- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT RNA polymerase inhibition plays an important role in the regulation of transcription in response to environmental changes and in the virus-host relationship. Here we present the
high-resolution structures of two such RNAP-inhibitor complexes that provide the structural bases underlying RNAP inhibition in archaea. The Acidianus two-tailed virus encodes the RIP factor
that binds inside the DNA-binding channel of RNAP, inhibiting transcription by occlusion of binding sites for nucleic acid and the transcription initiation factor TFB. Infection with the
Sulfolobus Turreted Icosahedral Virus induces the expression of the host factor TFS4, which binds in the RNAP funnel similarly to eukaryotic transcript cleavage factors. However, TFS4
allosterically induces a widening of the DNA-binding channel which disrupts trigger loop and bridge helix motifs. Importantly, the conformational changes induced by TFS4 are closely related
to inactivated states of RNAP in other domains of life indicating a deep evolutionary conservation of allosteric RNAP inhibition. SIMILAR CONTENT BEING VIEWED BY OTHERS THE HOST RNA
POLYMERASE II C-TERMINAL DOMAIN IS THE ANCHOR FOR REPLICATION OF THE INFLUENZA VIRUS GENOME Article Open access 05 February 2024 STRUCTURAL BASIS OF NIPAH VIRUS RNA SYNTHESIS Article Open
access 06 March 2025 PRE-INITIATION AND ELONGATION STRUCTURES OF FULL-LENGTH LA CROSSE VIRUS POLYMERASE REVEAL FUNCTIONALLY IMPORTANT CONFORMATIONAL CHANGES Article Open access 17 July 2020
INTRODUCTION RNA polymerases (RNAPs) are the engines of transcription and an important target for the regulation of gene expression relevant for health and disease. Gene-specific factors
regulate transcription dependent on promoter DNA sequence motifs, while global repression or attenuation of all RNAs is enabled by regulators that directly interact with RNAP independently
of the template DNA1,2. The underlying mechanisms of RNAP inhibition are not only of academic interest but also in the context of the emerging antibiotic resistance crisis, since many
frontline antibiotics including Rifampicin are RNAP inhibitors3. By exploring the structural bases of RNAP inhibition we not only learn about the mechanisms of transcription, but also inform
the rational design of novel antibiotics4. In this context, viral transcription factors that inhibit the host RNAP are a rich hunting ground. Viruses are the most abundant pathogens found
in nature and important drivers of evolution because of their ability to facilitate horizontal gene transfer5. Once viruses have infected a cell, they are subjected to similar needs: to
bypass cellular immunity and to rewire the host gene expression machinery for their own benefit—chiefly to produce virus particles. Well studied bacteriophages like T7 prevent the expression
of host genes by inhibiting the host transcription machinery, and replace it with virus-encoded components that exclusively transcribe viral genes. The bacteriophage T7 factors Gp2 and P7
target the RNAP β′ subunit directly and not only interfere with σ70 binding but also alter the conformational dynamics of RNAP during open complex formation and DNA melting, an essential
step during early transcription initiation6. Inhibition of host transcription is an obvious strategy for viruses that encode their own RNAP, but archaeal viruses are entirely dependent on
the host transcription machinery, as their genomes do not encode recognisable RNAP subunit genes. Hence, viral promoters utilise host promoter motifs which are active in in vitro
transcription experiments using host RNAP and general initiation factors7. We have recently characterised the molecular mechanisms of the highly toxic RNAP inhibitor RIP (RNAP Inhibitory
Protein, aka ORF145) encoded by the Acidianus two-tailed virus (ATV) that infects crenarchaea including _Sulfolobus_ species7. RIP has undergone a fascinating functional diversification; it
is evolutionary related to a capsid protein ORF131, but it binds RNAP with high affinity and efficiently inhibits transcription initiation and elongation7. But the global inhibition of RNAP
is not restricted to virally-encoded factors, as a plethora of cellular factors bind directly to and inhibit RNAP in response to unfavourable environmental conditions including stress. The
host encoded and constitutively expressed Gfh1 (Gre-factor homologue-1)8 and DksA (DnaK suppressor A)9 are inhibitors of bacterial RNAP that enable the fine-tuning of transcription, and
belong to the group of factors that act through the nucleotide triphosphates (NTPs) entry funnel of RNAP, or secondary channel10. These also include positive regulators such as the
eukaryotic RNAPII TFIIS11, archaeal TFS12,13 and bacterial GreA/B14,15 that enhance elongation and resolve stalled and backtracked transcription elongation complexes (TEC) by transcript
cleavage. We have previously described the molecular mechanisms of the archaeal TFS paralogue TFS4 as RNAP inhibitor16. TFS4 destabilises RNAP–DNA complexes and inhibits catalysis by
decreasing the affinity for NTP substrates16. Intriguingly, TFS4 cannot be detected in the cell during normal growth conditions, but it is strongly upregulated in response to viral infection
with STIV (Sulfolobus Turreted Icosahedral Virus)17. We have applied single particle cryo-electron microscopy (cryo-EM) to characterise the structural bases of RNAP inhibition in archaea.
Here, we present the high-resolution structures of the inhibitors RIP and TFS4 in complex with the 13-subunit RNAP from _Sulfolobus acidocaldarius_. Our results reveal intricate interaction
networks between inhibitors and RNAP that rationalise their modes of action. RIP binds to the RNAP clamp inside the DNA-binding channel and interferes with transcription by occluding DNA and
transcription factors binding sites. Intriguingly, the C-terminal tail of RIP interacts with the RNAP similarly to the B-linker of the basal factor TFB (homologous to TFIIB in eukaryotes).
In contrast, TFS4 interacts with RNAP through the funnel. Rather than interfering with the binding of transcription factors or nucleic acids, TFS4 inhibits RNAP by inducing large-scale
conformational changes that result in the opening of the DNA-binding channel, and the disruption of active site motifs including the bridge helix and the trigger loop. As similar structural
perturbations occur in other inhibited states of RNAP, our results demonstrate that the allosteric inhibition of the RNA polymerase is evolutionary conserved across all domains of life.
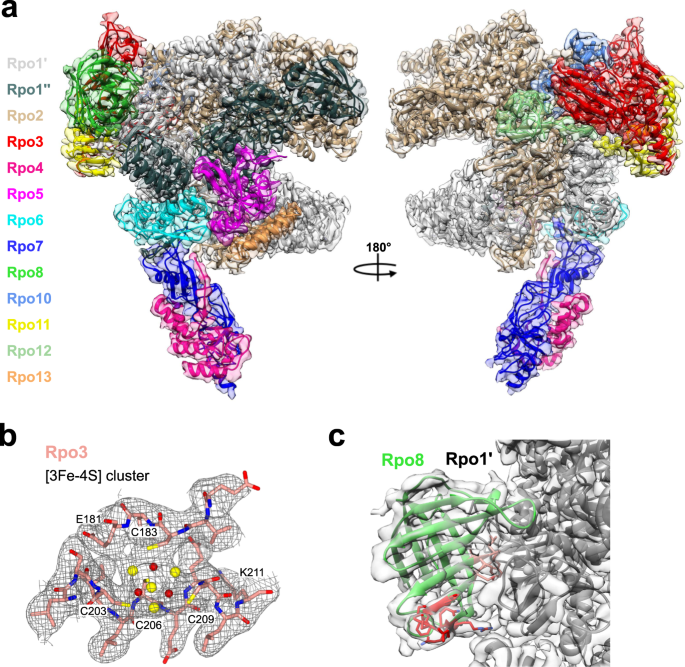
RESULTS THE CRYO-EM STRUCTURE OF THE SACI APO-RNAP We first determined the cryo-EM structure of the apo-RNAP from _Sulfolobus acidocaldarius_ (Saci) at 2.9 Å resolution (Table 1 and
Supplementary Fig. 1). The Saci RNAP consists of 13 subunits (Rpo1–13, Fig. 1a) with a molecular weight of 405 kDa. The active site environment is well resolved, with a clearly defined
bridge helix, trigger loop, and the catalytic aspartate triad (see below). The overall structure displays a strong correspondence both at sequence and structure level to crenarchaeal _S.
solfataricus_18 and _S. shibatae_19 RNAPs and eukaryotic RNAPII20 (Supplementary Figs. 2 and 3). The Saci RNAP structure includes eight conserved metal centres, six zinc coordinating motifs,
a redox-inactive18 cubane [3Fe-4S] iron-sulphur cluster, as well as the catalytic magnesium ion (MgA) that is characteristic for multisubunit, double-psi beta barrel (DPBB) RNAPs (Fig. 1b
and Supplementary Figs. 2, 3, and 4a). The high resolution enabled us to identify five prolines in the _cis_ configuration and an additional zinc finger domain not previously reported
(Supplementary Fig. 4a). Compared to previously published archaeal RNAPs, our cryo-EM map revealed a sequence register issue in the Rpo8 model starting from loop \({{{{\upbeta }}}}\)5–6.
Interestingly, the chain retracing led to the repositioning of the conserved GGLLM motif21 at the interface with Rpo1′, as seen in all RPB8 subunits of eukaryotic RNAPI, II, and III (Fig. 1c
and Supplementary Methods and Supplementary Fig. 4b, c). ATV RIP ADOPTS A FIVE-HELICAL BUNDLE STRUCTURE In order to gain insights into the structural basis of RIP function and to identify
the accurate binding mode of RIP, we solved the cryo-EM structure of the RNAP–RIP complex (Fig. 2b). A 10-fold excess of bacterially expressed recombinant RIP was incubated with Saci RNA
polymerase and crosslinked with BS3 (see “Methods” section). BS3 is a mild crosslinker that showed lower propensity to induce aggregation and precipitation compared to other known
crosslinkers during complex reconstitution carried out at 65 °C. Data were collected using the same approach used for the apo-RNAP and the processing provided a map with a final map
resolution of 3.3 Å (Fig. 2 and Supplementary Fig. 5, statistics reported in Table 1). The amino acid sequence of RIP suggests that it is a paralog of the viral P131/ORF131 capsid protein7.
The ancestral gene of ORF131 underwent gene duplication and speciation and the RIP paralogue acquired the novel function of tightly binding to RNAP and inhibiting its activity7. The
structure of RIP confirms this close relationship, as both RIP and ORF131 adopt a highly superimposable coil bundle structure with six α-helices and a rmsd of 1.7 Å (Fig. 2a). We prepared an
improved sequence alignment of RIP/ORF131 paralogues, informed by our cryo-EM structure, which allowed us to identify the sequence determinants and the structural features responsible for
the functional specialisation of RIP (Fig. 2a). The alpha-helical bundle of RIP is connected to a RIP-specific long C-terminal tail that is not conserved in any of the ORF131 paralogues
(Fig. 2a)7. We could resolve RIP encompassing residues 10–127 (of 145 aa) within the RNAP–RIP complex, and the secondary structure content of RIP is in good agreement with CD-spectra
recorded with free RIP which predicted a predominantly alpha-helical structure7. The first five α-helices are highly conserved between RIP and ORF131. The insertion of three additional
residues, L99–D100–T101, allows the α6* helix to fold partially back on α5, and project the C-terminal tail inside the DNA-binding channel of RNAP. The difference in the position and
orientation of the sixth α-helix is a crucial feature that allows the tail of RIP to adopt the correct position required for its interaction with RNAP (Fig. 2a, c). RIP FORMS A PLUG IN THE
DNA BINDING CHANNEL OF RNAP Our recent cross-linking/mass spectrometry studies suggested that RIP interacts with the RNAP clamp inside the DNA-binding channel of the archaeal RNAP7. The
cryo-EM structure of the RNAP–RIP complex provides the detailed structural basis for this interaction. The compact structure of RIP allows it to fit snugly in the DNA-binding channel of RNAP
between the Rpo1′ clamp, and the Rpo2 protrusion and lobe motifs, respectively, on each side of the channel (Fig. 2b). The C-terminal tail of RIP forms an intricate network of interactions
with the RNAP clamp and rudder motifs with an interface area of 1127 Å2 that includes both hydrogen bonds and hydrophobic interactions (Fig. 2c and Supplementary Fig. 7a, c). The RIP tail
makes a 90° bend at residue N117 and the region L114–M119 adopts an unusual L-shaped conformation that fits into a pocket between rudder and clamp (Fig. 2c). On the opposite side of the DNA
binding channel, RIP interacts with the RNAP Rpo2 protrusion and lobe motifs mainly via hydrophobic interactions and with a small interface area of 302 Å2. Taken together, the tight
interaction network between RIP and RNAP provides a structural rationale for the extreme salt-resistant binding that persists at up to 2 M NaCl7. The extremity of the RIP tail (128–145) that
was not resolved in the structure is enriched in negatively charged residues (6/18) that are not conserved in ORF131; these possibly mimic the negative charge of the DNA template
phosphodiester backbone. The importance of the C-terminal tail for RNAP binding is in good agreement with the previously published EMSA data, which showed that a C-terminal truncation (RIP
∆114–145) abrogated the RNAP binding and inhibitory activities of RIP, while not compromising the extreme heat-stability of the protein7. Some of the critical residues of Rpo1′ that are
involved in RIP binding including K238, R244, D241, H272, and R290 are strictly conserved among archaeal RNAPs but not with bacterial RNAPs (Supplementary Fig. 7d). This is in good agreement
with the observations that (i) RIP also inhibits the euryarchaeal _M. jannaschii_ RNAP in vitro, and (ii) the fact that recombinant RIP can be overexpressed in large quantities in _E. coli_
while being extremely toxic to _Sulfolobus acidocaldarius_7. The flexible RNAP clamp can adopt open and closed states which result in changes in the width of the DNA-binding channel in
response to the engagement of RNAP with the DNA template, and binding of initiation and elongation factors, respectively22. Similarly to other DPBB RNAPs, the archaeal enzyme cycles between
multiple distinct conformational states as RNAP progresses through the transcription cycle. We have previously applied smFRET to monitor the conformational changes of the _M. jannaschii_
RNAP in solution, and found that RIP strongly favoured one fixed closed conformation of the clamp7. The RIP–RNAP interaction network that involves both sides of the RNAP DNA-binding channel
provides a persuasive structural rationale for the nanomolar binding affinity and RIP’s ability to lock the clamp in a fixed conformation7. STRUCTURAL DETERMINANTS OF RIP INHIBITION The
structure and function of the archaeal preinitiation complex (PIC) is conserved with the eukaryotic RNAPII system; the combination of TBP and TFB is necessary and sufficient to enable RNAP
recruitment and start site-specific transcription initiation at basal levels23. TFE activates transcription by inducing conformational changes in RNAP and enhancing DNA strand separation
during the closed to open PIC transition24,25. Electrophoretic mobility shift assay (EMSA) experiments using the _Saccharolobus solfataricus_ RNAP, and initiation factors demonstrated that
RIP interferes with PICs by counteracting their formation and by destabilising preformed PICs7. A superposition of the RIP-RNAP structure with the closed PIC (from yeast, pdb 6gyk20) reveals
that the C-terminal tail of RIP (residues 117–123) and the helix \({{{{{\rm{\alpha }}}}}}\)4 (Fig. 2a) overlap with the TF(II)B residues 90–120 encompassing the B-linker and B-linker helix,
respectively, by adopting the same structure and equivalent binding mode to the RNAP rudder (Fig. 3a, b). Consequently, RIP inhibits the PIC by competing with TFB binding to RNAP. In
addition, RIP occludes the binding site to the nucleic acid scaffold in both closed and open PIC (pdb 5iyd26), and thus would prevent the loading of the DNA in the RNAP active site (Fig.
3c). In contrast to the strong inhibition of initiation factor-dependent and promoter-directed transcription initiation, RIP has a smaller effect on elongation, and EMSAs showed that RIP,
albeit in a limited fashion, can bind to TECs consisting of RNAP and a DNA/RNA scaffold7. Indeed, a superposition of the RNAP–RIP structure with a eukaryotic TEC (pdb 5oik27) reveals that
the downstream duplex DNA, and the unwound template DNA strand passes underneath RIP while the non-template strand (NTS) clashes with RIP (Fig. 3f, g). To evaluate the contribution of the
NTS clash to the inhibition, we carried out EMSA experiments with dual labelled components; RIP was radiolabelled using 32P while the DNA/RNA elongation scaffold was labelled with the
fluorescent dye Cy3 on the DNA template strand. The low mobility bands detected by the fluorescence signal in the upper panel of Fig. 3d correspond to bona fide TECs. The addition of 32P-RIP
counteracts the Cy3-TEC signal in a concentration-dependent manner. Vice versa, the addition of increasing amounts of Cy3-labelled nucleic acid scaffold leads to an increase in the signal
corresponding to free 32P-RIP. In summary, the EMSAs support a competitive binding mode of RIP and the DNA/RNA nucleic acid scaffold. This is congruent with a model in which RIP can
interfere with the DNA during loading, but not easily displace it once it is already loaded. RIP TEMPORAL EXPRESSION PATTERN SUPPORTS ROLE IN VIROID MATURATION The role of RIP for virus
fitness and function is still a matter of debate. As RIP directly interferes with nucleic acid binding and affects host and virus promoters alike, the early expression of RIP at high levels
appears problematic. However, the global attenuation of transcription could benefit the virus by preventing transcription-dependent host defence mechanisms including the CRISPR IIIb
system28. Alternatively, RIP could disengage RNAP from the actively transcribed viral genome aiding the DNA packaging into virus particles. The former would be associated with an expression
pattern during early stage, and the latter during a late infection stage. To address this question, we analysed the temporal gene expression of RIP over a time course of 72 h. Exponentially
growing _Acidianus_ cells were infected with ATV and samples taken at regular intervals; RIP protein levels were detected by immunoblotting using a polyclonal antibody raised against
recombinant RIP and compared to the expression levels of Alba, a chromatin protein that serves as control. In good agreement with its high toxicity, RIP could not be detected in _Acidianus_
cell extracts during the early and middle stages of infection but was strongly upregulated at the end of the time course (72 h p.i.) just prior to cell lysis that occurs 96 h of
post-infection (Fig. 3e). This suggests that RIP function is important during late infection, e.g., by dissociating RNAPs from the actively transcribed viral genome and thereby assisting
maturation of the virion particle. THE TFS4 CLEAVAGE FACTOR PARALOG EVOLVED INTO A RNAP INHIBITOR Unlike RIP, the TFS4 inhibitor is encoded by the host genome but only expressed in response
to viral infection. Like RIP, TFS4 binds tightly to RNAP and efficiently represses transcription16. In order to investigate the structural basis for TFS4 inhibition and compare it to RIP’s,
we solved the cryo-EM structure of the RNAP–TFS4 complex at 2.6 Å resolution (Fig. 4a, Supplementary Fig. 6, and Table 1 for statistics). The map has been further refined by multibody
refinement which allowed us to obtain a medium resolution map of the stalk at 3.8 Å, where all the known structural features were identified and correctly modelled inside the map
(Supplementary Fig. 6g, h and Table 1). Archaeal TFS paralogues have a domain configuration akin to RPA12, RPB9 and RPC11 of RNAPI, II, and III, respectively, composed of two zinc-ribbon
domains, ZRN and ZRC, which are connected by a long linker (Fig. 4e). As proven by the cryo-EM map, the TFS4 ZRN interacts with RNAP between the upper jaw and the lobe (Fig. 4a, b). The
N-terminal segment of the TFS4 linker forms two β-addition motifs (\({{{{{\rm{\beta }}}}}}3\) and \({{{{{\rm{\beta }}}}}}4\)) by providing one antiparallel and one parallel strand to two
β-sheets in the upper jaw of RNAP (Fig. 4b, e). The C-terminal segment (\({{{{{\rm{\beta }}}}}}\)5) of the linker, unexpectedly, packs on the TFS ZRC domain that interacts with the rim
helices of the funnel (Fig. 4b, e). The TFS4 ZRC binds in the NTPs-entry funnel of RNAP in a manner that is related to its eukaryotic homologues29,30,31, but without reaching through the
pore into the active site (Supplementary Fig. 8e). The chemical nature of the interactions between the ZRN and ZRC domains with RNAP is also different (Supplementary Fig. 7b, c). The
ZRN-jaw/lobe interactions are dominated by a network of hydrophobic interactions, whereas the ZRC-funnel interactions are facilitated by numerous hydrogen bonds and salt bridges which are
not evenly distributed at the interface. The ZRC surface is abundant in positively charged residues (R57, R65, R70, K76-78, R79, and R82), most of which are not conserved in other
TFS-related factors (Fig. 4c, e). This highly charged surface is balanced by interacting with two negatively charged patches formed by the lower alpha helix of the rim on one side (Rpo1′
S698, D702, and D705) and the upper jaw (Rpo1″ E301), which forms the internal wall of the funnel on the opposite side (Fig. 4b, d). ALLOSTERIC _MODUS OPERANDI_ OF TFS4 TFS4 overexpression
induces retardation of cell growth in vivo, and TFS4 is a potent inhibitor of RNAP in vitro, increasing the _K_M for substrate NTP binding by ~50-fold and, like RIP, destabilises PICs and
TECs16. Unlike RIP however, the binding sites of TFS4 with RNAP do not overlap with any of the RNAP interaction sites for the DNA or transcription initiation factors TBP, TFB, and TFE.
Rather, TFS4 allosterically inhibits the RNA polymerase by inducing conformational changes driven by the displacement of the upper jaw, encompassing Rpo1″, Rpo5, 6, and 13, and the Rpo1′
clamp head (Fig. 5a). The jaw displacement allows TFS4 linker to bind to a surface that is otherwise occluded in the apo-RNAP, and is stabilised within the RNAP structure by the replacement
of the two lost beta strands with two newly formed strands provided by TFS4 (Fig. 4 and enlargement in Fig. 5a). The superposition between the apo-bound and the TFS4-bound RNAP reveals a
swinging movement of the jaw in unison with the clamp head by 6.3° concomitant with the splaying of the DNA-binding channel by 5 Å (Supplementary Movie 1). Considering that the width of the
DNA double helix is 23.7 Å, the observed widening of the DNA-binding channel is likely to impair the close interactions of the DNA template with the RNAP, which provides the structural
rationale for TFS4 destabilisation of RNAP-nucleic acid interactions during either initiation or elongation16. The Rpo1′ bridge helix spans across the DNA binding channel and is anchored on
either side of it. The TFS4-induced opening stretches and bends the bridge helix by 3.8 Å and 2.2° leading to the loss of density between residues D808 to T813, likely due to the unwinding
of the helix in the TFS4-bound RNAP (Fig. 5b). The ensemble of bridge helix and trigger loop plays a key role in the substrate nucleotide binding in the active site and its translocation
cycle, i.e., the molecular mechanism underlying transcription elongation32. The TFS4 ZRC in the NTPs-entry pore clashes with the tip of the trigger loop resulting in its displacement, which
is confirmed by the replacement of the RNAP trigger loop map with the TFS4 ZRC density and loss of structural information between residues R86 to E95 of Rpo1″ in the RNAP–TFS4 cryo-EM map
(Fig. 5c). In summary, the changes induced by TFS4 binding include the (i) widening of the DNA-binding channel, (ii) melting of the bridge helix, and (iii) displacement of the trigger loop.
This three-pronged attack makes for a formidable intervention with the binding and catalytic mechanisms of RNAP. DISCUSSION As RNAPs are important therapeutic targets, the structural basis
and mechanisms of their inhibition have been studied in great detail in bacteria and eukaryotes—while nothing is known about archaeal RNAP. Arguing from first principles, inhibition can be
achieved by (i) preventing direct interactions between RNAP and basal factors, nucleic acid template, or substrate NTP, or (ii) via allosteric mechanisms that alter the structure or
conformation of RNAP in a way that abrogates catalysis. Our two novel structures of RNAP-inhibitor complexes both add new features and highlight common themes of intervening with RNAP
function. RIP is a small single-domain protein which binds to the inside of the RNAP clamp and rudder motifs, which is incompatible with the binding and function of the essential initiation
factor TFB. The C-terminal tail of RIP and the B-linker strand and B-helix motifs of TFB (and TFIIB) interact with RNAP in an identical fashion (Fig. 3b)33. RIP forms a plug in the
DNA-binding channel of RNAP, which stabilises and rigidifies the RNAP in the closed conformation. Such a binding mechanism interferes with the formation of the closed PIC, and it would
prevent the loading of the DNA into the RNAP active site during the transition from the closed to the open PIC (Fig. 3a–c). The binding of RIP is furthermore incompatible with elongation by
interfering with the stability of the DNA scaffold inside the DNA-binding channel (Fig. 3f). All of the observations above are in perfect agreement with published biochemical interaction
analyses, which suggested a competitive inhibition mechanism of RIP. The initiation factor TFE induces an opening of the RNAP clamp34, which stabilises the PIC and activates transcription25.
Somewhat counterintuitively, complete TFE-containing PICs are more sensitive to RIP as compared to minimal PIC lacking TFE7. The structure of the RNAP–RIP complex rationalises this
observation, as the TFE-induced opening the clamp34 makes the RIP binding site more accessible. The occlusion of the DNA-binding channel is a reliable and direct mechanism of inhibition
exploited by all domains of life. The bactoriaphage T7 Gp2 regulator binds within the DNA-binding channel of RNAP; it blocks the interaction between the downstream DNA and the RNAP
\({{{{{\rm{\beta }}}}}}\)′ jaw domain and effectively inhibits the open complex formation of _E. coli_ RNAP-\(\sigma\)70 (Fig. 6a)35. Among the eukaryotic transcription systems, the cellular
negative regulator MAF1 specifically inhibits RNAPIII in response to stress and nutrient deficiency. Similarly to RIP, MAF1 binds inside the DNA-binding channel of RNAPIII and occludes the
binding site of TFIIIB to the RNAP clamp and rudder motifs (Fig. 6b, c)36. What is singular about RIP and novel in the field is the unique ability of a viral protein to mimic the binding
mode, and thereby prevent the binding of the universally conserved host basal initiation factor TFB. Archaea, including Sulfolobales, encode functionally diversified paralogues of the
transcript cleavage elongation factor TFS16. Sso TFS1 and TFS4 share the same domain organisation and a 28% sequence identity and both bind competitively to RNAP, yet while TFS1 stimulates
elongation, TFS4 has evolved into a potent inhibitor of RNAP16. Although the structure of the RNAP-TFS1 complex has not been determined yet, it is very likely that the two conserved
carboxylate residues in the TFS1 ZRC penetrate deep into the active site through the NTPs-entry funnel like TFIIS11,16 as the molecular mechanisms of TFS1 and TFIIS are strictly
conserved12,13. However, our structure shows that the TFS4 ZRC binds in the RNAP funnel, but not as deep and in a different orientation compared to TFIIS (Fig. 7b and Supplementary Fig. 8e,
f). The TFS4 binding in the RNAP funnel is ensured by electrostatic interactions between the positively charged TFS4 ZRC domain and negatively charged patches on the rim helices, and the
upper jaw of the RNAP as shown in the interface analysis of TFS4-RNAP complex (Fig. 4d and Supplementary Fig. 7b). Two acidic patches face the highly positively charged surface of the TFS4
ZRC domain (Fig. 4b, c, e). Site directed mutagenesis has indicated that three consecutive lysine residues (K76/77/78) in the TFS4 ZRC domain contributed to RNAP binding and inhibition16,
however, the lysine residues are surface exposed and do not make any contacts with RNAP in the RNAP-TFS4 structure. We surmise that the lysine residues enable an initial binding inside the
negatively charged channel (Fig. 4c, d) that is followed by a rearrangement of the TFS4 ZRC domain inside the funnel resulting in the binding mode reflected in the RNAP-TFS4 structure.
Interestingly, the propensity of the ZRC domains to flip in and out of the NTPs-entry channel is a well characterised mechanism of regulation during transcription elongation observed with
RPA12 and RPC11 (RPC10 in human). Importantly, the inside funnel state of RPA12 and RPC10/11 is implicated in transcription termination of RNAPI and RNAPIII, respectively31,37,38,39. A
superimposition of the apo-RNAP and RNAP–TFS4 complex reveals global as well as local changes of RNAP structure that account for the efficient inhibition, with an overall opening of the RNAP
through the DNA-binding channel, and alteration of the bridge helix and trigger loop motifs in the active site (Figs. 5 and 7a). The NTPs-entry funnel provides a crucial orifice to the RNAP
active site that not only allows NTPs to enter, and the RNA 3′ terminus to exit the RNAP in backtracked TECs, but also serves as binding site for a plethora of transcription regulators10.
These include negative regulators such as the bacterial Gfh18,40 and DksA9,41 that have pleiotropic effects on transcription. During the stringent response induced by amino acids starvation,
DksA in conjunction with ppGpp is thought to alter and destibilise conformational transitions en route to the open complex of stringent promoters. The binding of ppGpp, even in the absence
of DksA, restricts the conformational flexibility and thereby inhibits RNAP42. Gfh1 inhibits all catalytic activities of RNAP enhancing pausing40,43,44. Like TFS1 and TFS4 in archaea, the
bacterial GreA, GreB, Gfh1, and DksA factors compete with each other for RNAP binding, and changes in their relative expression level is likely to be an integral part of their regulation. In
the case of TFS1 and 4, the former is constitutively expressed and engaged with RNAP, while the latter is exclusively expressed in response to viral infection17. TFS4 and Gfh1 are not
homologous but share intriguing functional properties that likely result from convergent evolution. Both bind to the RNAP funnel and allosterically inhibit RNAP which results in the widening
of the DNA binding channel and—albeit to a different degree—a distortion of the bridge helix and trigger loop in the active site (Fig. 7c). What is the origin of the conformational dynamics
of DPBB RNAP in all domains of life? The NTP translocation cycle of RNAP involves tightly coordinated conformational changes as it cycles through post-translocated and pre-translocated, and
of NTP pre-insertion and insertion states that have been captured by high-resolution X-ray structures of the bacterial RNAP45,46. As the archaeal RNAP progresses through the transcription
cycle, the flexible RNAP clamp adopts distinct conformational states, which alter the width of the DNA-binding channel34. Elongation is a discontinuous process interrupted by pausing and
termination, which involves inhibited states. Recently, it has been shown that the bacterial RNAP in the context of an RNA hairpin in elemental pause—which is likely preceeding
termination—has a widened DNA-binding channel, which is reminiscent of the archaeal TFS4-RNAP complex47. This is different from a closed RNAP clamp that is characteristic for processive
elongation complexes45,46. Equally, in the inactive dimeric form of RNAPI, the two macromolecules face each other with the DNA-binding channel of one molecule interacting with the stalk of
the other29. In this conformation, the RNAPI jaw and lobe modules have moved outwards and the DNA-binding channel is widened by 5 Å48 similarly to the inhibited state of the archaeal
TFS4-RNAP complex (Fig. 7c). Finally, eukaryotic RNAPI, II and III include RPA12, RPB9 and RPC10/11 subunits, respectively, that are paralogs of TFIIS but stably incorporated into the RNAP
rather than reversibly associated with it (Supplementary Fig. 8a–d). We posit that the allosteric changes induced by TFS4 are related to several inhibited states of DPBB RNAP, including
transcription effectors (like Gfh1), oligomerisation equilibria (RNAPI) and more general inhibited states of RNAPs associated with pausing and termination. In the structurally well
characterised cases discussed above, the DNA-binding channel is widened and the interactions with the template DNA weakened. The common denominator of inhibition is the ability to lock them
in a specific conformation. In a likely scenario, RNAP subunits and factors have evolved vertically by gene duplication and speciation (e.g., TFIIS, TFS1, and −4, RPA12, RPB9, and RPC11/10)
or by convergent evolution (e.g., GreA/B and Gfh1 relative to TFS1) to exploit the inherent conformational flexibility of RNAP to modulate and fine tune transcription. The biological
function of RIP for the ATV virus is still a matter of debate, but its tight regulation and strong induction during the very late stages of lytic infection supports a role in virus particle
assembly, possibly by dissociating transcribing RNAP from viral genomes. The strong and stable interactions between RIP and the RNAP clamp and rudder make RIP a lethal protein, and this
absolute mechanism to shut down RNAP is apparently the preferred option for viruses and bacteriophages like ATV and T76. In comparison, host-encoded factors such as TFS4 and Gfh1 seem to opt
for a more versatile approach by allosterically interfering with RNAP function. This provides an opportunity for the cell to temporarily pause transcription, mount additional defence
mechanisms, and eventually reactivate the gene expression programme once the favourable conditions have returned. This is the case for the well-studied Gfh1 which adopts an inactive
conformation during normal growth conditions, and undergoes a reversible conformational change upon acidification of the medium22. The plasmid-driven expression of TFS4 stops cell growth
similar to virus infection, but it is not known yet whether TFS4 is the sole agent to trigger the host growth retardation in response to infection16. The induction of a quiescent state is an
important preamble for persistence of many pathogenic bacteriophages49, hence, we hypothesise that TFS4 may play a similar role in archaea. The detailed structural bases and the mechanisms,
which underlie the inhibition of archaeal DPBB RNAP described in this manuscript, have the potential to aid the design of novel drugs targeting the RNAPs of bacterial pathogens and RNAPI in
cancer therapy50. These include small effector proteins that bind tightly to RNAP with high specificity, like RIP, and either deny access to the DNA binding channel or essential
transcription factors. Such effectors could be eventually delivered to their eukaryotic or bacterial targets by recombinant viruses or bacteriophages, respectively51. Moreover, agents that
bind to RNAP in a polydentate fashion like TFS4 and prevent conformational changes that are critical for catalysis are possibly more refractive to elicit the fast emergence of resistance
mutations. METHODS PROTEIN EXPRESSION AND PURIFICATION Saci RNA polymerase, 6×His tagged at the C-terminus of Rpo8, was expressed in Saci strain MW001 and purified according to established
protocols16. Cells were resuspended in buffer 50 mM Tris pH 8.0, 200 mM NaCl, 25 mM Imidazole, 1 mM DTT, 100 µM ZnSO4, 5% glycerol supplemented with one tablet of protease inhibitors
(cOmplete Tablets, Roche), 1 µl/ml DNase I, and 0.5 µl/ml RNase A. Cell suspension was sonicated for 30 min at 70% amplitude in pulse mode on ice, followed by centrifugation at 60,000 × _g_
at 10 °C for 30 min. Cell extract was first loaded onto Ni-column (the lysis buffer was supplemented with 250 mM Imidazole for the elution), followed by Heparin purification (the sample was
loaded at 50 mM NaCl buffer and eluted in gradient up to 1 M NaCl). Fractions containing DNA-free RNAP were pooled, concentrated in Amicon 100 kDa cutoff (Millipore), and loaded onto
Superose 6 10/300 column (GE Life Science). ATV RIP (ORF145), tagged at the N-terminus with a cleavable 6×His sequence, was over-expressed in BL21 pLyS by induction with 1 mM IPTG. After
lysis by sonication (6 min in pulse mode at 70% amplitude on ice) in buffer 50 mM Tris pH 8.0, 200 mM NaCl, 25 mM Imidazole, supplemented with one tablet of protease inhibitors (cOmplete
Tablets, Roche, the cell extract went through a precipitation heat step at 60 °C for 20 min, followed by centrifugation at 60,000 × _g_ at 10 °C. The cleared cell extract was purified in
Ni-column and the tag cleaved in dialysis at room temperature (rt) with TEV protease. The sample was further purified in Ni-column to remove the tag7. Sso TFS4 (without tag) was expressed in
BL21 pLysS by induction with 1 mM IPTG. Cells were resuspended in buffer 50 mM Tris pH 8.0, 50 mM NaCl, 1 mM DTT, 100 µM ZnSO4, 5% glycerol supplemented with 1 tablet of protease inhibitors
(cOmplete Tablets, Roche), 1 µl/ml DNase I, and 0.5 µl/ml RNase A, and sonicated for 6 min in pulse mode at 70% amplitude on ice. The cell lysate was incubated at 65 °C for 20 min to
promote bacterial proteins precipitation, and the cell extract cleared by centrifugation at 60,000 × _g_ at 10 °C. The sample was purified with HiTrap Q column (GE Life Science) using 1 M
NaCl for the elution in gradient, followed by Superose 12 10/300 column (GE Life Science)16. COMPLEX ASSEMBLY AND CRYO-ELECTRON MICROSCOPY DATA COLLECTION APO-RNAP The RNA polymerase was
crosslinked at 0.15 mg/ml in 200 µl of a buffer containing 20 mM Hepes pH 7.0, 200 mM NaCl, 5 mM MgCl2, 100 µM ZnSO4, 10% glycerol, 5 mM DTT, with 2 mM bis(sulfosuccinimidyl)suberate (BS3)
for 5 min at 65 °C. The reaction was quenched adding 150 mM NH4HCO3 at rt for 20 min. The sample was then diluted ten times in the same buffer without glycerol, filtered with a 0.22 µm
filter and concentrated up to 0.4 mg/ml in a concentrator with a cutoff of 100 kDa. Sample quality was firstly assessed by negative staining, then 3 µl of sample at 0.06 mg/ml was spotted on
a UltrAuFoil holey grid 300 mesh R1.2/1.3 (Quantifoil, Germany) covered with graphene oxide according to a protocol described by Cheng K. and co-workers52, and vitrified by plunging in
liquid ethane using Vitrobot Mark IV (Thermo Fisher Scientific, USA) at 4 °C and 94% humidity. Data were collected at eBIC National facility (Diamond Light Source, UK) using a Titan Krios
microscope (Thermo Fisher Scientific, USA) operated at 300 keV and equipped with a BioQuantum energy filter (Gatan, USA). The images were collected with a post-GIF K3 direct electron
detector (Gatan, USA) operated in super resolution mode, at a nominal magnification of 81,000, corresponding to a pixel size of 1.085 Å. The dose rate was set to 21 e− per pixel per second,
and a total dose of 44.46 e/Å2 was fractionated over 40 frames. An energy slit with a 20 eV width was used during data collection. Data were collected using EPU software (Thermo Fisher
Scientific, USA) with a nominal defocus range −1.0 to −2.5 μm. During the data collection microscope stage was tilted to −30° to overcome preferred orientations. RNAP/RIP COMPLEX Prior the
crosslinking procedure already described, the RNA polymerase was incubated with a 10-fold excess of RIP for 5 min at 65 °C. Grids covered with graphene oxide were prepared according to the
protocol described above, and data were collected in ISMB Birkbeck EM facility using a Titan Krios microscope operated at 300 keV and equipped with a BioQuantum energy filter. The images
were collected with a post-GIF K2 Summit direct electron detector (Gatan, USA) operated in counting mode, at a nominal magnification of 130,000 corresponding to a pixel size of 1.047 Å. The
dose rate was set to 5.84 e− per pixel per second, and a total dose of 45.5 e/Å2 was fractionated over 45 frames. An energy slit with a 20 eV width was used during data collection. Data were
collected using EPU software with a nominal defocus range −1 to −2.5 µm. During the data collection microscope stage was tilted to −30°. RNAP/TFS4 COMPLEX The RNA polymerase was incubated
and crosslinked as described above in presence of a 20 molar fold excess of TFS4. The sample at 0.4 mg/ml of concentration was applied twice on a UltrAuFoil holey grid 300 mesh R1.2/1.3 and
vitrified by plunging in liquid ethane using Vitrobot Mark IV at 4 °C and 94% humidity. Data were collected at eBIC National facility using a Titan Krios operated at 300 keV and equipped
with a BioQuantum energy. The images were collected with a post-GIF K3 direct electron detector (Gatan, USA) operated in super resolution mode, at a nominal magnification of 81,000,
corresponding to a pixel size of 1.085 Å. The dose rate was set to 21.221 e− per pixel per second, and a total dose of 45.2 e−/Å2 was fractionated over 40 frames. An energy slit with a 20 eV
width was used during data collection. Data were collected using EPU software with a nominal defocus range −1 to −2.5 µm. CRYO-ELECTRON MICROSCOPY DATA PROCESSING APO-RNAP The dataset of
1676 movie stacks was aligned, summed and 2× binned using MotionCor253, followed by CTF estimation using GCTF54. Relion 3.0 software55 was used for template-free particle picking (Laplacian
method) and all consequent image processing for this sample. Initially 1,286,432 particles were extracted and downscaled to a/pix of 4.5 Å. After multiple cycles of 2D and 3D classifications
423,157 best particles were selected and rescaled to the pixel size of 1.085 Å. The selected subset of particles was then refined and, after post-processing, subjected to four cycles of CTF
refinement to correct for the effect of stage tilt used during data collection on the initial CTF estimation. Three cycles of CTF refinement were sufficient for the correction, and the
fourth cycle didn’t provide any further improvement. The final cycle of 3D refinement and post-processing resulted a map with resolution of 2.88 Å as estimated using gold standard Fourier
Shell Correlation (FSC) with a 0.143 threshold (Supplementary Fig. 1 and Table 1). RNAP/RIP COMPLEX The 2130 movie stacks were aligned and summed using MotionCor2 followed by CTF estimation
in GCTF. Particles were picked using Relion 3.0 reference-based method. These 600,640 particles extracted from the micrographs were subjected to multiple rounds of 2D and 3D classifications
using cryoSPARC56. Multiple approaches were applied to identify the apo-RNAP but no additional species were found likely due to the relatively low number of particles available for the
search. The best 151,237 particles were rescaled to the original pixel size, and image processing continued using Relion 3.0. Particles were refined and subjected to three cycles of CTF
refinement. After a final 3D refinement and post processing step the resolution of 3.27 Å was estimated using gold standard FSC with 0.143 threshold (Supplementary Fig. 5 and Table 1).
RNAP/TFS4 COMPLEX The dataset of 1760 movie stacks was motion corrected and analysed with Relion 3.0 following the same routine described above for the apo-RNA polymerase sample. After
multiple rounds of 2D and 3D classifications, best 505,758 particles were selected out of 1,161,535, and subjected to two cycles of CTF refinement followed by particle polishing. Although
the large number of particles, we did not identify the apo-RNAP or any other intermediate conformational species, using either Relion 3.0 and Cryosparc. The last 3D refinement and
post-processing steps provided a map with an averaged resolution of 2.59 Å as estimated using gold standard FSC with 0.143 threshold. Analysis of the Euler angles distribution for this map
revealed a clear cluster of preferred orientations. Although it didn’t seem to affect map quality, we decided to prune the refined particles and repeat refinement and post-processing. This
step was carried out using an in-house script considering the tilt and rotation angles of the last refinement, and pruning the particles based on the CtfFigureOfMerit metadata up to 350,000
particles. The pruning provided a batch of 350,682 particles, which gave a map with resolution of 2.61 Å at the FSC of 0.143 (Supplementary Fig. 6 and Table 1). The local resolution of TFS4
map shown in Supplementary Fig. 6d resulted to be perfectly compatible with the local resolution of the RNA polymerase in the same area suggesting that the apomediate or intermediate
species, if present, represent all together less than the 5% of the dataset, which might explain the resulting single class from the 3D classification step. To improve map quality of the
stalk which is intrinsically a flexible arm protruding from the main body, we performed a multi-body refinement using the RNAP main domain and the stalk as moving bodies. To do that, we
generated two masks with soft edges for the large globular main domain and the stalk, respectively, to which we applied 20° of width for the rotation priors and five pixels for the
translation between the two bodies, a setup suggested for highly flexible bodies according to the protocol released by Nakane T and Sheres SHW57. The multibody refinement was the only
successful approach found improving the stalk resolution up to 3.75 Å (Supplementary Fig. 6g, h and Table 1). MODEL BUILDING AND REFINEMENT Local resolution was assessed using Relion 3.0
after post-processing, whereas map sharpening and model refinement were carried out in Phenix v1.15.2 and 1.19.258, including rounds of manual editing and refinement in Coot v0.8.9.159
(Table 1). Models of all RNAP subunits, RIP and TFS4 were prepared using Modeller34, then the RNAP complex was assembled using the homologous RNA polymerase from _Sulfolobus shibatae_ at 3.2
Å (pdb 4ayb19) and refined against the cryo-EM map of RNAP/TFS4 at 2.61 Å. The structure obtained was used to refine the model against the maps of the apo-bound and RIP-bound RNA
polymerase. Following the RNAP refinement, the initial models of RIP and TFS4, obtained using Modeller, were placed inside the extra-densities and refined accordingly. The C-terminal tail of
RIP and TFS4 linker, not predicted in the initial model, were built manually in Coot following map density. INTERFACES DATA ANALYSES AND SEQUENCE ALIGNMENTS The binding interfaces between
the RNA polymerase and RIP or TFS4 were analysed using two different programmes for consistency, PISA and LigPlot+60,61. As setup in LigPlot+ we used for the H-bonds and salt bridges a
maximum of 3.5 Å, for the hydrophobic interactions we used the maximum values of 3.5 Å. Structure-based alignments of all Saci RNAP subunits have been carried out with the Match-Align tool
in Chimera62 after superimposition of the _S. acidocaldarius_ apo-RNAP with _S. shibatae_ (4ayb19), _S. solfataricus_ (3hkz18), _T. kodakarensis_ (6kf963) and _S. cerevisiae_ (5vvs64)
followed by manual editing of the flexible loops. ATV RIP and P131/ORF131, and SMV-1 ORF122 and ORF114 have been aligned using PROMALS3D65. Alignment images throughout the text were prepared
in Espript366. ATV INFECTION AND BIOMASS SAMPLING Acidianus convivator strain AA9 was grown at 76 °C in 1× medium including sulphur67. The cell density was monitored with a Shimadzu
spectrophotometer (OD600) and the cell number determined with a Thoma counting chamber at different times points during the growth. Cells from 500 ml of exponentially growing A. convivator
were collected at OD600 = 0.08 by centrifugation at 3500 rpm for 30 min at 4 °C, and the pellet was suspended in 500 µl of 1× medium. A ATV virus preparation of 150 μl68 (titre of 1010
virions/ml) was added to the cell suspension, the mixture was incubated at 80 °C for 1 h and diluted with 500 ml of 1× medium including sulphur. Infected cells were grown at 76 °C and
samples taken at _t_ = 0, 6, 12, 18, 48 and 72 h post-infection. IMMUNODETECTION Cell pellets were sonicated (20% amplitude, 10 s pulse mode for 1 h) and protein content was measured using
the Qubit system (Invitrogen). For each sample, the equivalent of 9 μg of total protein was loaded on a 14% Tris-Tricine SDS-PAGE and blotted onto a 0.2 μm PVDF membrane. As primary
antibodies, we used a 1:1000 dilution of a rabbit polyclonal anti-RIP (Davids Biotechnology), and a 1:3000 dilution of a sheep polyclonal anti-Sso Alba (kindly provided by Malcom White,
University of St. Andrews, UK), the second target used as additional internal control to assess protein content of the samples. To visualise the two protein targets in different colours we
used two different secondary antibodies, the donkey anti-rabbit IgG was conjugated with Dylight 680 and used at 1:10,000 dilution (A120-208D6, Bethyl Laboratories, Cambridge, UK), the donkey
anti-sheep IgG was conjugated with Dylight 488 and used at 1:2000 dilution (A130-100D2, Bethyl Laboratories, Cambridge, UK). Fluorescent detection was carried out on Typhoon FLA 9500
scanner (GE Healthcare) for Alexa-488 and Alexa-680. EMSA EXPERIMENTS The DNA:RNA scaffold was prepared incubating the DNA template strand labelled with Cy3 (Cy3-TS83) with RNA (RNA14) at 76
°C for 5 min, then we added the non-template strand (NTS83) and we incubated for further 5 min at 76 °C. RIP was cloned into the pKA-vector carrying the phosphorylation site at the
N-terminus. It was expressed, purified and labelled with 32P according to published protocol7. In order to be able to use detectable signals of both fluorescent scaffold and radioactive RIP,
32P-RIP was mixed with cold RIP in to 3:1 ratio. We split the competition assay in two halves. In the first half assay we pre-incubated the scaffold (100 nM) with Saci RNAP (200 nM) in 15
µl of buffer 10 mM MOPS pH 6.5, 10 mM MgCl2, 200 mM NaCl, 7 mM DTT, 10% glycerol, 0.067 mg/ml BSA, and 0.1 mg/ml heparin at 65 °C for 5 min, then we added increasing amount of 32P-RIP (0.2–2
µM) and we incubated again at 65 °C for 5 min. The competition reactions of the second half assay were prepared by pre-incubating 32P-RIP (400 nM) with 200 nM Saci RNAP in 15 µl of the same
buffer at 65 °C for 5 min, then adding 0.1–1 µM scaffold and incubating for further 5 min at 65 °C. Samples were resolved on a 6% native PAGE and detected using Typhoon (GE). Oligo
sequences are reported in Supplementary table 1. REPORTING SUMMARY Further information on research design is available in the Nature Research Reporting Summary linked to this article. DATA
AVAILABILITY Structural data generated in the current study are available on the RCSB Protein Data Bank and the corresponding EM maps deposited on the Electron Microscopy Data Bank. PDB
accession codes: apo-RNA polymerase 7ok0, RNAP/RIP complex 7oq4, RNAP/TFS4 7oqy. EMDB accession codes: RNA polymerase EMD-12960, RNAP/RIP complex EMD-13026, RNAP/TFS4 EMD-13034. Source data
are provided with this paper. REFERENCES * Stepanova, E. V., Shevelev, A. B., Borukhov, S. I. & Severinov, K. V. Mechanisms of action of RNA polymerase-binding transcription factors that
do not bind to DNA. _Biofizika_ 54, 773–790 (2009). CAS PubMed Google Scholar * Sheppard, C. & Werner, F. Structure and mechanisms of viral transcription factors in archaea.
_Extremophiles_ 21, 829–838 (2017). Article CAS PubMed PubMed Central Google Scholar * Mosaei, H. & Harbottle, J. Mechanisms of antibiotics inhibiting bacterial RNA polymerase.
_Biochem. Soc. Trans._ 47, 339–350 (2019). Article CAS PubMed Google Scholar * Artsimovitch, I. & Vassylyev, D. G. Is it easy to stop RNA polymerase? _Cell Cycle_ 5, 399–404 (2006).
Article CAS PubMed Google Scholar * Forterre, P. The origin of viruses and their possible roles in major evolutionary transitions. _Virus Res._ 117, 5–16 (2006). Article CAS PubMed
Google Scholar * Bae, B. et al. Phage T7 Gp2 inhibition of Escherichia coli RNA polymerase involves misappropriation of sigma70 domain 1.1. _Proc. Natl Acad. Sci. USA_ 110, 19772–19777
(2013). Article ADS CAS PubMed PubMed Central Google Scholar * Sheppard, C. et al. Repression of RNA polymerase by the archaeo-viral regulator ORF145/RIP. _Nat. Commun._ 7, 13595
(2016). Article ADS CAS PubMed PubMed Central Google Scholar * Tagami, S. et al. Crystal structure of bacterial RNA polymerase bound with a transcription inhibitor protein. _Nature_
468, 978–982 (2010). Article ADS CAS PubMed Google Scholar * Perederina, A. et al. Regulation through the secondary channel–structural framework for ppGpp-DksA synergism during
transcription. _Cell_ 118, 297–309 (2004). Article CAS PubMed Google Scholar * Zenkin, N. & Yuzenkova, Y. New insights into the functions of transcription factors that bind the RNA
polymerase secondary channel. _Biomolecules_ 5, 1195–1209 (2015). Article CAS PubMed PubMed Central Google Scholar * Cheung, A. C. & Cramer, P. Structural basis of RNA polymerase II
backtracking, arrest and reactivation. _Nature_ 471, 249–253 (2011). Article ADS CAS PubMed Google Scholar * Hausner, W., Lange, U. & Musfeldt, M. Transcription factor S, a
cleavage induction factor of the archaeal RNA polymerase. _J. Biol. Chem._ 275, 12393–12399 (2000). Article CAS PubMed Google Scholar * Lange, U. & Hausner, W. Transcriptional
fidelity and proofreading in Archaea and implications for the mechanism of TFS-induced RNA cleavage. _Mol. Microbiol._ 52, 1133–1143 (2004). Article CAS PubMed Google Scholar * Laptenko,
O., Lee, J., Lomakin, I. & Borukhov, S. Transcript cleavage factors GreA and GreB act as transient catalytic components of RNA polymerase. _EMBO J._ 22, 6322–6334 (2003). Article CAS
PubMed PubMed Central Google Scholar * Yuzenkova, Y. et al. Control of transcription elongation by GreA determines rate of gene expression in Streptococcus pneumoniae. _Nucleic Acids
Res._ 42, 10987–10999 (2014). Article CAS PubMed PubMed Central Google Scholar * Fouqueau, T. et al. The transcript cleavage factor paralogue TFS4 is a potent RNA polymerase inhibitor.
_Nat. Commun._ 8, 1914 (2017). Article ADS PubMed PubMed Central CAS Google Scholar * Ortmann, A. C. et al. Transcriptome analysis of infection of the archaeon Sulfolobus solfataricus
with Sulfolobus turreted icosahedral virus. _J. Virol._ 82, 4874–4883 (2008). Article CAS PubMed PubMed Central Google Scholar * Hirata, A., Klein, B. J. & Murakami, K. S. The X-ray
crystal structure of RNA polymerase from Archaea. _Nature_ 451, 851–854 (2008). Article ADS CAS PubMed PubMed Central Google Scholar * Wojtas, M. N., Mogni, M., Millet, O., Bell, S.
D. & Abrescia, N. G. Structural and functional analyses of the interaction of archaeal RNA polymerase with DNA. _Nucleic Acids Res._ 40, 9941–9952 (2012). Article CAS PubMed PubMed
Central Google Scholar * Dienemann, C., Schwalb, B., Schilbach, S. & Cramer, P. Promoter distortion and opening in the RNA polymerase II cleft. _Mol. Cell_ 73, 97–106 (2019). Article
CAS PubMed Google Scholar * Briand, J. F. et al. Partners of Rpb8p, a small subunit shared by yeast RNA polymerases I, II and III. _Mol. Cell Biol._ 21, 6056–6065 (2001). Article CAS
PubMed PubMed Central Google Scholar * Grohmann, D. et al. The initiation factor TFE and the elongation factor Spt4/5 compete for the RNAP clamp during transcription initiation and
elongation. _Mol. Cell_ 43, 263–274 (2011). Article CAS PubMed PubMed Central Google Scholar * Werner, F. & Weinzierl, R. O. A recombinant RNA polymerase II-like enzyme capable of
promoter-specific transcription. _Mol. Cell_ 10, 635–646 (2002). Article CAS PubMed Google Scholar * Blombach, F. et al. Archaeal TFEalpha/beta is a hybrid of TFIIE and the RNA
polymerase III subcomplex hRPC62/39. _Elife_ 4, e08378 (2015). Article PubMed PubMed Central CAS Google Scholar * Blombach, F., Smollett, K. L., Grohmann, D. & Werner, F. Molecular
mechanisms of transcription initiation-structure, function, and evolution of TFE/TFIIE-Like factors and open complex formation. _J. Mol. Biol._ 428, 2592–2606 (2016). Article CAS PubMed
Google Scholar * He, Y. et al. Near-atomic resolution visualization of human transcription promoter opening. _Nature_ 533, 359–365 (2016). Article ADS CAS PubMed PubMed Central Google
Scholar * Bernecky, C., Plitzko, J. M. & Cramer, P. Structure of a transcribing RNA polymerase II-DSIF complex reveals a multidentate DNA-RNA clamp. _Nat. Struct. Mol. Biol._ 24,
809–815 (2017). Article CAS PubMed Google Scholar * Deng, L., Garrett, R. A., Shah, S. A., Peng, X. & She, Q. A novel interference mechanism by a type IIIB CRISPR-Cmr module in
sulfolobus. _Mol. Microbiol._ 87, 1088–1099 (2013). Article CAS PubMed Google Scholar * Engel, C., Sainsbury, S., Cheung, A. C., Kostrewa, D. & Cramer, P. RNA polymerase I structure
and transcription regulation. _Nature_ 502, 650–655 (2013). Article ADS CAS PubMed Google Scholar * Fernandez-Tornero, C., Bottcher, B., Rashid, U. J. & Muller, C. W. Analyzing RNA
polymerase III by electron cryomicroscopy. _RNA Biol._ 8, 760–765 (2011). Article CAS PubMed PubMed Central Google Scholar * Girbig, M. et al. Cryo-EM structures of human RNA polymerase
III in its unbound and transcribing states. _Nat. Struct. Mol. Biol._ 28, 210–219 (2021). Article CAS PubMed PubMed Central Google Scholar * Fouqueau, T., Zeller, M. E., Cheung, A. C.,
Cramer, P. & Thomm, M. The RNA polymerase trigger loop functions in all three phases of the transcription cycle. _Nucleic Acids Res._ 41, 7048–7059 (2013). Article CAS PubMed PubMed
Central Google Scholar * Kostrewa, D. et al. RNA polymerase II-TFIIB structure and mechanism of transcription initiation. _Nature_ 462, 323–330 (2009). Article ADS CAS PubMed Google
Scholar * Schulz, S. et al. TFE and Spt4/5 open and close the RNA polymerase clamp during the transcription cycle. _Proc. Natl Acad. Sci. USA_ 113, E1816–E1825 (2016). Article CAS PubMed
PubMed Central Google Scholar * Sheppard, C. et al. A non-bacterial transcription factor inhibits bacterial transcription by a multipronged mechanism. _RNA Biol._ 10, 495–501 (2013).
Article CAS PubMed PubMed Central Google Scholar * Vorlander, M. K. et al. Structural basis for RNA polymerase III transcription repression by Maf1. _Nat. Struct. Mol. Biol._ 27,
229–232 (2020). Article PubMed PubMed Central CAS Google Scholar * Neyer, S. et al. Structure of RNA polymerase I transcribing ribosomal DNA genes. _Nature_ 540, 607–610 (2016). Article
ADS CAS PubMed Google Scholar * Sharifi, S. & Bierhoff, H. Regulation of RNA polymerase I transcription in development, disease, and aging. _Annu. Rev. Biochem._ 87, 51–73 (2018).
Article CAS PubMed Google Scholar * Fernandez-Tornero, C. RNA polymerase I activation and hibernation: unique mechanisms for unique genes. _Transcription_ 9, 248–254 (2018). Article CAS
PubMed PubMed Central Google Scholar * Deighan, P. & Hochschild, A. Conformational toggle triggers a modulator of RNA polymerase activity. _Trends Biochem. Sci._ 31, 424–426 (2006).
Article CAS PubMed Google Scholar * Gourse, R. L. et al. Transcriptional responses to ppGpp and DksA. _Annu. Rev. Microbiol._ 72, 163–184 (2018). Article CAS PubMed PubMed Central
Google Scholar * Zuo, Y., Wang, Y. & Steitz, T. A. The mechanism of E. coli RNA polymerase regulation by ppGpp is suggested by the structure of their complex. _Mol. Cell_ 50, 430–436
(2013). Article CAS PubMed PubMed Central Google Scholar * Agapov, A., Esyunina, D. & Kulbachinskiy, A. Gre-family factors modulate DNA damage sensing by Deinococcus radiodurans RNA
polymerase. _RNA Biol._ 16, 1711–1720 (2019). Article PubMed PubMed Central Google Scholar * Esyunina, D., Agapov, A. & Kulbachinskiy, A. Regulation of transcriptional pausing
through the secondary channel of RNA polymerase. _Proc. Natl Acad. Sci. USA_ 113, 8699–8704 (2016). Article CAS PubMed PubMed Central Google Scholar * Vassylyev, D. G., Vassylyeva, M.
N., Perederina, A., Tahirov, T. H. & Artsimovitch, I. Structural basis for transcription elongation by bacterial RNA polymerase. _Nature_ 448, 157–162 (2007). Article ADS CAS PubMed
Google Scholar * Vassylyev, D. G. et al. Structural basis for substrate loading in bacterial RNA polymerase. _Nature_ 448, 163–168 (2007). Article ADS CAS PubMed Google Scholar *
Weixlbaumer, A., Leon, K., Landick, R. & Darst, S. A. Structural basis of transcriptional pausing in bacteria. _Cell_ 152, 431–441 (2013). Article CAS PubMed PubMed Central Google
Scholar * Fernandez-Tornero, C. et al. Crystal structure of the 14-subunit RNA polymerase I. _Nature_ 502, 644–649 (2013). Article ADS CAS PubMed Google Scholar * Rittershaus, E. S.,
Baek, S. H. & Sassetti, C. M. The normalcy of dormancy: common themes in microbial quiescence. _Cell Host Microbe_ 13, 643–651 (2013). Article CAS PubMed PubMed Central Google
Scholar * Ferreira, R., Schneekloth, J. S., Jr., Panov, K. I., Hannan, K. M. & Hannan, R. D. Targeting the RNA polymerase I transcription for cancer therapy comes of age. _Cells_ 9, 266
(2020). * Kortright, K. E., Chan, B. K., Koff, J. L. & Turner, P. E. Phage therapy: a renewed approach to combat antibiotic-resistant bacteria. _Cell Host Microbe_ 25, 219–232 (2019).
Article CAS PubMed Google Scholar * Cheng, K., Wilkinson, M., Chaban, Y. & Wigley, D. B. A conformational switch in response to Chi converts RecBCD from phage destruction to DNA
repair. _Nat. Struct. Mol. Biol._ 27, 71–77 (2020). Article CAS PubMed PubMed Central Google Scholar * Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for
improved cryo-electron microscopy. _Nat. Methods_ 14, 331–332 (2017). Article CAS PubMed PubMed Central Google Scholar * Zhang, K. Gctf: real-time CTF determination and correction. _J.
Struct. Biol._ 193, 1–12 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in
RELION-3. _Elife_ 7, e42166 (2018). * Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. _Nat.
Methods_ 14, 290–296 (2017). Article CAS PubMed Google Scholar * Nakane, T., Kimanius, D., Lindahl, E. & Scheres, S. H. Characterisation of molecular motions in cryo-EM
single-particle data by multi-body refinement in RELION. _Elife_ 7, e36861 (2018). * Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. _Acta Crystallogr.
D Struct. Biol._ 74, 531–544 (2018). Article CAS PubMed PubMed Central Google Scholar * Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. _Acta
Crystallogr. D Biol. Crystallogr_ 60, 2126–2132 (2004). Article PubMed CAS Google Scholar * Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state.
_J. Mol. Biol._ 372, 774–797 (2007). Article CAS PubMed Google Scholar * Laskowski, R. A. & Swindells, M. B. LigPlot+: multiple ligand-protein interaction diagrams for drug
discovery. _J. Chem. Inf. Model_ 51, 2778–2786 (2011). Article CAS PubMed Google Scholar * Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and
analysis. _J. Comput. Chem._ 25, 1605–1612 (2004). Article CAS PubMed Google Scholar * Jun, S. H. et al. Direct binding of TFEalpha opens DNA binding cleft of RNA polymerase. _Nat.
Commun._ 11, 6123 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Xu, J. et al. Structural basis for the initiation of eukaryotic transcription-coupled DNA repair.
_Nature_ 551, 653–657 (2017). Article ADS CAS PubMed PubMed Central Google Scholar * Pei, J., Tang, M. & Grishin, N. V. PROMALS3D web server for accurate multiple protein sequence
and structure alignments. _Nucleic Acids Res._ 36, W30–W34 (2008). Article CAS PubMed PubMed Central Google Scholar * Gouet, P., Robert, X. & Courcelle, E. ESPript/ENDscript:
extracting and rendering sequence and 3D information from atomic structures of proteins. _Nucleic Acids Res._ 31, 3320–3323 (2003). Article CAS PubMed PubMed Central Google Scholar *
Zillig, W. et al. Screening for sulfolobales, their plasmids and their viruses in Icelandic Solfataras. _Syst. Appl. Microbiol._ 16, 609–628 (1994). Article CAS Google Scholar *
Prangishvili, D. et al. Structural and genomic properties of the hyperthermophilic archaeal virus ATV with an extracellular stage of the reproductive cycle. _J. Mol. Biol._ 359, 1203–1216
(2006). Article CAS PubMed Google Scholar * Sadian, Y. et al. Molecular insight into RNA polymerase I promoter recognition and promoter melting. _Nat. Commun._ 10, 5543 (2019). Article
ADS CAS PubMed PubMed Central Google Scholar * Ehara, H. et al. Structure of the complete elongation complex of RNA polymerase II with basal factors. _Science_ 357, 921–924 (2017).
Article ADS CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We would like to thank Diamond Light Source for access to the cryo-EM facilities at the UK National electron
Bio-Imaging Centre (eBIC, proposal EM20287) funded by the Wellcome Trust, the Medical Research Council UK and the Biotechnology and Biological Sciences Research Council. Cryo-EM data for
this investigation were also collected at the ISMB EM facility at Birkbeck College, University of London with financial support from Wellcome (202679/Z/16/Z and 206166/Z/17/Z). We would like
to thank Dr. David Houldershaw for IT support and Giulia Zanetti for the script writing. We are very grateful to Jerome Gouge, Anthony Roberts and Christoph Müller for discussions, comments
and suggestions. Research in the RNAP laboratory at UCL is funded by a Wellcome Investigator Award in Science to FW (WT 207446/Z/17/Z) with the title Mechanisms and Regulation of RNAP
transcription. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * RNAP Laboratory, Institute for Structural and Molecular Biology, University College London, London, UK Simona Pilotto, Thomas
Fouqueau, Dorota Matelska & Finn Werner * Institute for Structural and Molecular Biology, Birkbeck College, London, UK Natalya Lukoyanova & Luis Miguel Díaz-Santín * Section of
Virology, Department of Infectious disease, Imperial College London, London, UK Carol Sheppard * Department of Microbiology, Institut Pasteur, Paris, France Soizick Lucas-Staat * Ivane
Javakhishvili Tbilisi State University, Tbilisi, Georgia David Prangishvili * School of Biochemistry, University of Bristol, Bristol, UK Alan C. M. Cheung Authors * Simona Pilotto View
author publications You can also search for this author inPubMed Google Scholar * Thomas Fouqueau View author publications You can also search for this author inPubMed Google Scholar *
Natalya Lukoyanova View author publications You can also search for this author inPubMed Google Scholar * Carol Sheppard View author publications You can also search for this author inPubMed
Google Scholar * Soizick Lucas-Staat View author publications You can also search for this author inPubMed Google Scholar * Luis Miguel Díaz-Santín View author publications You can also
search for this author inPubMed Google Scholar * Dorota Matelska View author publications You can also search for this author inPubMed Google Scholar * David Prangishvili View author
publications You can also search for this author inPubMed Google Scholar * Alan C. M. Cheung View author publications You can also search for this author inPubMed Google Scholar * Finn
Werner View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS S.P., N.L., L.M.D. and A.C. worked on cryo-EM data, T.F. and C.S. prepared proteins
and carried out cross-linking and Western blotting, S.L.S. and D.P. carried out virus infection experiments, D.M. contributed to the identification of Rpo8 mismatch, S.P. and F.W. wrote the
manuscript, and F.W. conceived and planned the project. CORRESPONDING AUTHOR Correspondence to Finn Werner. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing
interests. ADDITIONAL INFORMATION PEER REVIEW INFORMATION _Nature Communications_ thanks Sergei Borukhov and Hauke Hillen for their contribution to the peer review of this work. Peer
reviewer reports are available. PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY MOVIE 1 REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS
OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or
format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or
other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in
the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Pilotto, S., Fouqueau, T.,
Lukoyanova, N. _et al._ Structural basis of RNA polymerase inhibition by viral and host factors. _Nat Commun_ 12, 5523 (2021). https://doi.org/10.1038/s41467-021-25666-5 Download citation *
Received: 04 June 2021 * Accepted: 19 August 2021 * Published: 17 September 2021 * DOI: https://doi.org/10.1038/s41467-021-25666-5 SHARE THIS ARTICLE Anyone you share the following link with
will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative








