-NiO_x_-Y2O3 interfacial site enabled the catalyst to efficiently
catalyze the WGS reaction and largely improved its reaction efficiency. DISCUSSION In summary, the addition of Y2O3 was essential for the active Ni-NiO_x_-Y2O3 interface in WGS reaction.
Compared to pure Ni sample, the presence of Y2O3 tremendously improved the catalytic activity and stability, enabling efficient WGS reaction at medium temperature range. Based on
experimental investigation and DFT calculation, the Ni-NiO_x_-Y2O3 interface effectively dissociated H2O molecules and facilitated the reaction with associative mechanism. Y2O3 and other
rare earth metal oxide system should play an important role in catalytic reactions, in particular those with Lewis base molecules such as H2O and NH3. This will be a golden era for the
adventure of rare earth catalysis. METHODS SYNTHESIS OF CATALYSTS In a typical synthesis of Ni_a_Y_b_O_x_, 4 mmol nitrates (99%, Tianjin Kermal Factory) were added to 60 mL anhydrous
ethanol. The added mass of Ni(NO3)2·6H2O and Y(NO3)3·6H2O was according to the molar ratio of Ni to Y (1: 9, 3: 7, 5: 5, 7: 3, 9: 1 and 9.5: 0.5). The mixture was stirred at room temperature
for 10 minutes and then dispersed by ultrasound for 10 minutes. Then, N2 flow was used to take small droplets atomized by the atomizer into the high-temperature tubular furnace (450 °C) for
decomposition reaction, and the samples were pumped to the receiver for collection. The collected samples were dried overnight in an oven at 60 °C, and then calcined at 400 °C (2 °C min−1
of ramping rate). The obtained sample was named Ni_a_Y_b_O_x_, where a: b was the molar ratio of Ni and Y. Other composite catalysts (Ni9Ce1O_x_, Ni9Sm1O_x_ and Ni9Al1O_x_) and oxides (NiO
and Y2O3) followed the same synthetic method with Ni_a_Y_b_O_x_, where only nitrates were altered. N2 ADSORPTION-DESORPTION MEASUREMENTS The N2 adsorption-desorption measurements was
proceeded on a Builder SSA-4200 surface area analyzer at 77 K after degased the Ni_a_Y_b_O_x_ catalysts at 200 °C for 6 h under vacuum. The specific surface area (_S__BET_) was calculated
from Brunauer-Emmett-Teller (BET) method. TRANSMISSION ELECTRON MICROSCOPY (TEM) The TEM images were taken on a JEOL JEM-2100F microscope operating at 100 kV. The High-angle annular
dark-field scanning transmission electron microscopy (HAADF-STEM) images were obtained on a JEOL ARM200F microscope equipped with a probe-forming spherical-aberration corrector and Gatan
image filter (Quantum 965). The element mapping results were derived from the electronic energy-loss spectroscopy (EELS) analysis equipped with STEM model. SCANNING ELECTRON MICROSCOPY (SEM)
The SEM images were taken on a Zeiss SUPRA 55 scanning microscope with the acceleration voltage of 5.0 kV. X-RAY DIFFRACTION (XRD) Both ex situ and in situ modes were carried out on a
PANalytical X’pert3 powder diffractometer (40 kV, 40 mA) using Cu Kα radiation (λ = 0.15406 nm). The powder samples were placed inside a quartz holder and the diffraction angles (2_θ_)
ranged from 10 to 90°. For the in situ XRD experiments, 100 mg catalysts were loaded into an Anton Paar XRK900 in situ chamber. The XRD data was measured from 25 to 450 °C in 5% H2/Ar
mixture (30 mL min−1) or 2% CO/Ar mixture (30 mL min−1). X-RAY PHOTOELECTRON SPECTROSCOPY (XPS) XPS characterization was carried out on a Thermo scientific ESCALAB Xi+ XPS spectrometer with
Al Kα radiations, and with the C 1 _s_ peak at 284.8 eV as an internal standard for all the spectra. For the quasi in situ XPS experiments, 20 mg catalysts were loaded into an HPGC 300
reaction chamber made by Fermion instruments (Shanghai) Co., LTD. The XPS data was measured after the in situ WGS reaction (400 °C, 2%CO/~3%H2O in Ar, 15 mL min−1). X-RAY ABSORPTION FINE
STRUCTURE (XAFS) Y _K_-edge (17.038 keV) XAFS was performed at the BL01B1 beamline of Super Photon ring − 8 GeV (SPring-8, Japan). XAFS spectra (16.714–18.499 keV) were measured in
transmission mode using Si (311) DCM and ion chambers. Y2O3 was used for energy shift calibration. XAFS data was analyzed using Demeter software package (including Athena and Artemis,
version 0.9.26). Athena was used for data normalization and Artemis was used for EXAFS fitting. TEMPERATURE-PROGRAMMED REDUCTION The temperature-programmed reduction by H2 (H2-TPR) was
carried on a Builder PCSA-1000 instrument. 30 mg catalysts (20 − 40 mesh) were pretreated in air at 300 °C for 0.5 h and then purged with Ar. The final test was carried out in 5%H2/Ar (30 mL
min−1) from room temperature to 700 °C. The temperature-programmed reduction by CO (CO-TPR) was carried on an online mass spectrometer (TILON LC-D200M). 100 mg catalysts (20 − 40 mesh) were
pretreated by 5%H2/Ar (30 mL min−1) at 400 °C for 0.5 h and then purged with Ar. After the WGS reaction at 300 °C for 2 h, the catalyst was heated from room temperature to 800 °C in the
2%CO/Ar (30 mL min−1) to obtain the result of CO-TPR-1. Next, the sample were cooled to room temperature and switched to the retreatment gas (2%CO/~3%H2O in Ar at RT, 30 mL min−1; or ~3%H2O
in Ar at 300 °C, 30 mL min−1) for 0.5 h. Then, the catalyst was heated from room temperature to 800 °C to obtain the result of CO-TPR-2. IN SITU RAMAN SPECTROSCOPY All the Raman spectra were
collected from a LabRAM HR800 spectrometer (HORIBA JY) with 632.8 nm laser. Samples were placed on the micro-Raman reaction cell (Xiamen TOPS) and Raman shift ranged from 200 to 800 cm−1
with a spectral resolution of 2 cm−1. For the first mode, the samples were pretreated by 5%H2/Ar (30 mL min−1) at 400 °C for 0.5 h, and then cooled to room temperature. Next, WGS reaction
gas (2%CO/~3%H2O in Ar, 30 mL min−1) was fed and the samples were heated from room temperature to 400 °C. The second mode was the switch experiment. After pretreated in 5%H2/Ar (30 mL min−1)
at 400 °C, the samples were purged with two kinds of feeds (2%CO/Ar, 30 mL min−1; or ~3%H2O in Ar, 30 mL min−1) at 400 °C in turn. TEMPERATURE-PROGRAMMED SURFACE REACTION (TPSR) The TPSR
experiment was measured by the self-constructed device, using an online mass spectrometer (TILON LC-D200M) as the detector. The experiments were mainly divided into two parts. In the first
part, the H2O dissociation experiment, 100 mg catalysts (20 − 40 mesh) were pretreated by 5%H2/Ar (30 mL min−1) at 400 °C for 0.5 h and then purged with Ar. After WGS reaction at 300 °C for
2 h, the samples were heated from room temperature to 600 °C under the ~3%H2O/Ar atmosphere to get the heating process H2O dissociation result. The constant temperature H2O dissociation
experiment process was carried out after the WGS reaction. ~3%H2O/Ar was fed at a constant temperature (300, 350 and 400 °C) to collect the results. The second part was the reaction of CO
with surface OH. After the same pretreatment and in situ reaction process, Ar purge was carried out at 300 °C for 2 h. Then, the samples were cooled to room temperature and switched to the
2%CO/Ar. Next, the results were collected at constant temperature (250 °C). THE DENSITY FUNCTIONAL THEORY (DFT) CALCULATIONS The heterogeneous catalysis simulations were carried out using
the Vienna ab Initio simulation package55,56,57,58. The Perdew−Burke−Ernzerhof (PBE) exchange−correlation functional and the projector augmented-wave (PAW) pseudopotential were applied to
spin-unrestricted geometry optimizations58,59,60. The cutoff energy for the plane wave basis was set to 400 eV. The convergence threshold of the electronic self-consistency was specified as
1.0×10−6 eV, and the total energy change of the whole catalyst system between two ionic relaxation steps was designated as less than 0.02 eV. The cleaved Ni {111} surface comprised a 5-layer
slab (the bottom 2 layers were fixed) as well as a vacuum layer of 15 Å. The lattice parameters of the Ni {111} surface were a = b = 12.4592 Å, c = 23.1383 Å, α = β = 90° and γ = 120°, and
the number of Ni atoms was 125. Four oxygen atoms were positioned on the top layer of Ni to simulate NiO_x_, then Y3O4 cluster was loaded on NiO_x_ layer, shown as Supplementary Fig. 8. As a
group, the formula of the Y3O4/NiOx/Ni{111} model was Ni125O8Y3. The binding force between Y3O4 cluster and NiO_x_ was Y-O or Ni-O ionic bonds, and the NiO_x_ layer and Ni{111} was bound by
metallic bond. CATALYTIC PERFORMANCE MEASUREMENT The WGS activities of the Ni_a_Y_b_O_x_ catalysts were tested in a self-constructed fixed-bed flow reactor. The temperature controller
(UDIAN, XIAMEN YUDIAN AUTOMATION TECHNOLOGY CO., LTD.) was used in the reactor temperature control system. Both the actual temperature of the furnace temperature and the location of the
catalyst were detected. The gas path system consisted of two parts: CO was the standard gas, and H2O was generated by vaporization after quantitative pumping by water injection pumps. The
actual reaction gas content was 2%CO, 10%H2O, and the equilibrium gas was N2. During the test, 100 mg catalysts (20 − 40 mesh) were filled with gas hourly space velocity (GHSV) was 42,000
cm3 gcat −1 h−1. The catalysts were pretreated in a 5% H2/N2 at 400 °C. After a cooling system, the dry gas entered the gas analyzer (Gasboard-3100, Wuhan Sifang Corp), and then the
real-time CO, CO2 and CH4 contents were obtained. The CO conversion and CH4 selectivity were calculated through Eq. (1) and Eq. (2), respectively.
$${X}_{{{{{{\rm{CO}}}}}}}=\frac{{n}_{{{{{{\rm{CO}}}}}}}^{{{{{{\rm{in}}}}}}}-{n}_{{{{{{\rm{CO}}}}}}}^{{{{{{\rm{out}}}}}}}}{{n}_{{{{{{\rm{CO}}}}}}}^{{{{{{\rm{in}}}}}}}}\times 100\%$$ (1)
$${S}_{{{{{{{\rm{CH}}}}}}}_{4}}=\frac{{n}_{{{{{{{\rm{CH}}}}}}}_{4}}^{{{{{{\rm{out}}}}}}}}{{n}_{{{{{{{\rm{CO}}}}}}}_{2}+}^{{{{{{\rm{out}}}}}}}{n}_{{{{{{{\rm{CH}}}}}}}_{4}}^{{{{{{\rm{out}}}}}}}}\times
100\%$$ (2) $${{{{{\rm{TOF}}}}}}=\frac{{{{{{\rm{CO}}}}}}\; {{{{{\rm{converted}}}}}}\,({{{{{\rm{mol}}}}}})}{{{{{{\rm{Ni}}}}}}\; {{{{{\rm{atom}}}}}}\; {{{{{\rm{at}}}}}}\;
{{{{{\rm{interface}}}}}}\,\left({{{{{\rm{mol}}}}}}\right)\times {{{{{\rm{time}}}}}}\,({{{{{\rm{s}}}}}})}$$ (3) The stability test of the Ni9Y1O_x_ catalyst was conducted at 300 °C (GHSV =
420,000 cm3 gcat −1 h−1) for 100 h. The reaction rate was measured using 5 mg catalysts mixed with 200 mg quartz sand, and the CO conversion was controlled below 15%. The turnover frequency
(TOF) value were calculated according to the Eq. (3). The kinetic order of CO and H2O was measured by adjusting the content of CO and H2O in the mixed gas at constant temperature (240−280
°C). DATA AVAILABILITY The main data supporting the findings of this study are available within the article and its Supplementary Information. All other relevant source data are available
from the corresponding author upon reasonable request. REFERENCES * Tauster, S. J., Fung, S. C. & Garten, R. L. Strong metal-support interactions. Group 8 noble metals supported on
titanium dioxide. _J. Am. Chem. Soc._ 100, 170–175 (1978). Article CAS Google Scholar * Farmer, J. A. et al. Ceria maintains smaller metal catalyst particles by strong metal-support
bonding. _Science_ 329, 933–936 (2010). Article ADS CAS PubMed Google Scholar * Cargnello, M. et al. Control of metal nanocrystal size reveals metal-support interface role for ceria
catalysts. _Science_ 341, 771–773 (2013). Article ADS CAS PubMed Google Scholar * Du, X. et al. Size-dependent strong metal-support interaction in TiO2 supported Au nanocatalysts. _Nat.
Commun._ 11, 5811 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Wang, H. et al. Strong metal–support interactions on gold nanoparticle catalysts achieved through Le
Chatelier’s principle. _Nat. Catal._ 4, 418–424 (2021). Article CAS Google Scholar * Bahmanpour, A. M. et al. Cu-Al spinel as a highly active and stable catalyst for the reverse water gas
shift reaction. _ACS Catal._ 9, 6243–6251 (2019). Article CAS Google Scholar * Fan, R. et al. Synergistic catalysis of cluster and atomic copper induced by copper-silica interface in
transfer-hydrogenation. _Nano Res._ 14, 4601–4609 (2021). Article ADS CAS Google Scholar * Tang, H. L. et al. Classical strong metal-support interactions between gold nanoparticles and
titanium dioxide. _Sci. Adv._ 3, e1700231 (2017). Article ADS PubMed PubMed Central CAS Google Scholar * Wei, S. et al. The effect of reactants adsorption and products desorption for
Au/TiO2 in catalyzing CO oxidation. _J. Catal._ 376, 134–145 (2019). Article CAS Google Scholar * Matsubu, J. C. et al. Adsorbate-mediated strong metal-support interactions in
oxide-supported Rh catalysts. _Nat. Chem._ 9, 120–127 (2017). Article CAS PubMed Google Scholar * Xu, M. et al. TiO2–x-modified Ni nanocatalyst with tunable metal–support interaction for
water–gas shift reaction. _ACS Catal._ 7, 7600–7609 (2017). Article CAS Google Scholar * Xu, M. et al. Insights into interfacial synergistic catalysis over Ni@TiO2–x catalyst toward
water–gas shift reaction. _J. Am. Chem. Soc._ 140, 11241–11251 (2018). Article CAS PubMed Google Scholar * Herzing, A. A. et al. Identification of active gold nanoclusters on iron oxide
supports for CO oxidation. _Science_ 321, 1328–1331 (2008). Article ADS CAS Google Scholar * Guo, Y. et al. Uniform 2 nm gold nanoparticles supported on iron oxides as active catalysts
for CO oxidation reaction: structure–activity relationship. _Nanoscale_ 7, 4920–4928 (2015). Article ADS CAS PubMed Google Scholar * Lin, J. et al. Remarkable performance of Ir1/FeOx
single-atom catalyst in water gas shift reaction. _J. Am. Chem. Soc._ 135, 15314–15317 (2013). Article CAS PubMed Google Scholar * Zhang, S. et al. WGS catalysis and in situ studies of
CoO1–x, PtCon/Co3O4, and PtmCom′/CoO1–x nanorod catalysts. _J. Am. Chem. Soc._ 135, 8283–8293 (2013). Article CAS PubMed Google Scholar * Yu, Y. et al. Pretreatments of Co3O4 at moderate
temperature for CO oxidation at –80 °C. _J. Catal._ 267, 121–128 (2009). Article CAS Google Scholar * Lykhach, Y. et al. Counting electrons on supported nanoparticles. _Nat. Mater._ 15,
284–288 (2016). Article ADS CAS PubMed Google Scholar * Campbell, C. T. Catalyst–support interactions: electronic perturbations. _Nat. Chem._ 4, 597–598 (2012). Article CAS PubMed
Google Scholar * Fu, X. et al. Direct identification of active surface species for the water-gas shift reaction on a gold-ceria catalyst. _J. Am. Chem. Soc._ 141, 4613–4623 (2019). Article
CAS PubMed Google Scholar * Yan, H. et al. Construction of stabilized bulk-nano interfaces for highly promoted inverse CeO2/Cu catalyst. _Nat. Commun._ 10, 3470 (2019). Article ADS
PubMed PubMed Central CAS Google Scholar * Chen, A. et al. Structure of the catalytically active copper–ceria interfacial perimeter. _Nat. Catal._ 2, 334–341 (2019). Article CAS Google
Scholar * Si, R. & Flytzani-Stephanopoulos, M. Shape and crystal-plane effects of nanoscale ceria on the activity of Au-CeO2 catalysts for the water-gas shift Reaction. _Angew. Chem.
Int. Ed._ 47, 2884–2887 (2008). Article CAS Google Scholar * Kang, L. et al. Adsorption and activation of molecular oxygen over atomic copper(I/II) site on ceria. _Nat. Commun._ 11, 4008
(2020). Article ADS CAS PubMed PubMed Central Google Scholar * Kang, L. et al. The Electrophilicity of surface carbon species in the redox reactions of CuO-CeO2 catalysts. _Angew.
Chem. Int. Ed._ 60, 14420–14428 (2021). Article CAS Google Scholar * Ratnasamy, T. et al. Water gas shift catalysis. _Catal. Rev. Sci. Eng._ 51, 325–440 (2009). Article CAS Google
Scholar * Franchini, C. A. et al. Single-stage medium temperature water-gas shift reaction over Pt/ZrO2 – support structural polymorphism and catalyst deactivation. _Appl. Catal. B:
Environ._ 117–118, 302–309 (2012). Article CAS Google Scholar * Galletti, C. et al. Gold-supported catalysts for medium temperature-water gas shift reaction. _Top. Catal._ 52, 688–692
(2009). Article CAS Google Scholar * Ashok, J., Wai, M. H. & Kawi, S. Nickel-based catalysts for high-temperature water gas shift reaction-methane suppression. _ChemCatChem_ 10,
3927–3942 (2018). Article CAS Google Scholar * Lin, J. et al. Hydrogen production by water–gas shift reaction over bimetallic Cu–Ni catalysts supported on La-doped mesoporous ceria.
_Appl. Catal. A: Gen._ 387, 87–94 (2010). Article CAS Google Scholar * Ang, M. L. et al. Highly active Ni/xNa/CeO2 catalyst for the water–gas shift reaction: effect of sodium on methane
suppression. _ACS Catal._ 4, 3237–3248 (2014). Article CAS Google Scholar * Ang, M. L. et al. High-temperature water–gas shift reaction over Ni/xK/CeO2 catalysts: suppression of
methanation via formation of bridging carbonyls. _J. Catal._ 329, 130–143 (2015). Article CAS Google Scholar * Saw, E. T. et al. Bimetallic Ni–Cu catalyst supported on CeO2 for
high-temperature water–gas shift reaction: methane suppression via enhanced CO adsorption. _J. Catal._ 314, 32–46 (2014). Article CAS Google Scholar * Dongil, A. B. et al. Promoter effect
of sodium in graphene-supported Ni and Ni–CeO2 catalyst for the low-temperature WGS reaction. _Appl. Catal. A: Gen._ 505, 98–104 (2015). Article CAS Google Scholar * Chayakul, K.,
Srithanratana, T. & Hengrasmee, S. Catalytic activities of Re–Ni/CeO2 bimetallic catalysts for water gas shift reaction. _Catal. Today_ 175, 420–429 (2011). Article CAS Google Scholar
* Ashok, J. et al. Promotion of the water-gas-shift reaction by nickel hydroxyl species in partially reduced nickel-containing phyllosilicate catalysts. _ChemCatChem_ 8, 1308–1318 (2016).
Article CAS Google Scholar * Jin, Z. et al. Metal nanocrystal-embedded hollow mesoporous TiO2 and ZrO2 microspheres prepared with polystyrene nanospheres as carriers and templates. _Adv.
Funct. Mater._ 23, 2137–2144 (2013). Article CAS Google Scholar * Kuai, L. et al. A reliable aerosol-spray-assisted approach to produce and optimize amorphous metal oxide catalysts for
electrochemical water splitting. _Angew. Chem._ 126, 7677–7681 (2014). Article ADS Google Scholar * Li, Y., Fu, Q. & Flytzani-Stephanopoulos, M. Low-temperature water-gas shift
reaction over Cu- and Ni-loaded cerium oxide catalysts. _Appl. Catal. B_ 27, 179–191 (2000). Article Google Scholar * Yan, H. et al. Promoted Cu-Fe3O4 catalysts for low-temperature water
gas shift reaction: optimization of Cu content. _Appl. Catal. B_ 226, 182–193 (2018). Article CAS Google Scholar * Panagiotopoulou, P. & Kondarides, D. I. Effects of alkali promotion
of TiO2 on the chemisorptive properties and water–gas shift activity of supported noble metal catalysts. _J. Catal._ 267, 57–66 (2009). Article CAS Google Scholar * Barve, S. A. et al.
Microwave ECR plasma CVD of cubic Y2O3 coatings and their characterization. _Surf. Coat. Technol._ 204, 3167–3172 (2010). Article CAS Google Scholar * Narasimharao, K. & Al-Sultan, F.
S. Y2O3 modified Au-La2O3 nanorod catalysts for oxidative cracking of n-propane. _Fuel_ 280, 118599 (2020). Article CAS Google Scholar * Reddy, I. N. et al. Structural, optical, and XPS
studies of doped yttria for superior water splitting under visible light illumination. _J. Electroanal. Chem._ 848, 113335 (2019). Article CAS Google Scholar * Malvestuto, M. et al. X-ray
absorption study of the growth of Y2O3 on Si(001). _Phys. Rev. B_ 71, 075318 (2005). Article ADS CAS Google Scholar * Mironova-Ulmane, N. et al. Raman scattering in nanosized nickel
oxide NiO. _J. Phys. Conf. Ser._ 93, 012039 (2007). Article CAS Google Scholar * Sunny, A. & Balasubramanian, K. Raman spectral probe on size-dependent surface optical phonon modes
and magnon poperties of NiO nanoparticles. _J. Phys. Chem. C._ 124, 12636–12644 (2020). Article CAS Google Scholar * Bala, N. et al. Magnetic-order induced effects in nanocrystalline NiO
probed by Raman spectroscopy. _Phys. Rev. B_ 102, 024423 (2020). Article ADS CAS Google Scholar * Lai, W. et al. In situ Raman spectroscopic study towards the growth and excellent HER
catalysis of Ni/Ni(OH)2 heterostructure. _Int. J. Hydrog. Energy_ 46, 26861–26872 (2021). Article CAS Google Scholar * Pan, B. et al. Raman shift, Néel temperature, and optical band gap
of NiO nanoparticles. _Phys. Chem. Chem. Phys._ 22, 5735 (2020). Article CAS PubMed Google Scholar * Gokhale, A. A., Dumesic, J. A. & Mavrikakis, M. On the mechanism of
low-temperature water gas shift reaction on copper. _J. Am. Chem. Soc._ 130, 1402–1414 (2008). Article CAS PubMed Google Scholar * Kalamaras, C. M., Americanou, S. & Efstathiou, A.
M. “Redox” vs “associative formate with –OH group regeneration” WGS reaction mechanism on Pt/CeO2: Effect of platinum particle size. _J. Catal._ 279, 287–300 (2011). Article CAS Google
Scholar * Mudiyanselage, K. et al. Importance of the metal-oxide interface in catalysis: In situ studies of the water-gas shift reaction by ambient-pressure X-ray photoelectron
spectroscopy. _Angew. Chem. Int. Ed._ 52, 5101–5105 (2013). Article CAS Google Scholar * Aranifard, S., Ammal, S. C. & Heyden, A. On the importance of metal–oxide interface sites for
the water–gas shift reaction over Pt/CeO2 catalysts. _J. Catal._ 309, 314–324 (2014). Article CAS Google Scholar * Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid
metals. _Phys. Rev. B_ 47, 558–561 (1993). Article ADS CAS Google Scholar * Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a
plane-wave basis set. _Phys. Rev. B_ 54, 11169–11186 (1996). Article ADS CAS Google Scholar * Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the
liquid-metal-amorphous-semiconductor transition in germanium. _Phys. Rev. B_ 49, 14251–14269 (1994). Article ADS CAS Google Scholar * Kresse, G. & Joubert, D. From ultrasoft
pseudopotentials to the projector augmented-wave method. _Phys. Rev. B_ 59, 1758–1775 (1999). Article ADS CAS Google Scholar * Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized
gradient approximation made simple. _Phys. Rev. Lett._ 77, 3865–3868 (1996). Article ADS CAS PubMed Google Scholar * Blöchl, P. E. Projector augmented-wave method. _Phys. Rev. B_ 50,
17953–17979 (1994). Article ADS Google Scholar Download references ACKNOWLEDGEMENTS This work was financially supported from the National Science Foundation of China (no. 21771117,
21805167, 22075166), the Taishan Scholar Project of Shandong Province of China, the Young Scholars Program of Shandong University (grant nos. 11190089964158), EPSRC (EP/P02467X/1 and
EP/S018204/2), Royal Society (RG160661, IES\R3\170097, IES\R1\191035, IEC\R3\193038). We acknowledge SPring-8 (Japan) for the XAFS experiments conducted under the proposal no. 2021A1387 and
Dr. Hiroyuki Asakura from Kyoto University for helping with the XAFS measurement. We thank the Center of Structural Characterizations and Property Measurements at Shandong University for the
help on sample characterizations. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Key Laboratory for Colloid and Interface Chemistry, Key Laboratory of Special Aggregated Materials, School of
Chemistry and Chemical Engineering, Shandong University, Jinan, 250100, China Kai Xu, Han Yan, Wei-Wei Wang, Qing-Lu Meng, Wei-Peng Shao, Guo-Heng Ding & Chun-Jiang Jia * College of
Materials Science and Engineering, Hunan University, Changsha, 410082, China Chao Ma * Department of Chemical Engineering, University College London, Roberts Building, Torrington Place,
London, WC1E 7JE, UK Hao Gu & Feng Ryan Wang * Key Laboratory of Micro-Nano Powder and Advanced Energy Materials of Anhui Higher Education Institutes, Chizhou University, Chizhou,
247000, China Shan-Qing Li Authors * Kai Xu View author publications You can also search for this author inPubMed Google Scholar * Chao Ma View author publications You can also search for
this author inPubMed Google Scholar * Han Yan View author publications You can also search for this author inPubMed Google Scholar * Hao Gu View author publications You can also search for
this author inPubMed Google Scholar * Wei-Wei Wang View author publications You can also search for this author inPubMed Google Scholar * Shan-Qing Li View author publications You can also
search for this author inPubMed Google Scholar * Qing-Lu Meng View author publications You can also search for this author inPubMed Google Scholar * Wei-Peng Shao View author publications
You can also search for this author inPubMed Google Scholar * Guo-Heng Ding View author publications You can also search for this author inPubMed Google Scholar * Feng Ryan Wang View author
publications You can also search for this author inPubMed Google Scholar * Chun-Jiang Jia View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
C.-J.J. and F.R.W. supervised the work; K.X., H.Y., F.R.W. and C.-J.J. designed the experiments, analyzed the results and wrote the manuscript; K.X. and W.-W.W. performed the in situ XRD, in
situ Raman and quasi in situ XPS; S.-Q.L. performed the DFT calculation; K.X., Q.-L.M., W.-P.S. and G.-H.D. performed the catalysts preparation, catalytic tests and the TPR tests; C.M.
performed the aberration-corrected HAADF-STEM measurements and analyzed the results. H.G. and F.R.W. performed the XAFS experiments and analyzed the data. CORRESPONDING AUTHORS
Correspondence to Feng Ryan Wang or Chun-Jiang Jia. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature
Communications_ thanks Antonio Sepúlveda-Escribano, Jangam Ashok and the other, anonymous, reviewer for their contribution to the peer review of this work. Peer reviewer reports are
available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0
International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the
source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Xu, K., Ma, C., Yan, H. _et al._ Catalytically efficient Ni-NiO_x_-Y2O3 interface
for medium temperature water-gas shift reaction. _Nat Commun_ 13, 2443 (2022). https://doi.org/10.1038/s41467-022-30138-5 Download citation * Received: 18 September 2021 * Accepted: 28 March
2022 * Published: 04 May 2022 * DOI: https://doi.org/10.1038/s41467-022-30138-5 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable
link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative