- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT ABCG2 is a multidrug transporter that affects drug pharmacokinetics and contributes to multidrug resistance of cancer cells. In previously reported structures, the reaction cycle
was halted by the absence of substrates or ATP, mutation of catalytic residues, or the presence of small-molecule inhibitors or inhibitory antibodies. Here we present cryo-EM structures of
ABCG2 under turnover conditions containing either the endogenous substrate estrone-3-sulfate or the exogenous substrate topotecan. We find two distinct conformational states in which both
the transport substrates and ATP are bound. Whereas the state turnover-1 features more widely separated NBDs and an accessible substrate cavity between the TMDs, turnover-2 features
semi-closed NBDs and an almost fully occluded substrate cavity. Substrate size appears to control which turnover state is mainly populated. The conformational changes between turnover-1 and
turnover-2 states reveal how ATP binding is linked to the closing of the cytoplasmic side of the TMDs. The transition from turnover-1 to turnover-2 is the likely bottleneck or rate-limiting
step of the reaction cycle, where the discrimination of substrates and inhibitors occurs. SIMILAR CONTENT BEING VIEWED BY OTHERS RHODAMINE6G AND HŒCHST33342 NARROW BMRA CONFORMATIONAL
SPECTRUM FOR A MORE EFFICIENT USE OF ATP Article Open access 18 February 2025 THE NET ELECTROSTATIC POTENTIAL AND HYDRATION OF ABCG2 AFFECT SUBSTRATE TRANSPORT Article Open access 18 August
2023 ASYMMETRIC DRUG BINDING IN AN ATP-LOADED INWARD-FACING STATE OF AN ABC TRANSPORTER Article 20 December 2021 INTRODUCTION ABCG2 is an ABC transporter of broad substrate specificity
expressed in various tissues and tissue barriers1,2,3,4. Among its endogenous substrates are steroids, including estrone-3-sulfate (E1S), and the transporter has been reported to contribute
to renal excretion of uric acid5. ABCG2 also functions as a multidrug transporter and exports a wide range of xenobiotics and pharmaceuticals, thus strongly impacting their
pharmacokinetics1,2,3. Genetic polymorphisms of ABCG2 can lead to its dysfunction, which is associated with hyperuricemia and hypertension in humans6. Due to its broad substrate specificity,
ABCG2 contributes to multidrug resistance in cancer7,8. For example, ovarian tumor and medulloblastoma cells over-expressing ABCG2 have shown increased resistance to chemotherapeutic agent
including topotecan or mitoxantrone9,10. Considerable efforts have been directed at developing specific inhibitors of ABCG2 to counteract its activity in protecting tumor cells from
anti-cancer drugs11,12,13,14,15,16,17. At present, the clinical applicability of such compounds is limited by their insufficient specificity, toxicity, or poor oral availability18. A
detailed understanding of the mechanism of ABCG2 is therefore essential to advance the development of small-molecule modulators or inhibitors. A series of initial structures have revealed
the architecture of human ABCG2 and have provided insight into its binding of endogenous substrates as well as small-molecule inhibitors19,20,21. In the inward-open conformation, the pair of
transmembrane domains (TMDs) form a slit-like cavity (cavity 1) that serves as a substrate-binding pocket. Two phenylalanine side chains, one from each ABCG2 monomer, clamp substrates
between their phenyl rings21,22,23,24. This can rationalize that ABCG2 substrates are generally flat, polycyclic, and hydrophobic compounds20. Inhibitors can also bind in cavity 1, both
competing with substates for the binding pocket and interfering with the closing of the TMD dimer interface during the transport cycle19,20,21. Recent structural studies revealed that
exogenous substrates (cytotoxic drugs used to treat various cancers) bind as single copies at the same general location as the endogenous substrate E1S22,23. In addition to inward-open
structures, the structure of a variant with reduced catalytic activity, ABCG2E211Q, revealed an ATP-bound conformation with a closed nucleotide-binding domain (NBD) dimer and a collapsed
translocation pathway. This was taken as evidence supporting an ATP-driven, peristaltic extrusion mechanism21. The common denominator of all published ABCG2 structures is that they
represented trapped states determined under conditions where the transporter was prevented from cycling through its catalytically relevant conformations. This was accomplished either by the
removal of ATP or transport substrates or by the addition of small-molecule inhibitors or externally binding inhibitory Fab fragments such as 5D3-Fab19,25. The strategy of locking
intermediate states or reducing conformational flexibility has been at the heart of most high-resolution structural studies of multidrug ABC transporters since the publication of the Sav1866
structure26. Mutation of the catalytically essential Walker-B glutamate has often been used to trap ATP-bound states, where the nucleotide is sandwiched between the NBDs21,27. Occasionally,
disulfide cross-linking was employed to trap intermediate states28,29. Such approaches were essential when using X-ray crystallography as a structural technique, since excessive
conformational dynamics interfere with the generation of well-ordered 3D crystals. On the downside, these conditions differed from the physiological environment, where substrates, ATP, ADP,
and Mg2+ are all present. As a result, the catalytic cycle of ABCG2 and other multidrug transporters is insufficiently understood. While conformational rigidity is also beneficial for
cryo-EM approaches, the single-particle nature of cryo-EM studies provides an opportunity to investigate multidrug ABC transporters under turnover conditions, where distinct conformations
are allowed to co-exist30. This approach is essential for understanding the transport mechanism because it reduces the chance of visualizing physiologically irrelevant conformational states
or intermediates. In the present study, we reconstitute human ABCG2 in lipid nanodiscs, which provide a near-native environment containing phospholipids and cholesterol. We apply turnover
conditions using two distinct substrates and find a single major turnover state for the endogenous steroid E1S and two states for the larger, exogenous drug topotecan. Our study reveals key
conformational changes that are essential for substrate recognition and translocation and allow ABCG2 to distinguish substrates from inhibitors. RESULTS STRUCTURES OF ABCG2 UNDER TURNOVER
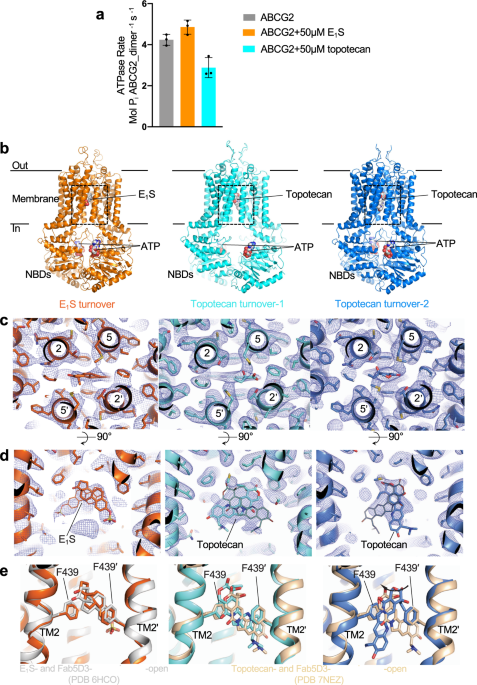
CONDITIONS We determined the ATPase activity of ABCG2 reconstituted in nanodiscs (Supplementary Fig. 1a) in the presence of E1S or topotecan (Fig. 1a). To ascertain that the two substrates
bound to nanodisc-reconstituted ABCG2, we measured the modulation of the ATPase rate compared to the absence of substrates. Compared to liposome-reconstituted ABCG2, the basal ATPase rate in
nanodiscs is markedly elevated, as was observed previously19,20,21. This rate was further increased in the presence of E1S, but decreased in the presence of topotecan (Fig. 1a). While the
absolute values are higher than in liposomes, this observation, combined with the structural data, suggests that the two substrates indeed bound to nanodisc-reconstituted ABCG2. To mimic
turnover conditions, we incubated ABCG2 with 5 mM ATP, 0.5 mM ADP, 5 mM MgCl2, and either 200 µM E1S or 100 µM topotecan, and applied the mixtures to EM grids. We collected single-particle
cryo-EM data of these two samples, which allowed us to determine three distinct high-resolution structures, one from the E1S-bound ABCG2 and two from topotecan-bound ABCG2. Both the NBDs and
TMDs were well resolved in all three structures (Supplementary Fig. 1b). Despite the fact that ABCG2 is a homodimer, we refined all maps in C1 symmetry and did not observe significant
structural differences between the two monomers. The turnover sample containing E1S revealed a single, well-defined conformation resolved at 3.4 Å resolution (Supplementary Fig. 2). In
contrast, the turnover sample containing topotecan revealed two well-ordered structures, which we termed turnover-1 and turnover-2 states, resolved at 3.1 Å and 3.4 Å, respectively
(Supplementary Fig. 3). The conformation of the E1S-bound structure was very similar to turnover-2 from the topotecan-containing sample (Supplementary Table 1). We, therefore, refer to the
E1S-bound structure as turnover-2 as well. The TMDs in the turnover states adopted inward-facing conformations but differed in their degree of opening towards the cytoplasmic side of the
membrane. Turnover-1 is a more open conformation, whereas turnover-2 is a more closed conformation. All three structures revealed densities for transport substrates and two bound
nucleotides, which were interpreted as bound ATP molecules (Fig. 1b). This choice was made because the relevant density in turnover-2 suggests bound ATP, while modeling ADP would leave
unexplained, additional density (Supplementary Fig. 4). For the turnover-1 state, the density of bound nucleotides is weaker because there are fewer contacts with the protein. While it would
in principle be possible to interpret the density as a mixture of ADP and ATP, we built ATP because it did not violate the observed density features and because the concentration of ATP in
the solution is ten times higher compared to that of ADP. While we assume that a closed conformation similar to that observed in the structure of the ATP-bound ABCG2_E211Q_ variant is
present under turnover condition21, we did not observe a defined class of such particles in our turnover samples. This suggests that this closed conformation is not a low-energy state of the
wild-type protein under turnover conditions. We also did not observe a collapsed apo-state similar to that reported recently for apo-ABCG222. In the topotecan-bound ABCG2 sample, the
dominant class (88% of the ordered particles) belonged to the turnover-1 state, whereas 12% particles belonged to the turnover-2 state (Supplementary Fig. 3). This suggests that turnover-1
is the lowest-energy state of ABCG2 in the presence of topotecan, whereas turnover-2 is the lowest-energy state in the presence of E1S. In all turnover structures, the leucine gate (formerly
called leucine plug), which separates cavity 1 from cavity 2, is closed (Supplementary Fig. 5a, b). E1S AND TOPOTECAN BINDING SITE We initially expected that under turnover conditions,
transport substrates would either be too mobile to be observed or not present at all. To our surprise, we found strong densities for bound substrates lodged in cavity 1 on the twofold
molecular symmetry axis of ABCG2 (Fig. 1c, d). Substrate recognition and binding are similar as in the previously reported, inward-open structures21,22,23. The polycyclic cores of E1S and
topotecan are sandwiched between the F439 side chains of the two ABCG2 protomers (Fig. 1c, d). Only one substrate molecule can be fitted inside the density, as fitting two molecules would
introduce serious steric clashes (Fig. 1d). The density suggests a certain degree of flexibility of bound substrates, in particular for topotecan (Fig. 1d). In the structure containing E1S
(turnover-2), the substrates could be fit in two orientations related approximately by a 180° rotation. Due to the C1 processing of the data, one orientation is more prominent. When compared
to the previously reported structure of E1S- and 5D3-Fab-bound ABCG2 in an inward-open conformation, E1S appeared slightly shifted (~1 Å) towards the leucine gate and thus towards the
external side of the transporter (Fig. 1e). In the topotecan-containing turnover structures, the shape of the density feature covering the drug is different in turnover-1 versus turnover-2.
In turnover-1, topotecan can be fitted in two orientations related approximately by a 180° rotation (Fig. 1d middle), similar to what was observed in an earlier structure of topotecan- and
5D3-Fab-bound, inward-open ABCG223, and with no significant shift of bound topotecan in the direction of the leucine gate. In turnover-2, the topotecan density was narrower in the horizontal
dimension and longer in the vertical dimension, probably reflecting a rotation of the drug compared to the turnover-1 state (Fig. 1d right). The changes in EM density could be visualized by
3D variability analysis of the final particles using the program cryoSPARC (Suppl. movies 1 and 2)31,32. COUPLED NBD AND TMD MOTIONS ABC transporters use the energy of ATP binding and
hydrolysis to translocate substrates across the membrane via alternating access mechanism33,34,35. In the course of the ATPase cycle, the NBDs cycle between closed and open conformations to
hydrolyze ATP or to release ADP and phosphate. This drives conformational changes in the TMDs. The separation of the NBDs in our turnover-1 and turnover-2 structures is smaller than that in
the fully inward-open, topotecan-bound ABCG2 structure23, but larger than that in the ATP-bound structure of ABCG2E211Q (Fig. 2a)21. A comparison of these four structures shows that as the
NBDs approach each other, so do the cytoplasmic sides of the TMDs (Fig. 2a). The TMDs rotate relative to the NBDs (Supplementary Fig. 5c). Compared to the inward-open conformation, access to
cavity 1 from the inner leaflet of the membrane is reduced in turnover-1, as the gap between transmembrane helices TM1 and TM5’ of the opposing ABCG2 monomer is narrower (Fig. 2b, c). In
turnover-2, the gap is even smaller, causing the entrance to cavity 1 from within the membrane to be completely sealed and even the opening to the cytosol to be reduced. As a result, a
larger substrate such as topotecan could not enter into, or exit from, cavity 1 without spreading of the cytoplasmic sides of the TMDs (Fig. 2c). The degree of NBD closing and TMD gap
narrowing can be measured by comparing the distances of the coupling helices (CpH), which are located in the first intracellular loop of the TMDs and transmit conformational changes at the
NBD-TMD interface. In turnover-1, the coupling helices are moved towards each other by ~1 Å compared to the fully inward-open, topotecan- and 5D3-Fab-bound structure (Fig. 2d). In
turnover-2, the distance between the coupling helices is reduced by another 4.0 Å. The distances between the TMDs have shifted accordingly (Supplementary Fig. 6). This reveals a strict
coupling between NBD and TMD closing. In addition to the motion of the NBDs as full domains, we observed a rotation of the RecA-like and the helical sub-domains relative to each other, which
caused the helical sub-domain to further approach the opposite NBD. Superimposing the RecA-like domains and measuring the relative angles of the α-helical domains revealed rotations of 2°,
19°, and 24° between the fully inward-open conformation and turnover-1, turnover-2, and the ATP-bound closed state (Supplementary Fig. 7). A comparison of the topotecan-bound turnover-1 and
turnover-2 structures revealed a key NBD segment contributing to functionally relevant NBD–NBD and NDB–TMD domain contacts. As ABCG2 transitions from turnover-1 to turnover-2, the segment
between Q181 and V186 undergoes a conformational change and facilitates key inter-domain contacts (Fig. 3a, b). V186 is the first residue of the VSGGE motif (equivalent to LSGGQ more
commonly found in ABC transporters)36. This motif is among the most conserved in ABC transporters and pins bound nucleotides against the Walker-A and Walker-B motifs of the opposing NBD. We
now found that in turnover-2, Q181 and F182 facilitated contacts between TM1 of one ABCG2 monomer and TM5’ of the other (Fig. 3a, b), thus helping stabilize the TMD domain closure. In
addition, the arginine residue R184 provides a direct contact, in the form of a cation-π interaction, with the adenine moiety of the ATP molecule bound to the opposite NBD (Fig. 3d). Neither
of these contacts is formed in the turnover-1 structure (Fig. 3c). We analyzed the role of R184 by mutating it to an alanine (Supplementary Fig. 8a, b). We reconstituted the purified
ABCG2_R184A_ variant in proteoliposomes and performed ATPase and transport assays (Fig. 3e, f). We found that in line with its location in the structure, both the ATPase and transport
activities of ABCG2_R184A_ were greatly reduced. As was observed for WT ABCG2, the ATPase rate of ABCG2_R184A_ was stimulated by E1S and topotecan (Fig. 3e). Furthermore, the E1S transport
rate was similarly reduced for WT ABCG2 and ABCG2_R184A_ in the presence of topotecan, suggesting that the mutation of R184 to alanine neither interfered with substrate binding nor abolished
the NBD-TMD coupling (Supplementary Fig 9a). Given that the EC50 values of E1S- and topotecan-induced stimulation of the ATPase activity of ABCG2 were similar (15.7 µM and 11.7 µM,
respectively, Supplementary Table 2)21,23, the reduction of the initial E1S transport rate by ~50% in the presence of topotecan is in line with the structural data in that topotecan and E1S
bind and compete for the same binding pocket both in WT ABCG2 and in the ABCG2R184A variant (Supplementary Fig. 9b). THE ROLE OF R482 The single-nucleotide polymorphisms R482G and R482T,
originally cloned from drug-resistant cancer cell lines37,38, have been reported to alter the substrate specificity of ABCG239. Cells expressing ABCG2R482G were found to efficiently extrude
rhodamine-123 or doxorubicin but were less resistant to topotecan, an anticancer drug that inhibits topoisomerase7,38,40,41. It was, therefore, suggested that R482 influenced substrate
transport and ATP hydrolysis, but not substrate binding42,43. R482 is located in TM3 and does not directly contact bound drugs21,22,23. Rather, it contacts TM2 which contains the key residue
F439 that interacts with substrates in cavity 119,20,21. Intriguingly, we found that the side chain of R482 adopts distinct conformations in the turnover-1 and turnover-2 structures (Fig.
4). In turnover-1, the side chain points towards the cytoplasmic side of the membrane, where the guanidinium group forms a hydrogen bond with the side-chain of S443 of TM2 (Fig. 4b and c,
left). In contrast, the R482 side chain is rotated towards the external side of the membrane in turnover-2, where the guanidinium group contacts side chains and main-chain atoms from TM2 and
TM4 in the direct vicinity of F439 (Fig. 4b, c, right). This suggests a structural role for R482 in facilitating the conformational changes required to transition from turnover-1 to the
turnover-2 state (visualized in Suppl. movie 2). The impact of mutations of R482 on drug recognition is therefore likely of an allosteric nature. STERIC CLASHES OF INHIBITORS IN TURNOVER-2
STATE Our structures provide additional insight into the mechanism of small-molecule inhibitors of ABCG2. Previous structures revealed that the inhibitors MZ29 (a derivative of Ko143) and
MB136 (a derivative of tariquidar) bound to cavity 1 of inward-open ABCG2, which was bound to 5D3-Fab20. We find that while the size of cavity 1 in our turnover-1 structure is slightly
smaller than that of the fully inward-open conformation, only minor steric clashes would exist with the tert-butyl group of MZ29. In contrast, the strongly reduced size of cavity 1 in the
turnover-2 conformation would lead to major steric clashes with bound inhibitor molecules. Figure 5 reveals that whereas there is sufficient space in turnover-2 for bound substrates (E1S or
topotecan), the inhibitors MZ29 and MB136 could not fit. The concerted motions of TM1, TM2, and TM5 reduce the volume of cavity 1 by approximately one-third, from ~1300 Å3 in turnover-1 to
~830 Å3 in turnover-244, which is incompatible with the binding of the inhibitor molecules. We conclude that with small-molecule inhibitors bound, ABCG2 may be able to adopt the conformation
of turnover-1, but could not adopt the conformation of the turnover-2 state. Unlike in B-subfamily ABC transporters, where longer intracellular loops allow additional flexibility, binding
of inhibitors to ABCG2 prevents the TMDs to close at the cytoplasmic side and consequently the NBDs from forming a closed dimer arrangement, which is required for ATP hydrolysis. This can
explain why ABCG2 has a strongly reduced ATPase rate in the presence of inhibitors20, whereas ABCB1 does not20,29,45,46. DISCUSSION In contrast to earlier structural studies of ABCG2, we
could not predict whether the transporter would adopt a single conformation or multiple distinct conformations under turnover conditions. We also could not predict which state would
correspond to the lowest energy state in the transport cycle. Our finding that two distinct inward-open conformations containing both a bound transport substrate and two ATP molecules
corresponded to the lowest energy states was nevertheless unexpected. This arrangement was often considered to be of higher energy or even transient in ABC transporters, and thus expected to
quickly convert to intermediates where either the transport substrate was absent, or ATP had been hydrolyzed47,48. Indeed, an analysis of bovine ABCC1, another multidrug resistance protein,
captured a post-hydrolysis state under turnover conditions featuring a closed NBD dimer and a (slightly) outward-open substrate translocation pathway. This resembled a previously reported
structure of a catalytically impaired ABCC1 variant containing a Walker-B glutamate to glutamine mutation47. In contrast to bovine ABCC1, a bacterial ABC transporter of peptides, TmrAB, was
found to adopt multiple inward-open conformations under turnover conditions. These varied in how widely the NBDs were separated, the degree of inward opening of the translocation pathway,
and whether bound nucleotides or substrates could be identified48. Our findings with human ABCG2 are distinct from both of those studies, which most likely reflects the mechanistic diversity
within the ABC transporter superfamily34. By combining previously reported structures of trapped states and our turnover structures presented here, we can now propose a structure-based
hypothesis of the complete transport cycle of ABCG2 (Fig. 6). Only conformational states that are required for transport are included in this scheme. Our turnover structures have a key role
in this mechanistic scheme. Given that E1S did not promote the turnover-1 state, we concluded that ABCG2 can directly proceed to turnover-2 when smaller or endogenous substrates are bound.
We anticipate a similar situation with urate, an even smaller molecule than E1S. For these substrates, turnover-2 appears to immediately precede the rate-limiting step of the transport
cycle. In contrast, turnover-1 was the dominant class in the topotecan turnover sample. This suggests that the transition from turnover-1 to turnover-2 may be the step where exogenous
compounds are “tested” for their suitability as transport substrates. Because topotecan is larger than E1S, the transition from turnover-1 to turnover-2 is slower. We conclude that the
degree of TMD and NBD closing in the lowest energy intermediate correlates with the shape and size of ABCG2 substrates. More generally, compounds that bind in cavity 1 and allow ABCG2 to
adopt the turnover-2 conformation may be transported, even if their transport rates are slower than that of E1S. In contrast, in the presence of strongly bound inhibitors such as Ko143 and
its derivatives13,20, ABCG2 cannot advance to the turnover-2 conformation. Our results further assign a key role to the segment 181-184 of ABCG2. It was previously reported that the
polypeptide segment leading up to the LSGGQ motif appeared to have a role in stabilizing TMD-TMD interfaces in B-subfamily ABC transporters, and the term X-loop was introduced to account for
this role26. Rather than an aromatic residue to stabilize the adenine moiety of bound nucleotides, as observed in the A-loops of most ABC transporters27,49,50,51, ABCG2 relies on R184 from
the opposite protomer to assume this function. R184, therefore, appears to have a dual role in the turnover-2 state: It pioneers the contact between the two NBDs while simultaneously
strengthening the binding of ATP through a cation-π interaction with the adenine moiety. The initial NBD-NBD contact is probably important because the signature motif is not yet close enough
to interact with the ribose and triphosphate moieties of the ATP molecule. Our mechanistic scheme indicates two distinct levels of ATPase activity, a basal and a stimulated level (Fig. 6).
In the absence of substrates, ABCG2 displays a basal ATPase rate that generates 0.65 molecules of phosphate per ABCG2 dimer per second. In the presence of E1S, the ATPase rate increases to
2.4. We compared this value with the initial E1S transport rate, which is 0.1 molecules E1S per ABCG2 dimer per second. If ABCG2 hydrolyzes two molecules of ATP in one productive transport
cycle, the net futile ATPase rate in the presence of E1S is 2.2 molecules phosphate per ABCG2 dimer per second, which is 3.4 times higher than in the absence of E1S. We conclude that
substrates not only increase the ATPase rate of ABCG2 due to productive transport, but also increase the basal/futile ATPase rate. This suggests that ABCG2 may require multiple attempts
(each consuming ATP) before substrate extrusion is successfully accomplished. In both turnover states described here, the leucine gates are closed. This is notable in the case of turnover-2,
where not only the NBDs are semi-closed, but the cytoplasmic ends of the TMDs are also partially closed. This suggests that the opening of the leucine gate may only occur once the
cytoplasmic ends of the TM helices are further pushed together and peristaltic pressure is exerted on bound substrate. We speculate that at this point, ABCG2 must adopt an outward-facing
conformation, in which the leucine gates are opened and substrate can escape via cavity 2 into the extracellular environment. Such a conformation may be transient in nature given that we
have not observed it in our turnover samples. Further studies are required to capture ABCG2 or an appropriate ABCG2 variant in an outward-open conformation. In conclusion, our results
provide key insight into the transport cycle of ABCG2 and help understand how small-molecular compounds act as substrates or inhibitors. Our study may also have predictive value for
understanding the mechanisms of other ABC transporters, in particular of the G-subfamily. Given their diversity with respect to mechanistic details, it is quite possible that ABC
transporters belonging to other subfamilies will reveal distinct conformations or intermediate states most strongly populated under turnover conditions. METHODS EXPRESSION AND PURIFICATION
OF ABCG2 Human wild type ABCG2 (Uniprot: Q9UNQ0) or ABCG2 _R184A_ containing an N-terminal Flag-tag was expressed in HEK293-EBNA (Thermo Fisher Scientific) cells by transient transfection19.
Cells were incubated at 37°C for 48–60 h before harvesting. Harvested Cell were lysed using a Dounce homogenizer and solubilized with 1% DDM, 0.1% CHS (cholesteryl hemisuccinate) (w/v)
(Anatrace), 40 mM HEPES buffer pH 7.5, 150 mM NaCl, 10% (v/v) glycerol, 1 mM PMSF (phenylmethylsulfonyl fluoride), 2 μg ml−1 DNaseI (Roche), and protease inhibitor cocktail (Sigma). Lysed
cells were centrifuged at 100,000 _g_ and the supernatant was incubated with anti-Flag M2 affinity agarose gel (Sigma). ABCG2 was eluted with Flag peptide (Sigma) and applied to a Superdex
200 increase 10/300 column (GE Healthcare) in 40 mM HEPES, pH 7.5, 150 mM NaCl, 0.026% DDM and 0.0026% CHS (w/v). Peak fractions were collected for further use. ABCG2-NANODISC PREPARATION
Membrane scaffold protein (MSP) 1D1 was expressed in _E. coli_ and purified as described52. ABCG2 reconstitution in nanodiscs was performed as following methods19. In brief, brain polar
lipid (BPL, Avanti Polar Lipids) and CHS (cholesteryl hemisuccinate) were mixed at a 4:1 (w/w) ratio. Lipids were solubilized with a 3x molar excess of sodium cholate using a sonic bath.
Lipids were then mixed with MSP1D1 and detergent-purified ABCG2 at a molar ratio of 100:5:0.2 (lipid:MSP:ABCG2). Detergent was removed by addition of Bio-Beads SM-2 (Biorad) and incubated at
4 °C overnight. Biobeads were removed and the sample was centrifuged at 100,000 _g_ for 30 min. The supernatant was loaded on a Superdex 200 increase column and fractions containing
nanodisc-reconstituted ABCG2 were collected for Cryo-EM grid preparation or for functional assays. ABCG2-LIPOSOME PREPARATION ABCG2-containing proteoliposomes were prepared as
described19,53. In brief, BPL was mixed with cholesterol (Chol) at a 4:1 (w/w) ratio. Liposomes were resuspended in transport buffer (25 mM HEPES pH 7.5, 150 mM NaCl) and extruded using a
400 nm polycarbonate filter (Avanti Polar Lipids). To reconstitute ABCG2, liposomes were destabilized with 0.17% (v/v) Triton X100. Detergent-purified ABCG2 was mixed with liposomes at a
100:1 (w/w) lipid: protein ratio. Detergent was then removed using Bio-Beads, added in multiple batches. Proteoliposomes were collected using centrifugation at 100,000 _g_. The
proteoliposome pellet was resuspended in transport buffer at a final lipid concentration of 10 mg ml−1. ATPASE AND TRANSPORT ASSAYS ATPase assays were performed at 37 °C in the presence of 2
mM ATP and 10 mM MgCl2. Where indicated, E1S or topotecan was added at 50 μM. The concentration of inorganic phosphate released by the hydrolysis of ATP was measured by tracking absorbance
at 850 nm following the classical molybdate method54. ATPase rates were determined using linear regression in GraphPad Prism v8 and v9. For transport assays, proteoliposomes in transport
buffer (25 mM HEPES pH 7.5, 150 mM NaCl) were extruded through a 400 nm polycarbonate filter. MgCl2 (5 mM) and E1S (50 μM, containing mixtures of 3H-E1S and 1H-E1S) were added in the
presence or absence of topotecan and the samples were incubated for 5 min at 30 °C. Transport reactions were initiated by adding ATP (2 mM) and stopped by adding an aliquot to ice-cold
transport buffer containing unlabeled E1S (100 μM). Sample was filtered with a Multiscreen vacuum manifold (MSFBN6B filter plate, Millipore) and washed three times. Radioactivity trapped on
the filters was measured with the microplate scintillation counter (Perkin Elmer 2450 Microbeta2). Data were analyzed and curves were plotted using the nonlinear regression Michaelis–Menten
analysis and initial rates were calculated using the data points from the fitting curve (GraphPad Software, La Jolla, California, USA). CRYO-EM SAMPLE PREPARATION All grids were prepared
using a Vitrobot (FEI), with the environmental chamber set at 100% humidity and 4 °C. Nanodisc-reconstituted ABCG2 (1 mg ml−1) was incubated with 5 mM ATP, 5 mM MgCl2, 0.5 mM ADP, 100 μM
topotecan or 200 μM E1S at room temperature for 10 min. 3.5 μl sample was applied on glow-discharged Quantifoil carbon grids (300 meshes, R 1.2/1.3 copper) immediately after incubation.
Grids were blotted for 2.5 s with blot force 1 and flash-frozen in a mixture of liquid ethane and propane. CRYO-EM DATA ACQUISITION Cryo-EM data of ABCG2-topotecan turnover sample was
collected with a 300 keV Titan Krios (FEI) transmission electron microscope (TEM) equipped with a Gatan BioQuantum 1967 filter and a Gatan K3 camera. Images were recorded with three
exposures per hole using EPU 2 in super-resolution mode with a 20 eV slit width of the energy filter and at a nominal magnification of 130,000x, resulting in a calibrated super-resolution
pixel size of 0.33 Å. Defocus was set to vary from −0.6 to −2 μm. Each image was dose fractionated to 40 frames with 1.01 s total exposure time. The dose was 1.45 e−/Ų/frame (Total dose 58
e−/Ų). The super-resolution micrographs were down-sampled twice by Fourier cropping (to a pixel size of 0.66 Å), drift-corrected and dose-weighted using MotionCor255. Micrographs were
visually inspected, and bad micrographs were removed manually. The topotecan turnover dataset was composed of 24,177 movies from two sessions after removing bad micrographs. Cryo-EM data of
ABCG2-E1S turnover sample was collected on a 300 keV Titan Krios TEM equipped with a K2 Summit direct electron detector and a Quantum-LS energy filter (GIF) (20 eV zero-loss filtering; Gatan
Inc.). The microscope was operated with a nominal magnification of 165,000× (the actual magnification 60,975×). Automated data collections were performed using SerialEM with seven exposures
per hole using image-shift and coma-free setup56. Dose-fractionated movies were recorded in counting mode with a physical pixel size of 0.82 Å and the defocus was set in a range from −0.8
to −2.8 μm. Each movie was dose fractionated to 40 frames with 10 s total exposure time. The total dose was 50 e− /Å2. The recorded movie stacks were initially processed by MotionCor2 (FOCUS
integrated), including pre-processing procedures such as the gain-normalization, motion correction, and dose weighting57,58. The entire dataset of ABCG2-E1S turnover sample from two EM
sessions was composed of 15,460 movies. Cryo-EM data collection statistics in this study are presented in Supplementary Table 3. IMAGE PROCESSING The ABCG2-E1S turnover data were processed
separately with a similar procedure until 2D classifications (Supplementary Fig. 2). The aligned micrographs were manually sorted in FOCUS57 and imported to CryoSPARC 2.131. The contrast
transfer function (CTF) and defocus values were estimated on the dose-weighted micrographs using patch-based CTF in CryoSPARC. Images with poor quality were discarded. Defocus was calibrated
with a range of −0.8 to –2.8 μm. Images with a CTF-estimated resolution of better than 5.2 Å were selected, resulting in a total of 11,343 micrographs. A 2D template was produced by initial
2D classifications with manually picked particles. Particles of the full dataset were auto-picked with 2D templates. After multiple rounds of 2D classification, particles were combined for
3D classification. The merged data with 645,803 particles were subjected to three rounds of 3D classification to classify distinct conformations. A 3D reference was generated from ab-initio
3D reconstructions. Each round was followed by a heterogenous refinement. As shown in Supplementary Fig. 2, several setting parameters were tested and applied (Round 1: 2 classes, 0.1
similarity, force hard-classification; Round 2: 2 classes, 0.8 similarity, force hard-classification; Round 2: 3 classes, 0.1 similarity, force hard-classification), resulting in a particle
subset containing 221,160 particles. The best-resolved 3D class was further subjected to 3D nonuniform refinement (CryoSPARC). The map was refined with C1 symmetry by local refinement using
a soft-mask. The overall resolution was 3.4 Å at FSC = 0.143 cutoff. A local-resolution map was generated by MonoRes59. For the topotecan turnover sample, the drift corrected, dose-weighted
micrographs were imported in CryoSPARC v2.1531. Contrast transfer function (CTF) parameters were estimated with GCTF60. The calculated defocus parameters of the micrographs were −0.3 to −2.7
μm. 1,534 particles were manually picked from selected micrographs to generate a 2D template for auto-picking. 2,623,169 particles were picked and extracted from 23,574 micrographs. After
10 rounds of classifications, 1,486,797 particles belonging to ‘good’ 2D classes were selected. An initial model was generated in Ab-Initio Reconstruction and used as a reference for the
first round of classification. The selected particles from 2D classification were subjected to 3D classification with 4 classes and binned 3×. A good class from the first round of 3D
classification with 750,851 particles was selected. This class was refined to 3.8 Å in Homogeneous refinement. The class was subjected to a second round of 3D classification with three
classes and binned 3×. A good class with 455,604 particles was selected, and it was refined to 3.45 Å in Homogeneous refinement. A third round of 3D classification was done with 455,604
particles and three classes. An excellent class with 311,273 particles was selected, and it was refined to 3.34 Å in Homogeneous refinement. To check if resolution can be further improved by
3D classification, a fourth round of 3D classification was done with three classes and binned 3×. Two good classes with 299,139 particles were selected and refined to 3.28 Å in Homogeneous
refinement. The 299,139 particles subset was subjected to Global CTF Refinement followed by Local CTF refinement. Further 3D variability analysis showed there was heterogeneity in one
dimension for this particle subset. To deal with the heterogeneity, a fifth-round of 3D classification was performed with 3 classes and no bin. A minor class with 78,323 particles was
isolated with the NBDs in a semi-closed conformation. Further homogeneous, local refinement and post-processing of the minor 3D class resulted in a 3.4 Å map named topotecan turnover-2, with
automatically determined B factor of −92.8 Å2. The other two major classes with 220,816 particles showed NBD open conformation, thus they were combined. Further homogenous refinement, local
refinement, and post-processing resulted in a 3.1 Å map named topotecan turnover-1, with automatically determined B factor of −106.5 Å2. The final maps were refined in C1 symmetry and were
directly used for modeling. Local resolution maps were generated using CryoSPARC v2.15. The data processing pipeline is shown in Supplementary Fig. 3. A 3D variability analysis (3DVA) of the
topotecan turnover sample was performed with particle stacks that contributed to high-resolution 3D maps in cryoSPARC v2.1531. The aim was to explore heterogeneity in single-particle
cryo-EM data sets. A mask without lipids belt, three eigenvectors and 4 Å low-pass filtered were applied for 3DVA, generating simple linear “movies” of volumes. 3DVA outputs were visualized
with 3D Variability Display tool in cryoSPARC32. We observed both turnover-1 and turnover-2 conformations in the topotecan turnover sample, whereas we only observed the turnover-2
conformation in the E1S turnover sample. To avoid bias derived from reference map when doing 3D classification, we re-classified particles of the E1S turnover sample into 6-10 classes using
turnover-1 as a reference map. We expected to classify different conformations in this way. We observed some minor classes with separated NBDs. However, none of these classes produced EM
maps of sufficient resolution to identify secondary structures. We can therefore exclude that the turnover-1 state with well-defined features is present in meaningful amounts in the E1S
turnover sample. Turnover-2 is the only well-defined state of the E1S turnover sample. MODEL BUILDING AND REFINEMENT Coot 0.9 was used for all model building steps61. ABCG2-MZ29 model (PDB
6FFC) was docked into turnover maps and used as the reference for manual rebuilding20. The coordinate of topotecan was from ABCG2-topotecan-Fabs structure (PDB 7NEZ). E1S coordinate was from
ABCG2-E1S-Fab (PDB 6HCO). Cholesterol coordinate was from ABCG2-MZ29-Fab (PDB 6HIJ) and ATP coordinate was ABCG2E211Q-ATP (PDB: 6HBU)21. The ligands were fitted into the EM density in Coot
0.9. We generated the restraints in eLBOW of Phenix62. Both topotecan and E1S turnover structures were refined against their final maps respectively in real space refinement of Phenix63. In
the final refinement, reciprocal-space refinement of the B factors and minimization global refinements were applied together with standard geometry, rotamer, Ramachandran plot, Cβ,
non-crystallographic symmetry and secondary structure restraints. The quality of final model was assessed by MolProbity64. The refinement statistics are in Supplementary Table 3. For model
validation, we applied 0.3 Å random shifts to final models using phenix_pdb_tools65. The scrambled model was refined against one of the unfiltered half maps (half map A). The Fourier shell
correlation between refined scrambled model and half map A was plotted as FSCwork. The Fourier shell correlation between refined scrambled model and half map B was plotted as FSCfree. The
overlay between the FSCwork and FSCfree indicated no over-fitting presence. FIGURE PREPARATION Figures were prepared with PyMOL (The PyMOL Molecular Graphics System, Version 2.4.0
Schrödinger, LLC), GraphPad Prism v8, v9, and UCSF ChimeraX66. Movies were prepared with UCSF Chimera67. The volume of cavity 1 in ABCG2 turnover structures was determined using POCASA44.
DATA AVAILABILITY Atomic coordinates for the E1S-bound turnover-2 structure and the topotecan-bound turnover-1 and turnover-2 structures are deposited with the Protein Data Bank under
accession codes 7OJ8, 7OJH, and 7OJI, respectively. The cryo-EM density maps for the three structures were deposited in the Electron Microscopy Data Bank under accession codes EMD-12939
(E1S-bound turnover-2 state), EMD-12951 (topotecan-bound turnover-1 state) and EMD-12952 (topotecan-bound turnover-2 state). Source Data for Figs. 1a, 3e and 3f, Supplementary Figures 1a,
2e, 3f, 3g, 8a, 9a, and 9b are provided with this paper in the Source Data file. Raw EM data are available upon request. A Life Sciences Reporting Summary for this article is available.
Source data are provided with this paper. REFERENCES * Krishnamurthy, P. et al. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. _J. Biol.
Chem._ 279, 24218–24225 (2004). Article CAS PubMed Google Scholar * Kerr, I. D., Haider, A. J. & Gelissen, I. C. The ABCG family of membrane-associated transporters: you don’t have
to be big to be mighty. _Br. J. Pharm._ 164, 1767–1779 (2011). Article CAS Google Scholar * Enokizono, J., Kusuhara, H., Ose, A., Schinkel, A. H. & Sugiyama, Y. Quantitative
investigation of the role of breast cancer resistance protein (Bcrp/Abcg2) in limiting brain and testis penetration of xenobiotic compounds. _Drug Metab. Dispos._ 36, 995–1002 (2008).
Article CAS PubMed Google Scholar * Maliepaard, M. et al. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. _Cancer
Res._ 61, 3458–3464 (2001). CAS PubMed Google Scholar * Matsuo, H. et al. Common defects of ABCG2, a high-capacity urate exporter, cause gout: a function-based genetic analysis in a
Japanese population. _Sci. Transl. Med._ 1, 5ra11 (2009). Article PubMed CAS Google Scholar * Ishikawa, T., Aw, W. & Kaneko, K. Metabolic interactions of purine derivatives with
human ABC transporter ABCG2: genetic testing to assess gout risk. _Pharmaceuticals_ 6, 1347–1360 (2013). Article PubMed PubMed Central CAS Google Scholar * Gottesman, M. M., Fojo, T.
& Bates, S. E. Multidrug resistance in cancer: role of ATP-dependent transporters. _Nat. Rev. Cancer_ 2, 48–58 (2002). Article CAS PubMed Google Scholar * Szakacs, G., Paterson, J.
K., Ludwig, J. A., Booth-Genthe, C. & Gottesman, M. M. Targeting multidrug resistance in cancer. _Nat. Rev. Drug Discov._ 5, 219–234 (2006). Article CAS PubMed Google Scholar *
Morfouace, M. et al. ABCG2 transporter expression impacts group 3 medulloblastoma response to chemotherapy. _Cancer Res._ 75, 3879–3889 (2015). Article CAS PubMed PubMed Central Google
Scholar * Maliepaard, M. et al. Overexpression of the BCRP/MXR/ABCP gene in a topotecan-selected ovarian tumor cell line. _Cancer Res._ 59, 4559–4563 (1999). CAS PubMed Google Scholar *
Rabindran, S. K., Ross, D. D., Doyle, L. A., Yang, W. & Greenberger, L. M. Fumitremorgin C reverses multidrug resistance in cells transfected with the breast cancer resistance protein.
_Cancer Res._ 60, 47–50 (2000). CAS PubMed Google Scholar * Rabindran, S. K. et al. Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C.
_Cancer Res._ 58, 5850–5858 (1998). CAS PubMed Google Scholar * Allen, J. D. et al. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro
and in mouse intestine by a novel analogue of fumitremorgin C. _Mol. Cancer Ther._ 1, 417–425 (2002). CAS PubMed Google Scholar * Puentes, C. O. et al. Solid phase synthesis of
tariquidar-related modulators of ABC transporters preferring breast cancer resistance protein (ABCG2). _Bioorg. Med. Chem. Lett._ 21, 3654–3657 (2011). Article CAS PubMed Google Scholar
* Pick, A., Klinkhammer, W. & Wiese, M. Specific inhibitors of the breast cancer resistance protein (BCRP). _ChemMedChem_ 5, 1498–1505 (2010). Article CAS PubMed Google Scholar *
Kohler, S. C. & Wiese, M. HM30181 derivatives as novel potent and selective inhibitors of the breast cancer resistance protein (BCRP/ABCG2). _J. Med. Chem._ 58, 3910–3921 (2015). Article
PubMed CAS Google Scholar * Miyata, H. et al. Identification of febuxostat as a new strong ABCG2 inhibitor: potential applications and risks in clinical situations. _Front. Pharm._
7(2016). * Weidner, L. D. et al. The inhibitor Ko143 is not specific for ABCG2. _J. Pharm. Exp. Ther._ 354, 384–393 (2015). Article CAS Google Scholar * Taylor, N. M. I. et al. Structure
of the human multidrug transporter ABCG2. _Nature_ 546, 504–509 (2017). Article ADS CAS PubMed Google Scholar * Jackson, S. M. et al. Structural basis of small-molecule inhibition of
human multidrug transporter ABCG2. _Nat. Struct. Mol. Biol._ 25, 333 (2018). -+. Article CAS PubMed Google Scholar * Manolaridis, I. et al. Cryo-EM structures of a human ABCG2 mutant
trapped in ATP-bound and substrate-bound states. _Nature_ 563, 426–430 (2018). Article ADS CAS PubMed PubMed Central Google Scholar * Orlando, B. J. & Liao, M. ABCG2 transports
anticancer drugs via a closed-to-open switch. _Nat. Commun._ 11, 2264 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Kowal, J. et al. Structural basis of drug
recognition by the multidrug transporter ABCG2. _J. Mol. Biol_. 166980 (2021). * Gose, T. et al. ABCG2 requires a single aromatic amino acid to “clamp” substrates and inhibitors into the
binding pocket. _FASEB J._ 34, 4890–4903 (2020). Article CAS PubMed Google Scholar * Zhou, S. et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a
molecular determinant of the side-population phenotype. _Nat. Med._ 7, 1028–1034 (2001). Article CAS PubMed Google Scholar * Dawson, R. J. & Locher, K. P. Structure of a bacterial
multidrug ABC transporter. _Nature_ 443, 180–185 (2006). Article ADS CAS PubMed Google Scholar * Kim, Y. & Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound,
outward-facing conformation. _Science_ 359, 915–919 (2018). Article ADS CAS PubMed Google Scholar * Korkhov, V. M., Mireku, S. A. & Locher, K. P. Structure of AMP-PNP-bound vitamin
B12 transporter BtuCD-F. _Nature_ 490, 367–372 (2012). Article ADS CAS PubMed Google Scholar * Alam, A. et al. Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1.
_Proc. Natl Acad. Sci. USA_ 115, E1973–E1982 (2018). Article CAS PubMed PubMed Central Google Scholar * Frank, G. A. et al. Cryo-EM analysis of the conformational landscape of human
P-glycoprotein (ABCB1) during its catalytic cycle. _Mol. Pharm._ 90, 35–41 (2016). Article CAS Google Scholar * Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A.
cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. _Nat. Methods_ 14, 290–296 (2017). Article CAS PubMed Google Scholar * Punjani, A & Fleet, DJ. 3D
variability analysis: resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. _J. Struct. Biol_. 107702 (2021). * Beis, K. Structural basis for the
mechanism of ABC transporters. _Biochem. Soc. Trans._ 43, 889–893 (2015). Article CAS PubMed Google Scholar * Locher, K. P. Mechanistic diversity in ATP-binding cassette (ABC)
transporters. _Nat. Struct. Mol. Biol._ 23, 487–493 (2016). Article CAS PubMed Google Scholar * Rees, D. C., Johnson, E. & Lewinson, O. ABC transporters: the power to change. _Nat.
Rev. Mol. Cell Biol._ 10, 218–227 (2009). Article CAS PubMed PubMed Central Google Scholar * Hollenstein, K., Frei, D. C. & Locher, K. P. Structure of an ABC transporter in complex
with its binding protein. _Nature_ 446, 213–216 (2007). Article ADS CAS PubMed Google Scholar * Miyake, K. et al. Molecular cloning of cDNAs which are highly overexpressed in
mitoxantrone-resistant cells: Demonstration of homology to ABC transport genes. _Cancer Res._ 59, 8–13 (1999). CAS PubMed Google Scholar * Doyle, L. A. et al. A multidrug resistance
transporter from human MCF-7 breast cancer cells. _Proc. Natl Acad. Sci. USA_ 95, 15665–15670 (1998). Article ADS CAS PubMed PubMed Central Google Scholar * Robey, R. W. et al.
Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. _Br. J. Cancer_ 89, 1971–1978 (2003). Article CAS PubMed PubMed Central Google Scholar *
Honjo, Y. et al. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. _Cancer Res._ 61, 6635–6639 (2001). CAS PubMed Google
Scholar * Komatani, H. et al. Identification of breast cancer resistant protein/mitoxantrone resistance/placenta-specific, ATP-binding cassette transporter as a transporter of NB-506 and
J-107088, topoisomerase I inhibitors with an indolocarbazole structure. _Cancer Res._ 61, 2827–2832 (2001). CAS PubMed Google Scholar * Pozza, A., Perez-Victoria, J. M., Sardo, A. &
Ahmed-Belkacem, A. & Di Pietro, A. Purification of breast cancer resistance protein ABCG2 and role of arginine-482. _Cell. Mol. Life Sci._ 63, 1912–1922 (2006). Article CAS PubMed
Google Scholar * Ejendal, K. F., Diop, N. K., Schweiger, L. C. & Hrycyna, C. A. The nature of amino acid 482 of human ABCG2 affects substrate transport and ATP hydrolysis but not
substrate binding. _Protein Sci._ 15, 1597–1607 (2006). Article CAS PubMed PubMed Central Google Scholar * Yu, J., Zhou, Y., Tanaka, I. & Yao, M. Roll: a new algorithm for the
detection of protein pockets and cavities with a rolling probe sphere. _Bioinformatics_ 26, 46–52 (2010). Article PubMed CAS Google Scholar * Alam, A., Kowal, J., Broude, E., Roninson,
I. & Locher, K. P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. _Science_ 363, 753 (2019). -+. Article ADS CAS PubMed PubMed Central
Google Scholar * Nosol, K. et al. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. _Proc. Natl Acad. Sci. USA_ 117, 26245–26253 (2020).
Article CAS PubMed PubMed Central Google Scholar * Wang, L. et al. Characterization of the kinetic cycle of an ABC transporter by single-molecule and cryo-EM analyses. _Elife_ 9 (2020).
* Hofmann, S. et al. Conformation space of a heterodimeric ABC exporter under turnover conditions. _Nature_ 571, 580–583 (2019). Article CAS PubMed PubMed Central Google Scholar *
Johnson, Z. L. & Chen, J. ATP binding enables substrate release from multidrug resistance protein 1. _Cell_ 172, 81–89 (2018). e10. Article CAS PubMed Google Scholar * Shintre, C. A.
et al. Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. _Proc. Natl Acad. Sci. USA_ 110, 9710–9715 (2013). Article ADS CAS PubMed
PubMed Central Google Scholar * Ambudkar, S. V., Kim, I. W., Xia, D. & Sauna, Z. E. The A-loop, a novel conserved aromatic acid subdomain upstream of the Walker A motif in ABC
transporters, is critical for ATP binding. _FEBS Lett._ 580, 1049–1055 (2006). Article CAS PubMed Google Scholar * Ritchie, T. K. et al. Reconstitution of membrane proteins in
phospholipid bilayer nanodiscs. _Methods Enzymol._ 464, 211–231 (2009). Article CAS PubMed PubMed Central Google Scholar * Geertsma, E. R., Mahmood, N. A. B. N., Schuurman-Wolters, G.
K. & Poolman, B. Membrane reconstitution of ABC transporters and assays of translocator function. _Nat. Protoc._ 3, 256–266 (2008). Article CAS PubMed Google Scholar * Chifflet, S.,
Torriglia, A., Chiesa, R. & Tolosa, S. A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: application
to lens ATPases. _Anal. Biochem._ 168, 1–4 (1988). Article CAS PubMed Google Scholar * Li, X. M. et al. Electron counting and beam-induced motion correction enable near-atomic-resolution
single-particle cryo-EM. _Nat. Methods_ 10, 584 (2013). -+. Article CAS PubMed PubMed Central Google Scholar * Mastronarde, D. N. SerialEM: A program for automated tilt series
acquisition on tecnai microscopes using prediction of specimen position. _Microsc. Microanal._ 9, 1182–1183 (2003). Article ADS Google Scholar * Biyani, N. et al. Focus: The interface
between data collection and data processing in cryo-EM. _J. Struct. Biol._ 198, 124–133 (2017). Article CAS PubMed Google Scholar * Li, X. et al. Electron counting and beam-induced
motion correction enable near-atomic-resolution single-particle cryo-EM. _Nat. Methods_ 10, 584–590 (2013). Article CAS PubMed PubMed Central Google Scholar * Vilas, J. L. et al.
MonoRes: automatic and accurate estimation of local resolution for electron microscopy maps. _Structure_ 26, 337–344 (2018). e4. Article CAS PubMed Google Scholar * Zhang, K., Gctf &
Real-time, C. T. F. determination and correction. _J. Struct. Biol._ 193, 1–12 (2016). Article CAS PubMed PubMed Central Google Scholar * Emsley, P., Lohkamp, B., Scott, W. G. &
Cowtan, K. Features and development of Coot. _Acta Crystallogr. Sect. D Biol. Crystallogr._ 66, 486–501 (2010). Article CAS Google Scholar * Moriarty, N. W., Grosse-Kunstleve, R. W. &
Adams, P. D. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. _Acta Crystallogr. D. Biol. Crystallogr._ 65, 1074–1080
(2009). Article CAS PubMed PubMed Central Google Scholar * Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. _Acta Crystallogr. D Struct. Biol._ 74,
531–544 (2018). Article CAS PubMed PubMed Central Google Scholar * Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. _Acta Crystallogr D.
Biol. Crystallogr_ 66, 12–21 (2010). Article CAS PubMed Google Scholar * Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. _Acta
Crystallogr D. Biol. Crystallogr_ 66, 213–221 (2010). Article CAS PubMed PubMed Central Google Scholar * Goddard, T. D. et al. UCSF ChimeraX: Meeting modern challenges in visualization
and analysis. _Protein Sci._ 27, 14–25 (2018). Article CAS PubMed Google Scholar * Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. _J.
Comput Chem._ 25, 1605–1612 (2004). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS This research was supported by the Swiss National Science Foundation through
the National Centre of Competence in Research (NCCR) TransCure. We thank the Scientific Center for Optical and Electron Microscopy (ScopeM) at ETH Zürich for technical support. We thank K.
Goldie, R. Irobalieva, L. Kováčik and A. Fecteau-Lefebvre for technical support with EM data collection. AUTHOR INFORMATION Author notes * Dongchun Ni Present address: Laboratory of
Biological Electron Microscopy, Institute of Physics, SB, EPFL, Lausanne, Switzerland * Henning Stahlberg Present address: Laboratory of Biological Electron Microscopy, Institute of Physics,
SB, EPFL, and Dep. Fund. Microbiol., Faculty of Biology and Medicine, University of Lausanne, Lausanne, Switzerland AUTHORS AND AFFILIATIONS * Institute of Molecular Biology and Biophysics,
Department of Biology, ETH Zürich, Zürich, Switzerland Qin Yu, Julia Kowal, Ioannis Manolaridis, Scott M. Jackson & Kaspar P. Locher * Center for Cellular Imaging and NanoAnalytics
(C-CINA), Biozentrum, University of Basel, Basel, Switzerland Dongchun Ni & Henning Stahlberg Authors * Qin Yu View author publications You can also search for this author inPubMed
Google Scholar * Dongchun Ni View author publications You can also search for this author inPubMed Google Scholar * Julia Kowal View author publications You can also search for this author
inPubMed Google Scholar * Ioannis Manolaridis View author publications You can also search for this author inPubMed Google Scholar * Scott M. Jackson View author publications You can also
search for this author inPubMed Google Scholar * Henning Stahlberg View author publications You can also search for this author inPubMed Google Scholar * Kaspar P. Locher View author
publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Q.Y. and I.M. expressed and purified wild-type ABCG2. Q.Y. cloned, expressed, and purified the
ABCG2R184A variant. S.M.J. and I.M. reconstituted ABCG2 in lipid nanodiscs and prepared EM grids of the E1S turnover sample with the help of J.K. Q.Y. reconstituted ABCG2 in lipid nanodiscs
and prepared EM grids of topotecan turnover sample with the help of J.K. Q.Y. reconstituted ABCG2 into liposomes and carried out all functional experiments. D.N. collected and processed
cryo-EM data and determined the structure of the E1S turnover sample supervised by H.S. J.K. collected cryo-EM data of the topotecan turnover sample. Q.Y. processed EM data of the topotecan
turnover ABCG2 sample and determined the turnover-1 and turnover-2 structures with the help of J.K. D.N. built and refined initial model of the E1S turnover structure. K.P.L. and Q.Y. built,
refined, and validated the structures. K.P.L. and Q.Y. wrote the manuscript with input from all authors. All authors contributed to revision of the manuscript. CORRESPONDING AUTHOR
Correspondence to Kaspar P. Locher. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PEER REVIEW INFORMATION _Nature Communications_
thanks the anonymous reviewers for their contributions ot the peer review of this work. Peer review reports are available. PUBLISHER’S NOTE Springer Nature remains neutral with regard to
jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE SUPPLEMENTARY MOVIE 1 SUPPLEMENTARY MOVIE 2
SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation,
distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and
indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to
the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will
need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE
CITE THIS ARTICLE Yu, Q., Ni, D., Kowal, J. _et al._ Structures of ABCG2 under turnover conditions reveal a key step in the drug transport mechanism. _Nat Commun_ 12, 4376 (2021).
https://doi.org/10.1038/s41467-021-24651-2 Download citation * Received: 01 March 2021 * Accepted: 24 June 2021 * Published: 19 July 2021 * DOI: https://doi.org/10.1038/s41467-021-24651-2
SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to
clipboard Provided by the Springer Nature SharedIt content-sharing initiative