- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Understanding the molecular events controlling melanoma progression is of paramount importance for the development of alternative treatment options for this devastating disease.
Here we report a mechanism regulated by the oncogenic SOX2-GLI1 transcriptional complex driving melanoma invasion through the induction of the sialyltransferase ST3GAL1. Using in vitro and
in vivo studies, we demonstrate that ST3GAL1 drives melanoma metastasis. Silencing of this enzyme suppresses melanoma invasion and significantly reduces the ability of aggressive melanoma
cells to enter the blood stream, colonize distal organs, seed and survive in the metastatic environment. Analysis of glycosylated proteins reveals that the receptor tyrosine kinase AXL is a
major effector of ST3GAL1 pro-invasive function. ST3GAL1 induces AXL dimerization and activation that, in turn, promotes melanoma invasion. Our data support a key role of the ST3GAL1-AXL
axis as driver of melanoma metastasis, and highlight the therapeutic potential of targeting this axis to treat metastatic melanoma. SIMILAR CONTENT BEING VIEWED BY OTHERS SUPPRESSION OF
HEPARAN SULFATION RE-SENSITIZES YAP1-DRIVEN MELANOMA TO MAPK PATHWAY INHIBITORS Article Open access 07 July 2022 STAT3 PROMOTES MELANOMA METASTASIS BY CEBP-INDUCED REPRESSION OF THE MITF
PATHWAY Article 15 December 2020 LIMK2 PROMOTES MELANOMA TUMOR GROWTH AND METASTASIS THROUGH G3BP1-ESM1 PATHWAY-MEDIATED APOPTOSIS INHIBITION Article 16 March 2023 INTRODUCTION Malignant
melanoma is the most aggressive and treatment-resistant form of skin cancer. Its aggressiveness is based on the highly metastatic potential of melanoma cells even at early stage of the
disease. Although recent treatment advances using targeted therapy and immune checkpoint inhibitors achieved long-term survival in a small subset of patients with advanced melanomas,
metastatic melanoma still remains an incurable disease1. Therefore, identification and targeting of key drivers of melanoma metastasis are crucial steps towards an effective control of tumor
progression. Aberrant glycosylation, in particular increased sialylation, is an important determinant of malignant phenotype, as it directly impacts on key processes supporting tumor
progression and metastasis, including cell adhesion, motility, invasion, and immune evasion2,3,4,5,6,7. Enhanced sialic acid levels increase resistance to apoptosis and modulate the function
of immune cells8, as well as alter tumor cell–cell interaction, promoting cell detachment from a site of origin. Because sialylated glycoconjugates regulate adhesion and promote motility,
they may also be important for the colonization and metastatic potential. Although aberrant glycosylation has been associated with melanoma progression9,10, the biological significance and
mechanism(s) controlling this post-translational modification during disease pathogenesis remain for the most poorly understood. Recent studies from our and other groups have shown an
interplay between the transcription factor GLI1, the final effector of the Hedgehog (HH) pathway, and the pluripotency transcription factor SOX2 in several types of cancer including
melanoma11,12,13. Here we identify a set of genes co-regulated by SOX2-GLI1 contributing to aberrant O-glycosylation in metastatic melanoma. Using in vitro and in vivo assays, we demonstrate
that ST3GAL1, a β-galactoside-α-2,3-sialyltransferase-1 that catalyzes the transfer of sialic acid from cytidine monosphosphate (CMP)-sialic acid to galactose-containing substrates, is a
crucial driver of melanoma invasion and metastasis downstream the SOX2-GLI1 transcriptional complex. In addition, biochemical and functional studies reveal that the receptor tyrosine kinase
AXL is a major mediator of the pro-invasive effects of ST3GAL1 in melanoma. Our study reveals a functional ST3GAL1-AXL axis driving melanoma metastasis and suggests that inhibition of this
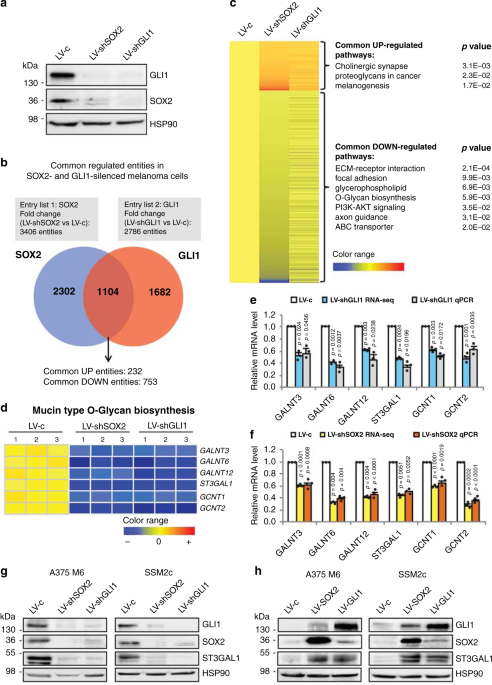
axis may become in the future a promising therapeutic strategy for metastatic melanoma. RESULTS O-GLYCOSYLATION IS A COMMON REGULATED PATHWAY OF SOX2 AND GLI1 To identify genes co-regulated
by both SOX2 and GLI1, we performed whole-transcriptome analysis in patient-derived SSM2c cells, which were obtained from a subcutaneous metastasis11,14, after genetic silencing of either
SOX2 or GLI1 with specific shRNAs (Fig. 1a). Three pairs of total RNAs from each condition were generated for library preparations and subsequent RNA-sequencing (RNA-seq). All samples had
over 50 million reads with over 93% of the reads mapping to the human genome (obtained from the UCSC Genome Browser assembly ID:hg19). Using differential expression analysis (fold change
(FC) > 1.5, false discovery rate (FDR) < 0.1 as standard cut-offs), we identified 1104 entities commonly altered in both SOX2- and GLI1-knocked down cells (Fig. 1b and Supplementary
Data 1). Grouping of genes into enrichment clusters revealed by DAVID functional annotation tool (https://david.ncifcrf.gov/) identified O-Glycan biosynthesis pathway (_p_ = 5.9E-3) among
the top-7 pathways significantly downregulated in both SOX2-and GLI1-depleted melanoma cells (Fig. 1c). As aberrant glycosylation has been associated with melanoma progression and
immunosuppression4,9,10,15, we examined in detail the expression of genes that are components of the O-linked glycan biosynthetic pathway. RNA-seq results coupled with quantitative real-time
PCR (qPCR) validation identified six different glycosyltransferases significantly downregulated in both SOX2- and GLI1-silenced cells: the _N_-acetylgalactosaminyltransferases _GALNT3_,
_GALNT6_ and _GALNT12_, the sialyltransferase _ST3GAL1_ and the _N_-acetylglucosaminyltransferases _GCNT1_ and _GCNT2_ (Fig. 1d–f). Data mining of transcriptomic datasets from independent
clinical cohorts revealed consistently higher levels of ST3GAL1 in melanoma compared to nevi and in metastatic cases compared to primary melanomas (Supplementary Fig. 1). Western blotting
analysis confirmed a strong reduction of ST3GAL1 protein level in both SOX2- and GLI1-depleted A375 M6 and SSM2c cells (Fig. 1g), and a consistent increase of ST3GAL1 after ectopic
expression of these transcription factors (TFs; Fig. 1h). Based on these results, we focused on defining the functional role of the sialyltransferase ST3GAL1 in melanoma metastasis. ST3GAL1
EXPRESSION CORRELATES WITH MELANOMA PROGRESSION The β-galactoside-α-2,3-sialyltransferase-1 ST3GAL1 catalyzes the transfer of sialic acid from cytidine monosphosphate (CMP)-sialic acid to
galactose-containing substrates and is associated with cancer progression and drug resistance16,17. Data mining of publicly available transcriptomic datasets of several types of skin cancer
(GSE7553)18 revealed that _ST3GAL1_ is preferentially upregulated in malignant melanomas compared to basal cell carcinoma (BCC) or squamous cell carcinoma (SCC; Fig. 2a). In addition, a
transcriptomic dataset of human nevi, primary, and metastatic melanomas (GSE46517)19 showed that _ST3GAL1_ is strongly associated with melanoma progression, with higher _ST3GAL1_ mRNA levels
in primary melanomas compared to nevi and in metastatic compared to primary melanomas (Fig. 2b). Moreover, in silico data analysis showed that ST3GAL1 is altered in 23% of human melanomas
(missense mutations, gene amplification, or mRNA upregulation; Fig. 2c). Consistently with transcriptomic data, immunohistochemical analysis of human melanoma tissue microarrays showed
higher proportion of metastatic cases with medium/high ST3GAL1 staining compared to primary melanomas (_p_ = 0.0198) or nevi (_p_ = 0.0007; Fig. 2d, e and Supplementary Table 1). ST3GAL1
staining in melanoma cells was observed in the cytoplasm with a perinuclear granular pattern (Fig. 2d, high magnification). In human normal skin ST3GAL1 staining was confined to sparsed
cells with weak cytoplasmic expression (Fig. 2d). Accordingly, western blot analysis showed that ST3GAL1 was expressed at various levels in metastatic melanoma cell lines, but barely present
in normal human epidermal melanocytes (NHEM; Fig. 2f). Altogether, these data indicate that ST3GAL1 is consistently expressed in human melanomas and is associated with tumor progression.
These results prompted us to investigate the role of ST3GAL1 in the pathobiology of melanoma and address its functional regulation by SOX2 and GLI1. ST3GAL1 REGULATES MELANOMA INVASIVENESS
To investigate the role of ST3GAL1 in melanoma progression, we silenced ST3GAL1 in a lung metastatic clone derived from A375 cells (A375 M6)20 and in SSM2c cells using two independent
ST3GAL1 shRNAs (LV-shST3GAL1.1 and LV-shST3GAL1.2), which target the _3_′_-UTR_ or the _CDS_ region of ST3GAL1 respectively (Fig. 3a). We first assessed whether ST3GAL1 plays a role in
melanoma cell invasiveness using in vitro migration and invasion assays. In the scratch assay, motility and wound closure of scratched area were significantly reduced in ST3GAL1 depleted
cells in a time-dependent manner, with a rate of reduction of 30–40% in LV-shST3GAL1.1 and LV-shST3GAL1.2 A375 M6 cells compared to scramble, respectively (Fig. 3b), and of ~80–90% in
LV-shST3GAL1.1 and LV-shST3GAL1.2 SSM2c cells (Fig. 3c). In addition, ST3GAL1 knockdown strongly reduced the ability of A375 M6, or nearly abrogated that of SSM2c cells, to invade through
Matrigel-coated Boyden chambers in a transwell invasion assay (Fig. 3d, e). Expression of the EMT markers N-cadherin, Vimentin, SNAI1 (Snail), and SNAI2 (Slug) decreased upon ST3GAL1
depletion, suggesting that ST3GAL1 regulates the transition to a more mesenchymal phenotype (Fig. 3a). Consistent with these results, ectopic expression of ST3GAL1 in the metastatic cell
line MeWo and in patient-derived metastatic cells M51 (Fig. 3f), both of which express low levels of ST3GAL1 (Fig. 2f), accelerated wound closure and enhanced invasion in vitro (Fig. 3g–j),
with concomitant increased expression of EMT markers (Fig. 3f). Modulation of ST3GAL1 expression had no significant effect on melanoma cell growth, although ST3GAL1 overexpression slightly
decreased the fraction of early apoptotic cells only in MeWo cells, without affecting late apoptosis (Supplementary Fig. 2). Altogether these results indicate that ST3GAL1 endows melanoma
cells with metastatic potential. ST3GAL1 IS REQUIRED FOR IN VIVO MELANOMA METASTASIS We first examined how depletion of ST3GAL1 impacts on the ability of melanoma cells to survive and
proliferate in a metastatic environment, using a model based on intracardiac instillation of melanoma cells10. We injected A375 M6 cells stably transduced with lentiviral particles carrying
one of the two independent shRNA against ST3GAL1 (LV-shST3GAL1.1 or LV-shST3GAL1.2) or scramble control (LV-c) and a luciferase reporter vector (Fig. 4a). Effective ST3GAL1 silencing with
both shRNAs was confirmed by qPCR and western blotting (Fig. 4b, c). We performed intracardiac injection of transduced A375 M6 in athymic nude mice and assessed for colonization by in vivo
bioluminescence imaging (BLI) at days 7 and 14 post-injection. A successful intracardiac injection was indicated on day 1 by images showing systemic bioluminescence distributed throughout
the animal. Mice injected with either LV-shST3GAL1.1 or LV-shST3GAL1.2 displayed significantly reduced seeding ability compared to mice injected with LV-c in multiple organs (Fig. 4d–f).
Mice injected with A375 M6 cells expressing both ST3GAL1 shRNAs showed fewer and much smaller metastatic lesions in the lungs (Fig. 4g, h). We next determined whether modulation of ST3GAL1
affects metastasis formation in vivo using a xenograft model of metastasis. We injected A375 M6 cells stably transduced with GFP/luciferase reporter and LV-shST3GAL1.1, LV-ST3GAL1 or
scramble control (LV-c) into the flank of athymic nude mice, and monitored mice for primary tumor growth, presence of circulating melanoma cells (CTCs) in the blood, and metastasis (Fig. 5a,
b). We observed no significant difference in tumor growth between LV-c, LV-shST3GAL1.1, and LV-ST3GAL1, except a slight tumor growth delay in mice injected with A375 M6 cells transduced
with LV-shST3GAL1 (Fig. 5c). When local tumors reached the same tumor volume (21–24 days post-injection; Fig. 5c, d), primary tumors were surgically resected, and the ability of cells to
enter the blood stream (CTCs) and give rise to distant metastases was examined. Silencing of ST3GAL1 substantially reduced the frequency of circulating melanoma cells in the blood (Fig. 5e)
and strongly inhibited the metastatic potential of A375 M6 cells, as shown by the reduced distribution of luminescence in organs such as lungs (Fig. 5f, g). Forty-five days post-injection
mice were killed and lungs and main organs dissected for histology and immunohistochemistry. Mice injected with LV-shST3GAL1 exhibited significantly reduced number of lung micrometastases
compared to LV-c (Fig. 5h, i). Ectopic expression of ST3GAL1 did not affect the number of CTCs nor the number of micrometastases per lung compared to control (Fig. 5e–i), although mice
injected with melanoma cells transduced with LV-ST3GAL1 exhibited higher number of macrometastases (Supplementary Fig. 3). These data provide evidence that ST3GAL1 silencing drastically
impairs the ability of aggressive melanoma cells to enter the blood stream, colonize distal organs and seed, survive and proliferate in the metastatic environment. SOX2 AND GLI1 CO-REGULATE
_ST3GAL1_ GENE TRANSCRIPTION Both SOX2 and GLI1 positively modulate ST3GAL1 expression (Fig. 1), thus we sought to investigate whether the effect of SOX2 and GLI1 was a result of a direct
transcriptional control on _ST3GAL1_. In silico analysis of ST3GAL1 regulatory regions (RRs) (obtained from the UCSC Genome Browser assembly ID:hg38) using the TFBIND bioinformatics software
(http://tfbind.hgc.jp) identified several putative SOX2- and GLI-binding sites (BS) possessing the reported consensus sequences (SOX2-BS: wwTGnwTw)21 (GLI-BS: GACCACCCA)22,23. Chromatin
immunoprecipitation assay (ChIP) of SOX2 and GLI1 followed by qPCR using different sets of primers spanning the putative BS within the selected RRs revealed co-occupancy of SOX2 and GLI1 at
a distal enhancer element located at ~10 kb upstream the transcription start site (TSS) of ST3GAL1 (enhancer II), with a 3–8-fold enrichment in ST3GAL1 signal over ChIP with non-specific IgG
in two different melanoma cell types (Fig. 6a–c). Meanwhile, negative control and primers spanning _ST3GAL1_ promoter showed no significant enrichment (Fig. 6b, c). To confirm the ability
of SOX2 and GLI1 to control ST3GAL1 expression, we cloned the ST3GAL1 enhancer II construct encompassing putative SOX2-BS and GLI-BS (−12,453/−11,457 bp from TSS; Fig. 6d and Supplementary
Fig. 4) into a luciferase reporter vector, and tested the effects of SOX2 and GLI1 on transcriptional activation of ST3GAL1 in SSM2c cells. We observed a strong induction of the luciferase
activity upon ectopic expression of SOX2, which was reverted after depletion of GLI1, and viceversa (Fig. 6e, f). As GLI1 directly regulates the expression of SOX211 and SOX2 appears to
modulate that of GLI1 (Fig. 1g, h), we assessed whether the observed induction of ST3GAL1 transcription could depend on a sequential regulation of the two TFs expression by overexpressing
SOX2 in cells depleted for GLI1 and viceversa. Luciferase assays showed that SOX2 overexpression partially compensates for the absence of GLI1 (Supplementary Fig. 5a), similarly to what
happens after ectopic expression of GLI1 in SOX2-silenced cells (Supplementary Fig. 5b), suggesting that both TFs are required for the induction of ST3GAL1 transcription. This was also
confirmed at both mRNA and protein levels (Fig. 6g and Supplementary Fig. 5c–f). Next, disruption of the putative GLI-BS (−12,187/−12,178 bp from TSS) or of the two putative SOX2-BS (SBS1 at
−12,212/−12,204 bp and SBS2 at −11,944/−11,936 bp from TSS) through site-directed mutagenesis (Fig. 6d) further corroborates our hypothesis of a direct transcriptional regulation of
_ST3GAL1_ by both SOX2 (through its binding at SBS2) and GLI1 (Fig. 6h). The slight transactivation of ST3GAL1 GBSmut enhancer II by GLI1 was completely abrogated when GLI1 was overexpressed
in cells depleted for SOX2 (Supplementary Fig. 5g), similarly to that of SBS2mut enhancer II by SOX2 (Supplementary Fig. 5h). However, while SOX2-induced transcriptional activation of
ST3GAL1 occurs even in absence of a functional GBS, disruption of SBS2 appeared to reduce transactivation by GLI1 (Fig. 6h). This effect was still abrogated when GLI1 was overexpressed in
SOX2-depleted cells (Supplementary Fig. 5g), suggesting that GLI1 regulates ST3GAL1 both directly and indirectly through SOX2. In support of this regulation, a positive correlation between
the expression of _SOX2_ and _ST3GAL1_ mRNA was observed in a panel of commercial and patient-derived melanoma cells (Supplementary Fig. 6). Next, we asked whether ST3GAL1 could mediate
SOX2- and GLI1-induced melanoma invasiveness. We first silenced ST3GAL1 in A375 M6 cells overexpressing either SOX2 or GLI1. Our results showed that both GLI1 and SOX2 increased melanoma
cell invasion and that depletion of ST3GAL1 was able to suppress the effects of both SOX2 and GLI1 (Fig. 6i–k). Consistently, ectopic expression of ST3GAL1 in SOX2- or GLI1-depleted SSM2c
cells was able to rescue the decrease in melanoma cell invasion produced by SOX2 or GLI1 silencing (Supplementary Fig. 5i–k). Overall, these data reveal ST3GAL1 as a key downstream mediator
that contributes to the aggressive behavior of melanoma cells induced by SOX2 and GLI1. AXL MEDIATES THE PRO-INVASIVE EFFECTS OF ST3GAL1 IN MELANOMA To identify putative mediators of ST3GAL1
in metastatic melanoma cells, we performed proteomic analysis of sialylated proteins. To this end, we prepared whole cell extracts of A375 M6 transduced with either LV-ST3GAL1 or
LV-shST3GAL1.1 and enriched for sialylated proteins using MAL lectin affinity chromatography (Fig. 7a). Mass spectrometric analysis led to the identification of more than 300 proteins, 43 of
which showed a significant increased sialylation level (fold change ≥1.5, _p_ value < 0.05) in the enriched fraction of ST3GAL1 sample (Supplementary Data 2). After removing contaminant
unglycosylated proteins (i.e. ribosomal proteins, tubulins, and heat shock proteins), gene ontology analysis with DAVID functional annotation tool revealed enrichment in biological processes
relevant to metastasis such as cell motility, cell migration, and locomotion control mechanisms (Fig. 7b). Proteins identified as MAL-bound that are involved in the aforementioned cellular
processes are shown in Supplementary Table 2. To validate our proteomic analysis, we examined the sialylated state of integrin β4 (ITGB4), that has been recently reported as
ST3GAL1-sialylated target24, and of 3 receptor tyrosine kinases (RTKs) that have been linked to melanoma progression: AXL receptor (also known as UFO)25, nerve growth factor receptor (NGFR)
(known also as CD271)26, and epidermal growth factor receptor (EGFR)27,28, that are expressed in melanoma cells at variable levels (Supplementary Fig. 7a). Immunoprecipitation with MAL-II
lectin, which specifically recognizes α2,3-linked sialic acid residues, followed by western blot showed increased integrin-β4, AXL, NGFR, and EGFR levels in ST3GAL1 expressing cells (Fig.
7c; in agreement with our proteomic analysis). Remarkably, ST3GAL1 did not affect sialylation of integrin-α5, as already reported24, corroborating the reliability of our MS results (Fig. 7c
and Supplementary Data 2). We next asked whether the increased sialylation of the identified RTKs by ST3GAL1 could be relevant for their biological activation in melanoma. We employed the
human phospho-RTK assay in A375 M6 to profile the relative phosphorylation levels of 49 different activated transmembrane proteins that become phosphorylated at tyrosine residues in their
catalytic domain, such as AXL, EGF, and NGF receptor family proteins. Ectopic expression of ST3GAL1 increased phosphorylation status of AXL compared to scrambled LV-c cells, without altering
that of EGFR or NGFR (Fig. 7d). This was confirmed by immunoprecipitation of AXL, EGFR, or NGFR followed by immunoblotting with anti-phospho-Tyrosine (Fig. 7e). Further, ectopic expression
of ST3GAL1 in melanoma cells increased the phosphorylation of AXL at residue Tyr702 in the catalytic kinase domain (Fig. 7f), which becomes autophosphorylated in response to AXL
activation29, suggesting that ST3GAL1-mediated increased sialylation of AXL could be responsible for its activation. To further understand whether sialylation could induce AXL dimerization
and activation, we stimulated serum-starved A375 M6 cells with Gas6 and analyzed AXL dimerization. AXL appeared to exist mainly as monomer, whereas Gas6 stimulation induced its partial
dimerization only in presence of endogenous ST3GAL1 (Fig. 7g). Consistent with the dimerization result, A375 M6 cells depleted for ST3GAL1 showed no AXL phosphorylation at Tyr702 in response
to ligand stimulation (Fig. 7h). These data suggest that sialylation induces AXL dimerization and activation, likely increasing the affinity of AXL for its ligand Gas6. The AXL receptor
kinase is known to play a critical role in cell proliferation, survival, cancer stem cell maintenance, and metastasis in several cancers25, and has been associated with melanoma cell
motility, invasion, and drug resistance30,31,32. Thus, it represents a reasonable candidate for mediating the pro-invasive effects of ST3GAL1. As expected, ectopic expression of ST3GAL1 in
A375 M6 and MeWo cells triggered a significant increase in melanoma cell invasion. Of note, concomitant silencing of AXL in these cells was able to fully revert the pro-invasive effects
mediated by ST3GAL1 (Fig. 8a–d), as demonstrated by the reduction of Vimentin and TFs SNAI1 and SNAI2 (Fig. 8a), in contrast to what observed after depletion of either NGFR (Supplementary
Fig. 7b–d) or EGFR (Supplementary Fig. 7e–g). In addition, overexpression of AXL or its stimulation by Gas6 enhanced the invasive phenotype only in presence of endogenous ST3GAL1
(Supplementary Fig. 8), suggesting that sialylation represents the key effector of this pathway. AXL impacted the migratory ability of A375 M6 ST3GAL1-overexpressing cells but not that of
MeWo cells, suggesting that additional mediators could be involved in this process, possibly NGFR (Supplementary Fig. 9). Overall, our data suggest that sialylated AXL is a major mediator of
the pro-metastatic role played by ST3GAL1 in melanoma. To further support the relevance of the SOX2/GLI1-ST3GAL1-AXL axis in melanoma progression, we performed single-cell analysis in cells
derived from melanoma brain metastasis PDX models (M12, M15, and M27) as well as normal melanocyte cultures (Supplementary Figs. 10 and 11). These data show that _ST3GAL1, GLI1, SOX2_, and
_AXL_ are co-expressed in melanoma cells with similar or higher levels in tumor samples (M) vs. normal (N), except M12 (Fig. 8e–g and Supplementary Data 3). Furthermore, protein levels of
GLI1, SOX2, ST3GAL1, and AXL were analyzed in short-term cultures of primary and metastatic melanomas. Western blots showed that primary melanoma cells express lower levels of SOX2, GLI1,
ST3GAL1, and AXL than metastatic melanoma cells, and that the latters harbor a stronger activation of SOX2/GLI1-ST3GAL1-AXL axis (Fig. 8h). In addition, analysis of a cohort of 481 human
melanoma samples from The Cancer Genome Atlas (TCGA) showed that expression of _ST3GAL1_ is significantly increased in BRAF mutant melanomas compared to wild type cases, and that of _AXL_ is
increased in TP53 mutant melanomas (Supplementary Fig. 12). These data support the existence of both non-genetic (the GLI1-SOX2 transcriptional mechanism) and genetic mechanisms controlling
the ST3GAL1-AXL axis. DISCUSSION Malignant melanoma is one of the most lethal skin cancers worldwide and one of the malignancies with the highest potential to metastasize. Sialylation, the
addition of sialic acid from a donor substrate to terminal positions of glycoprotein and glycolipid carbohydrate groups, has been shown to play major roles in tumor cell invasiveness and
tumor progression. Here we show that the sialyltransferase ST3GAL1 is transcriptionally regulated by both GLI1 and SOX2 in melanoma and is a crucial driver of melanoma cell invasiveness and
melanoma metastasis in vivo. Our work indicates that the receptor tyrosine kinase AXL is a major mediator of the pro-invasive effects of ST3GAL1 in melanoma (Fig. 8i). Our study suggests
that targeting ST3GAL1-linked processes may become in the future a promising therapeutic approach to prevent or treat established melanoma metastasis. Aberrant glycosylation is one of the
hallmarks of cancer with altered gene expression signatures of sialyltransferases. Mounting evidence demonstrated that the alteration of sialylation and the levels of sialyltransferase
activities are relevant for cancer invasion and metastasis33. ST3GAL1 has been shown to exert a tumor promoting effect in a PyMT mouse model of breast cancer34. ST3GAL1 is crucial for the
maintenance of glioblastoma growth, through the regulation of stemness traits16. Furthermore, ST3GAL1 promotes cell migration and invasion, and confers resistance to paclitaxel in ovarian
cancer17. However, no data are available on the role of ST3GAL1 in melanoma. Our study indicates that ST3GAL1 expression levels correlate with melanoma progression. Indeed, we found that
ST3GAL1 is expressed at low levels in normal skin and nevi, and its expression increases during melanoma progression at both mRNA and protein levels. Both microarray dataset and
immunohistochemistry show that ST3GAL1 expression is significantly higher in metastatic compared to primary melanomas. In addition, ST3GAL1 is preferentially expressed in human melanomas
compared to other types of skin cancer, such as BCC or SCC, which is consistent with the higher potential to metastasize of melanomas respect to other types of skin cancer. Genetic silencing
of ST3GAL1 reduces melanoma cell migration and invasion in vitro, whereas ST3GAL1 overexpression has the opposite effects. Importantly, we show that ST3GAL1 silencing in a highly invasive
melanoma cell line reduces the number of melanoma cells able to enter the blood stream (circulating melanoma cells) and drastically diminishes metastatic dissemination to the lungs. In
addition, ST3GAL1 depletion inhibits seeding of melanoma cells in several organs, suggesting that sialylation by ST3GAL1 may be required for the ability of melanoma cells to enter the blood
stream, form metastases, and survive in a metastatic environment. In agreement with the identified role of ST3GAL1 in melanoma progression, ST3GAL1 is expressed at higher levels in
metastastic compared to primary melanoma cases. Together our in vitro and in vivo data point toward several promoting effects of ST3GAL1 in melanoma progression, and provide strong evidence
that silencing of ST3GAL1 inhibits the metastatic process in vivo. These effects appear not related to growth advantage, because modulation of ST3GAL1 expression does not impact on the
growth of subcutaneous melanoma xenografts. Our data also indicate that overexpression of ST3GAL1 does not endow melanoma cells with increased ability to metastatize. This might be explained
with that fact that level of endogenous ST3GAL1 is sufficient to explicate its function. However, we found that melanoma cells overexpressing ST3GAL1 give rise to larger metastases compared
to control cells, suggesting that melanoma cells with high ST3GAL1 expression may have a better chance to strive and survive in “host” tissues. Here we identified the transcription factors
SOX2 and GLI1 as positive regulators of ST3GAL1. This is in agreement with previous studies showing that SOX2 and GLI1 are both involved in melanoma progression. For instance, SOX2 has been
shown to contribute to melanoma metastasis35 and to be highly expressed in melanomas compared to nevi36. In addition, SOX2 regulates survival, self-renewal, and tumorigenicity of human
melanoma-initiating cells11,37 and participates in dormancy regulation of melanoma tumor-repopulating cells38. SOX2 has been recently related to the acquisition of an aggressive oxidative
tumor phenotype endowed with enhanced drug resistance and metastatic ability39. Besides SOX2, the GLI transcription factors GLI1 and GLI2 have been suggested to be involved in melanoma
progression and metastasis40,41,42 and to be responsible for maintenance of melanoma-initiating cells14. Our data indicate that the transcriptional regulation of ST3GAL1 by SOX2 is direct,
whereas GLI1 acts both directly and indirectly via SOX2. This result is consistent with a previous report demonstrating that SOX2 is a target of GLI1 in melanoma11. In addition, our results
suggest the existence of reciprocal transcriptional regulation between SOX2 and GLI1. This regulatory loop might further contribute to potentiate the transcriptional activation of ST3GAL1.
Our in vitro data support the biological relevance of the regulation of ST3GAL1 by SOX2 and GLI1. Indeed, ST3GAL1 appears to mediate the effects of SOX2 and GLI1 on melanoma cell invasion.
Furthermore, ectopic expression of ST3GAL1 rescues the effect of SOX2 and/or GLI1 depletion on melanoma cell invasiveness. Interestingly, other members of the O-glycan biosynthesis pathway
appear to be regulated by both SOX2 and GLI1, suggesting that these two TFs may promote melanoma progression through the induction of aberrant glycosylation. Sialylation is a
post-translational modification found in a large variety of proteins, including cell surface receptors and adhesion molecules. Sialylation is responsible for increased protein stability and
activity, and proteins assembly. MS analysis in a melanoma cell line derived from a metastasis (A375 M6) identified several putative ST3GAL1 substrates, with enrichment in proteins involved
in cell migration, locomotion, motility, and adhesion, all processes relevant for tumor metastasis. These proteins included the tyrosine kinase receptors AXL, NGFR, and EGFR, integrin β4 and
β-catenin. However, only AXL is strongly activated in its catalytic domain by ST3GAL143. In agreement with this finding, our data indicate that AXL is the main downstream mediator of
ST3GAL1 function in a subgroup of human melanomas, given that genetic silencing of AXL rescues the increase in invasion induced by ectopic expression of ST3GAL1. Consistently, our data rule
out the involvement of EGFR in mediating ST3GAL1-dependent cell migration and invasiveness and narrow that of NGFR in ST3GAL1-dependent melanoma cell migration. However, we cannot exclude
the possibility that other RTK or proteins can play a role in mediating ST3GAL1 functions. AXL is a member of the TAM family of receptor tyrosine kinases. AXL is specifically activated by
the binding of its ligand Gas6, which induces AXL homodimerization and subsequent autophosphorylation of multiple tyrosine residues in the cytoplasmic kinase domain44. Our data suggest that
ST3GAL1 is required for ligand-dependent AXL dimerization and consequent autophosphorylation at residue Tyr702. Thus, we hypothesize that sialylation might be responsible for increasing the
affinity of AXL for its ligand Gas6. Indeed, several glycosylation sites have been identified in the two Ig-like domains for ligand binding45. Clinically, the expression of AXL has been
shown to correlate with a poor prognosis and to increase metastatic risk in several tumors, where it is implicated in cancer proliferation, survival, apoptotic evasion, and invasion, as well
as in the acquisition of a stem-cell phenotype and in drug resistance25. Accumulating evidence indicate that AXL is a marker of invasive phenotype in melanoma, linked to the absence of
microphthalmia-associated transcription factor (MITF) and the expression of an EMT signature46,47. Moreover, AXL expression has been also related with resistance to MAPK pathway
inhibitors32,46,48, and has been reported to desensitize melanoma cells to chemotherapy by preventing p53 activation49. However, AXL has been shown to have a dual regulatory function on
melanoma cell invasion, given that both inhibition or overexpression enhance invadopodia formation and activity, due to compensatory mechanisms by ERBB3 signaling pathway31. Taken together,
our study reveals a functional SOX2/GLI1-ST3GAL1-AXL axis in a subgroup of melanomas involved in cancer progression, and highlights the therapeutic potential of targeting ST3GAL1 to prevent
or treat metastatic melanoma. The biosynthetic machinery involved in aberrant glycosylation has the potential to become a promising target for the development of anti-cancer drugs50.
However, very few of these agents have the features required to translate into clinical trials. Our work provides the rationale for designing small molecules against ST3GAL1 and for drug
repurposing to identify effective ST3GAL1 inhibitors for treatment of a subset of melanomas. METHODS CELL CULTURES Normal human epidermal melanocytes (NHEM) were purchased from PromoCell
(Heidelberg, Germany). Human melanoma cell lines A375, SK-Mel-2, SK-Mel-5, SK-Mel-28, MeWo, and HEK-293T were obtained from ATCC, whereas 501-Mel, A2058 and SK-Mel-197 were kindly provided
by Dr. Laura Poliseno (CNR, Pisa, Italy). A375 M6 cells were isolated from lung metastases after tail-vein injection in SCID bg/bg mice of A375 cells20. Patient-derived SSM2c and M51 cells
were obtained from metastatic melanomas14,51 (Supplementary Table 3). All cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Euroclone, Milan, Italy) containing 10% fetal
bovine serum (FBS), 1% penicillin/streptomycin (PS), and 1% Glutamine, and maintained at 37 °C in a 5% CO2 incubator. Short-term (passage 1–2) patient-derived melanoma cells Me-14, Me-16,
Me-17, Me-25, Me-28, Me-32, Me-41, Me-42, and Me-44 were obtained from patients with primary or metastatic melanomas. Fresh tissue samples were enzymatically digested with 20 μg/ml DNase I
(Roche Applied Science, Basel, Switzerland) and 1 mg/ml collagenase A in Dulbecco’s modified Eagle’s medium (DMEM)-F12 (Euroclone, Milan, Italy) and filtered. Cells were grown in DMEM-F12
supplemented with 10% FBS, 2% PS, 1% Glutamine, and 5 ng/ml epidermal growth factor (EGF; Life Technologies, Paisley, UK). All cells were recently authenticated by DNA fingerprinting
analysis and regularly tested by PCR to exclude Mycoplasma contamination. The use of melanoma samples was approved by the Ethic Committee of the University Hospital of Careggi with protocol
numbers N. BIO.13.009 and N. BIO.14.026. All the subjects gave their written informed consent to participate in the study. PLASMIDS AND VIRAL PRODUCTION Lentiviruses for gene knockdown were
produced in HEK-293T cells by cotransfecting lentiviral vector, dR8.74 packaging plasmid (Addgene #22036) and pMD2.G envelope plasmid (Addgene #12259). shRNA vectors used were: pLKO.1-puro
(scramble, LV-c; Addgene #8453), pLKO.1-puro-shGLI1 (targeting sequence 5′-CCTGATTATCTTCCTTCAGAA-3′), pLKO.1-puro-shSOX2.1 (targeting sequence 5′-CTGCCGAGAATCCATGTATAT-3′),
pLKO.1-puro-ST3GAL1.1 (LV-shST3GAL1.1) targeting the 3′ untranslated region (3′-UTR) of ST3GAL1 (targeting sequence 5′-AGAGACTTGAGTGGCGATTAC-3′) and pLKO.1-puro-shST3GAL1.2 (LV-shST3GAL1.2)
targeting the coding region (CDS) of ST3GAL1 (targeting sequence 5′-GATGCAGACTTTGAGTCTAAC-3′), pLKO.1-puro-AXL.1 (LV-AXL.1) targeting the 3′-UTR of AXL (targeting sequence 5′-
CTTTAGGTTCTTTGCTGCATT-3′) and pLKO.1-puro-shAXL.2 (LV-shAXL.2) targeting the CDS of AXL (targeting sequence 5′-GATTGCCATTGAGAGTCTAGC-3′); pLKO.1-puro-NGFR.1 (LV-shNGFR.1) targeting the
3′-UTR (targeting sequence 5′-GCACTGTAGTAAATGGCAATT-3′) and pLKO.1-puro-NGFR.2 (LV-shNGFR.2) targeting the CDS of NGFR (targeting sequence 5′-CCTCCAGAACAAGACCTCATA-3′); pLKO.1-puro-shEGFR.1
(LV-shEGFR.1) targeting the 3′-UTR (targeting sequence 5′-GAGAATGTGGAATACCTAAGG-3′) and pLKO.1-puro-shEGFR.2 (LV-shEGFR.2) targeting the CDS of EGFR (targeting sequence
5′-GCCACAAAGCAGTGAATTTAT-3′). When unspecified, gene silencing was performed by co-infection of melanoma cells with both CDS- and 3′-UTR-targeting lentiviruses. Lentiviruses for gene
overexpression were produced in HEK-293T by co-transfection of CSGW vector, CSGW-SOX252 (cloned into the BglII-NotI restriction sites of CSGW vector using the following primers: SOX2-F
5′-ATGTACAACATGATGGAGACGG-3′ and SOX2-R 5′- TCACATGTGTGAGAGGGGC-3′) or CSGW-GLI1 (cloned into the BglII-NotI restriction sites of CSGW vector using the following primers: GLI1-F 5′-
ATGTTCAACTCGATGACCCCAC-3′ and GLI1-R 5′-TTAGGCACTAGAGTTGAGGAA-3′), pCMV-dR8.91 packaging plasmid (gift from Silvestro Conticello) and pCMV-VSV-G envelope plasmid (Addgene #8454).
Retroviruses for gene overexpression were also produced in HEK-293T by cotransfecting pBABE-puro (Addgene #1764), pBABE-ST3GAL1 (cloned into the BamHI/SnaBI restriction sites of pBABE-puro
vector using the following primers: ST3GAL1-F 5′-ATGGTGACCCTGCGGAAGAGG-3′ and ST3GAL1-R 5′-TCATCTCCCCTTGAAGATCCGGA-3′) or pWZL-Neo-Myr-Flag-AXL (Addgene #20428) with pUMVC packaging plasmid
(Addgene #8449) and pCMV-VSV-G envelope plasmid (Addgene #8454). RNA EXTRACTION AND REAL-TIME QPCR Total RNA was extracted with RNeasy mini kit (Qiagen) and quantified by using Nanodrop
8000. After DNase I treatment (Roche Diagnostics), 1 μg of RNA was reverse transcribed with High-Capacity RNA-to-cDNA™ Kit (Applied Biosystems) according to manufacturer’s instructions.
Quantitative real-time PCR was carried out at 60 °C using FastStart SYBR Green Master (Roche Diagnostics) in a Rotorgene-Q (Qiagen). Primers were designed by using Primer3. Primer sequences
are reported in Supplementary Table 4. RNA SEQUENCING For the RNA-seq analysis cDNA libraries were prepared following Illumina RNA-Seq preparation protocol. The cDNA fragments were amplified
by polymerase chain reaction (PCR) and sequenced at both ends using an Illumina Genome Analyzer IIx. RNA-seq data were analyzed using MAP-RSEQ (http://bioinformaticstools.mayo.edu/), a
high-throughput package designed by the Mayo Clinic Bioinformatics Program for these purposes. PATIENT-DERIVED XENOGRAFT CELL CULTURES AND SCRNA-SEQ Four cell types were used for single-cell
RNA sequencing. Normal human neonatal epidermal melanocytes were purchased from Lifeline Cell Technology (FC-0019). These were briefly propagated in culture using the DermaLife M Melanocyte
Medium with complete supplements (Lifeline Cell Technology, FC-0027). Melanoma patient-derived xenograft lines M12, M15, and M27 were grown in DMEM with 10% FBS. All PDXs were derived from
the brain metastases of Mayo Clinic patients. These cells were harvested and sent to Mayo Clinic’s Genome Analysis Core Facility where the cells were prepared for single-cell RNA sequencing.
Tissue acquisition for M12, M15, and M27 melanoma brain metastases was approved by IRB and PDXs development by IACUC. All the subjects gave their written informed consent to participate in
the study. For scRNA-seq cells were dissociated to single cells using a combination of centrifugation and straining, and resuspended to desired concentration based on cell type and
sequencing goal. Whole live cells were washed twice in 1X PBS + 0.04% BSA and immediately single cell sorted. Cells were first counted and measured for viability using either the Vi-Cell XR
Cell Viability Analyzer (Beckman-Coulter) or a basic hemocytometer and light microscopy. Based on the desired number of cells to be captured for each sample, a volume of live cells was mixed
with the cDNA master mix. The cell suspension master mix, Gel Beads and partitioning oil were added to a Chromium Single Cell B chip. The filled chip was loaded into the Chromium
Controller, where each sample was processed and the individual cells within the sample were captured into uniquely labeled Gel Beads-In-Emulsion (GEMs). The GEMs were collected from the chip
and taken to the bench for reverse transcription, GEM dissolution, and cDNA clean-up. The resulting cDNA contained a pool of uniquely barcoded molecules. A portion of the cleaned and
measured pooled cDNA continued on to library construction, where standard Illumina sequencing primers and a unique i7 Sample index were added to each cDNA pool. All cDNA pools and resulting
libraries were measured using Qubit High Sensitivity assays (Thermo Fisher Scientific), Agilent Bioanalyzer High Sensitivity chips (Agilent) and Kapa DNA Quantification reagents (Kapa
Biosystems). Libraries were sequenced at 60,000 fragment reads per cell following Illumina’s standard protocol using the Illumina cBot and HiSeq 3000/4000 PE Cluster Kit. The flow cells were
sequenced as 100 × 2 paired end reads on an Illumina HiSeq 4000 using HiSeq 3000/4000 sequencing kit and HCS v3.3.52 collection software. Base-calling was performed using Illumina’s RTA
version 2.7.3. The single-cell RNA-seq dataset was analyzed using Monocle353. The gene expression matrices of all 4 libraries were combined and genes expressed in <3 cells were filtered
out. Cells with the number of genes detected <500, or total UMI fewer than 1000 or more than 102,784 (three standard deviations above the mean), or having mitochondrial content >50%
were discarded. After these initial filtering steps the gene counts were log-transformed after adding a pseudocount, and each gene was normalized to a Gaussian distribution with mean of 0
and variance of 1. The dimensionality of the data was reduced by principal component analysis (PCA) and only the first 100 components were used for downstream analysis. For visualization and
clustering purpose the data were further projected into two-dimensional space using the UMAP algorithm [https://arxiv.org/abs/1802.03426v2]. Cells were then clustered based on the UMAP
projection using the Leiden community detection algorithm [https://www.nature.com/articles/s41598-019-41695-z]. To determine marker genes unique to each cluster, we used the top_markers
function of Monocle3, where 1000 cells from each cluster were randomly selected as reference set for marker significance testing. MICROARRAY ANALYSES Publicly available gene expression data
were: GSE46517 (31 primary and 73 metastatic melanomas); GSE7553 (4 normal skin, 11 squamous cell carcinoma, 15 basal cell carcinoma, 2 melanoma in situ and 54 malignant melanomas); GDS1375
(17 nevi and 45 malignant melanomas); GDS3966 (31 primary and 52 metastatic melanomas). All data sets were profiled on Affymetrix U133 platform. IN VITRO INVASION AND WOUND-HEALING ASSAYS
Cell invasion was performed using 24-well Corning transwell membranes pre-coated with Matrigel (0.4 mg/ml; Becton Dickinson). Briefly, A357 M6 (50,000 cells/well), SSM2c, SK-Mel-28, MeWo,
and M51 (100,000 cells/well) were suspended in serum-free medium supplemented with 500 μg/ml Mitomycin C (Sigma-Aldrich), and seeded over the Matrigel coating. Medium supplemented with 20%
FBS was used as a chemo-attractant. After 30 h invaded cells were stained with Diff Quick (MediCult Italia S.p.A) and counted. For would-healing, 30,000 melanoma cells were seeded on a plate
using Culture 2 well silicone inserts (IBIDI) and let to adhere overnight in complete medium until they reached a confluence of ~90%. The inserts were then removed, medium replaced by
serum-free DMEM added with 0.5 mg/ml mitomycin C, and the quality of the covered area was evaluated. The cell-free scratches were imaged at indicated different time points after insert
removal, using a LEICA DFC450C microscope with ×4 objective lens, until complete wound closure. The measure of cell-free scratch was performed with Image J software and the relative
migration rate calculated as the mean of the relative percentage of covered area compared to the T0 area for each well. INTRACARDIAC METASTASIS MODEL IN VIVO To assess the effect of ST3GAL1
depletion on melanoma cell seeding and survival in a metastatic environment, we used a model of intracardiac instillation of melanoma cells. A375 M6 cells were first transduced with LV-c,
LV-shST3GAL1.1, or LV-shST3GAL1.2, and then stably transfected with a luciferase reporter plasmid (pGL4.51[luc2/CMV/Neo] Vector). Cells were resuspended in DMEM at the concentration of 5 ×
105 cells per 150 μl, aliquoted into Eppendorf tubes and maintained on ice until injection. On day 0, female athymic nude 8-week-old mice were anesthetized by exposure to isoflurane and
injected into the left ventricle of the heart with 5 × 105 A375 M6 cells transduced as indicated above. A successful intracardiac injection was indicated on day 1 by images showing systemic
bioluminescence distributed throughout the animal. Only mice with evidence of a satisfactory injection were kept in the experiment. Assessment of metastases was performed weekly by measuring
bioluminescence produced by metastatic cells in the living mice. Briefly, the substrate luciferin was injected into the intraperitoneal cavity at a dose of 150 mg/kg body weight (30 mg/ml),
~15 min before imaging. Mice were anesthetized with 2.5% Avertin and placed on the imaging stage. Ventral images were collected for automatic exposure (3 min) using the Photon Imager system
(Biospace Lab). Analysis was performed using M3Vision software (Biospace Lab) by measurement of photon flux (measured in photons/s/cm2/steradian) with a region of interest (ROI) drawn
around the bioluminescence signal to be measured. Data were plotted using GraphPad PRISM and significance was determined by unpaired _t_ test. After killing mice, metastasis-bearing lungs
and other organs were dissected, fixed in 10% formalin and processed for histology. IN VIVO METASTASIS ASSAY To assess the role of ST3GAL1 in the formation of metastases we used a xenograft
model. A375 M6 cells were first stably transfected with a luciferase reporter plasmid (pGL4.51[luc2/CMV/Neo] Vector), and then co-transduced with LV-GFP and either LV-c, LV-shST3GAL1.1, or
LV-ST3GAL1. Cells were resuspended in DMEM at the concentration of 1.5 × 106 cells per 150 μl, aliquoted into Eppendorf tubes and maintained on ice until injection. On day 0, 8-week-old
female athymic nude mice were anesthetized by exposure to isoflurane and injected subcutaneously into the right flank (_n_ = 8 for each group). When primary tumors were palpable,
measurements were made with a caliper twice a week until resection. Tumor volume was calculated with the formula _V_ = _W_2 × _L_ × 0.5, where _W_ represents tumor width and _L_ the length.
Primary tumors were surgically resected when tumor volume reached 800 mm3. Assessment of metastases was performed weekly by measuring bioluminescence produced by metastatic cells in the
living mice by injecting the substrate luciferin, as described above. After killing mice, metastasis-bearing lungs and other organs were dissected, fixed in 10% formalin and processed for
histology. All animal protocols were approved by local ethic authorities (CeSAL, Centro Stabulazione Animali da Laboratorio) and by the Italian Ministry of Health and conducted in accordance
with Italian Governing Law (D.lgs 26/2014). In all in vivo experiments, mice were maintained at the animal facility (CeSAL) of the University of Florence (Italy). Animals were maintained in
a pathogen-free, temperature-controlled, 12 h light and dark cycle environment, and were fed ad libitum. Mice were housed in plastics cages (no more than four animals per cage to minimize
aggressiveness). FLOW CYTOMETRY For apoptosis analysis, cells were serum-starved for 48 h and apoptosis was measured using the Annexin V–phycoerythrin/7-AAD apoptosis kit (BD Biosciences,
San Diego, CA, USA) according to the manufacturer’s protocol. The number of Annexin V+/7-AAD- (early apoptosis) and Annexin V+/7-AAD+ (late apoptosis) labeled cells was detected with a
CytoFLEX S Flow Cytometer (Beckman Coulter) and analyzed using the CytExpert Software. For circulating melanoma cells analysis, 100 μl of blood was collected from mice at surgery of the
primary tumors by cardiac puncture through a syringe using heparin as anticoagulant54. Red blood cells (RBC) were removed by addition of RBC lysis solution (83 mg NH4Cl, 10 mg KHCO3, 18 μl
EDTA 5%, ddH2O to a final volume of 10 ml) followed by centrifugation at 1200 × _g_ for 10 min. Pellets were resuspended in 300 μl in PBS before flow cytometry. GFP+-cells were detected with
a CytoFLEX S Flow Cytometer. Sorting gates were drawn using A375 M6 GFP− and GFP+ cells as the negative or the positive control, respectively. Blood sample from healthy mice was also used
to exclude unspecific fluorescence signals from murine blood cells. Data were analyzed with FlowJo software (TreeStar Inc., Ashland, OR, United States) and normalized on number of events.
All FACS sorting gate strategies are provided in Supplementary Fig. 13. IMMUNOHISTOCHEMISTRY AND HISTOPATHOLOGICAL ANALYSIS Immunohistochemistry was performed on tissue microarrays (US
Biomax, Inc.) of formalin-fixed paraffin-embedded specimens of human nevi (_n_ = 24), malignant melanomas (_n_ = 56), and metastatic malignant melanomas (_n_ = 40). After antigen retrieval
(with citrate buffer pH 6.0), staining was performed with the UltraVision Detection System kit (Lab Vision, Fremont, CA, USA) following manufacturer’s instructions. Sections were incubated
overnight at 4 °C with rabbit anti-ST3GAL1 antibody (1:50 dilution; #PA5-21721, Invitrogen). AEC (3-amino-9-ethylcarbazole; Dako, Copenhagen, Denmark) was used as chromogen. Sections were
counterstained with hematoxylin. Each stained core section was scored by visual microscopy inspection as follows: 0, for no staining (negative); 1, for weak staining (low); 2, for moderate
staining (medium); and 3, for marked staining (high). Most of the cores showed expression in >75% of the tumor cells. Immunohistochemistry in formalin-fixed paraffin-embedded murine lung
sections was performed as reported above, using anti-GFP antibody (1:500 dilution; Santa Cruz Biotechnology). CHROMATIN IMMUNOPRECIPITATION For ChIP experiments, 3 × 106 SSM2c cells were
fixed with 1% formaldehyde for 15 min, and fixation stopped by adding 125 mM glycine for 5 min. Cells were lysed in Farnham lysis buffer (5 mM PIPES pH 8, 85 mM KCl, 0.5% NP-40) added with
protease inhibitors for 10 min. Nuclei were collected by centrifugation at 800 × _g_ for 10 min and then lysed in nuclear lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl pH 8) added with
protease inhibitors. Chromatin was sonicated to an average size of 200–600 bp with a SONOPULS Mini20 Sonicator (Bandelin), diluted with ChIP Dilution Buffer (10 mM Tris-HCl pH 8, 2 mM EDTA,
140 mM NaCl, 1% Triton X-100, 0.1% SDS) and incubated overnight with 20 μl protein G magnetic dynabeads and 3 μg of mouse anti-SOX2 (R&D System, #MAB2018) (1:1000), mouse anti-GLI1 (Cell
Signaling, #2643) (1:500), or normal mouse IgG (Santa Cruz Biotechnology, #sc-2025) (1:100) antibodies. Immunocomplexes were washed with increasing salt concentrations, DNA was eluted at 85
°C with 1% SDS and crosslinks reversed overnight at 65 °C with 200 mM NaCl. DNA was treated with 4 μg RNase A (Thermo Fisher Scientific) and 20 μg Proteinase K (Qiagen) at 37 °C for 2 h to
remove RNA and protein contaminations, and recovered with QIAquick PCR Purification Kit (Qiagen). qPCR was carried out at 60 °C using FastStart SYBR Green Master (Roche) in a Rotorgene-Q
(Qiagen). Primer sequences are reported in Supplementary Table 4. LUCIFERASE REPORTER ASSAY AND MUTAGENESIS A fragment of ST3GAL1 enhancer II (−12461/−11451bp from TSS) was PCR amplified
with KOD hot start DNA polymerase (Merck Millipore) and cloned into the pGL3Basic vector (Promega) using NheI-HindIII sites, to generate ST3GAL1 enh-luc reporter. Primers used were: ST3GAL1
enh F, 5′-AGTTGCCATCCCCTAAGGTAA-3′; ST3GAL1 enh R, 5′-AGGAACCATGATCCTTATGGA-3′. Mutations of ST3GAL1 enh-luc reporter were introduced using QuickChange II (Agilent Technologies) with the
following oligos: SBSmut1 F, 5′-TTTGTCAAAAATCAGCTCATGAAGGAATAGCTTTAATCCCTACTTGG-3′; SBSmut1 R, 5′-CCAAGTAGGGATTAAAGCTATTCCTTCATGAGCTGATTTTTGACAAA-3′; SBSmut2 F,
5′-GTTCTGGACTTTCTGTTTCATGAGTCAATAAAATTCTTTATATTACTCCACTTTAGGTCATAG-3′; SBSmut2 R, 5′-CTATGACCTAAAGTGGAGTAATATAAAGAATTTTATTGACTCATGAAACAGAAAGTCCAGAAC-3′; GBSmut F,
5′-ATGAATGAATAGCTTTAATCCCTACTTGGAGGACCAAGATAAAGAGATAAAT-3′; GBSmut R, 5′-ATTTATCTCTTTATCTTGGTCCTCCAAGTAGGGATTAAAGCTATTCATTCAT-3′. ST3GAL1 enh-luc reporters were used in combination with
_Renilla_ luciferase pRL-TK reporter vector (Promega) to normalize luciferase activities. The sequence of ST3GAL1 enhancer II is reported in Supplementary Fig. 4. PROTEIN EXTRACTION AND
WESTERN BLOT Cells were lysed in cold RIPA buffer (50 mM Tris-HCl pH 7.5, 1% NP-40, 150 mM NaCl, 5 mM EDTA, 0.25% NaDOC, and 0.1% SDS) supplemented with protease and phosphatase inhibitors
and centrifuged at 20,000 × _g_ for 20 min at 4 °C55. Supernatant was collected as whole cell extract (WCE). Equal amounts of protein were resolved by SDS-polyacrylamide gel electrophoresis,
transferred onto nitrocellulose membranes, and incubated for 1 h in blocking buffer at room temperature. Primary antibodies are reported in Supplementary Table 5. Blotted membranes were
developed by using SuperSignal West Femto (Thermo Fisher Scientific) and imaged with ChemiDoc Imaging Systems (Bio-Rad). DETECTION OF SIALYLATION BY LECTIN AFFINITY IMMUNOPRECIPITATION For
immunoprecipitation (IP) of sialylated proteins with MAL-II lectin, cells were lysed in cold IP buffer (0.5% NP-40, 100 mM NaCl, 5 mM EDTA, 10% glycerol, and 50 mM Tris-HCl pH 7.5)
supplemented with protease and phosphatase inhibitors, and supernatant was collected as WCE after centrifugation at 20,000×_g_ for 15 min at 4 °C. In all, 1 mg WCE was diluted with IP buffer
to a final volume of 500 μl and incubated for 3 h at room temperature with 10 μg of biotinylated Maackia Amurensis Lectin II (MAL-II; Vector Laboratories). Streptavidin-agarose beads
(Thermo Fisher Scientific) were then added and samples were incubated for additional 2 h at room temperature in costant rotation. Lectin-bounded sialylated proteins were collected after
brief centrifugation, washed with IP buffer, eluted from beads by boiling in SDS–PAGE sample buffer 2X and resolved by SDS-polyacrylamide gel electrophoresis. AXL ACTIVATION AND DIMERIZATION
In all, 600,000 A375 M6 cells were seeded in 60 mm dishes in complete medium to allow them to adhere overnight, serum-starved for 16 h and stimulated for 1 hr with 250 ng/ml of human
recombinant GAS6 (R&D Systems, 885-GSB-050) on ice. Cells were then crosslinked with 3 mM BS3 (Sigma-Aldrich, Cat.no. 82436-77-9) for 20 min on ice and quenched with 250 mM glycine for 5
min at 4 °C56. Pellets were lysed in ice-cold lysis buffer (140 mM NaCl, 10 mM EDTA, 10% glycerol, 1% NP-40, and 20 mM TRIS-HCl pH 8) supplemented with protease and phosphatase inhibitors,
and supernatant was collected as WCE after centrifugation at 16,000 × _g_ at +4 °C for 20 min. In total, 50 μg proteins were resolved on a 7% SDS-polyacrylamide gel, transferred overnight
onto a nitrocellulose membrane and incubated with anti-AXL antibody (Santa Cruz Biotechnology, sc-166269). MASS SPECTROMETRY ANALYSIS Purification of sialylated proteins for Mass
Spectrometry (MS) analysis was performed with MAL spin columns in the Qproteome Sialic Glycoprotein Kit (Qiagen) according to the manufacturer instructions. In total, 40 μg of sialylated
proteins enriched from LV-shST3GAL1 and from LV-ST3GAL1 were trypsin digested with a filter aided sample preparation (FASP) protocol57. After digestion, tryptic peptides were desalted using
C18 cartridges (Sep-pack, Waters) according to manufacturer’s instructions. Eluted peptides were dried under vacuum and suspended in 40 μL of CH3CN 3%/formic acid 0.1%. In all, 1 μL of
sample was used for each round of analysis. LC-MS/MS experiments were performed with a LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) coupled online to a nano-HPLC Ultimate
3000 (Dionex – Thermo Fisher Scientific). Samples were loaded into a 10-cm pico-frit column (75 μm I.D., 15 μm tip, New Objective) packed in-house with C18 material (Aeris Peptide 3.6 lm
XB-C18; Phenomenex). Each sample was repeatedly injected to obtain technical replicates of the experiment. Peptides were separated at a flow rate of 250 nL/min using a linear gradient of
CH3CN from 3% to 40% in 90 min. A data dependent acquisition (DDA) with a top4 method was used for the analysis: one full scan between 300 and 1700 _m/z_ at high resolution (60,000) in the
Orbitrap was followed by the CID fragmentation in the linear trap of the four most intense ions. Raw data files were analyzed with the software package MaxQuant and the search engine
Andromeda58,59, against the human section of the Uniprot database (version july 2018) concatenated with a database of contaminants commonly found in proteomics experiments. Trypsin was
selected as enzyme with two missed cleavages allowed; carbamidomethylation of cysteine residues was set as fixed modification and methionine oxidation as variable modification. Results were
filtered at a false discovery rate (FDR) of 0.01, both at the peptide and protein level. Only proteins identified with at least three independent peptides were considered as a significant
hit. The parameter “LFQ” calculated by the software was used to estimate the abundance of the proteins across the different samples. A Student’s _t_ test was performed to highlight proteins
with a significant (_p_ ≤ 0.05) different abundance in LV-shST3GAL1 and LV-ST3GAL1 samples. A fold change ≥1.5 was selected to classify proteins as potential substrates of ST3GAL1.
PHOSPHO-RECEPTOR TYROSINE KINASE PROTEOME ARRAY A Proteome Profiler Antibody Arrays Kit for human phospho-receptor tyrosine kinases (phospho-RTK, R&D System) was used to determine which
RTK is phosphorylated in presence of ST3GAL1. Briefly, A375 M6 cells transduced with scramble (LV-c) or LV-ST3GAL1 were lysed for 30 minutes in cold lysis buffer (R&D System)
supplemented with protease and phosphatase inhibitor cocktail (Sigma-Aldrich), and spun at 14,000 × _g_ for 5 min to remove debris. Array membranes were blocked with the provided blocking
buffer (R&D System) for 1 h, and then incubated overnight with the cell lysate at 4 °C on a rocking platform shaker. The day after, array membranes were washed three times in wash buffer
(R&D System), incubated for 2 h with Anti-Phospho-Tyrosine-HRP Detection Antibody (R&D System) and imaged on the ChemiDoc MP Imaging System. STATISTICAL ANALYSIS Data represent mean
± s.d or mean ± s.e.m. values calculated on at least three independent experiments. To assess normal distribution and homoscedasticity for each quantitative outcome in each group
Kolmogorov–Smirnov’s test and Bartlett’s test was used respectively. Statistical significance was assessed by two-tailed unpaired Student’s _t_ test, by ANOVA or Welch ANOVA and Tukey’s,
Dunnett’s or Games-Howell’s tests for multiple comparisons. For TCGA analysis statistical significance was determined using Welch’s _t_-test. Details on the number of independent experiments
or samples and statistical tests can be found in figure legends. _P_ value < 0.05 was considered statistically significant. REPORTING SUMMARY Further information on research design is
available in the Nature Research Reporting Summary linked to this article. DATA AVAILABILITY RNAseq and scRNAseq primary data generated in this manuscript are available from GEO under
accession numbers GSE159049 and GSE159597. The unique raw and normalized enriched MAL-bound proteins identified by MS are available as Supplementary Data 2. Public available microarray data
used in this study were obtained from GSE46517, GSE7553, GDS1375, and GDS3966. All the data supporting this study are available within the article, the Supplementary file, the Source Data
file, as indicated in the Reporting Summary for this article. A Reporting Summary for this article is available as a Supplementary Information file. Source data are provided with this paper.
REFERENCES * McDermott, D. et al. Durable benefit and the potential for long-term survival with immunotherapy in advanced melanoma. _Cancer Treat. Rev._ 40, 1056–1064 (2014). Article
PubMed Google Scholar * Bird-Lieberman, E. L. et al. Molecular imaging using fluorescent lectins permits rapid endoscopic identification of dysplasia in Barrett’s esophagus. _Nat. Med._
18, 315–321 (2012). Article CAS PubMed Google Scholar * Schultz, M. J., Swindall, A. F. & Bellis, S. L. Regulation of the metastatic cell phenotype by sialylated glycans. _Cancer
Metastasis Rev._ 31, 501–518 (2012). Article CAS PubMed PubMed Central Google Scholar * Christiansen, M. N. et al. Cell surface protein glycosylation in cancer. _Proteomics_ 14, 525–546
(2014). Article CAS PubMed Google Scholar * Pinho, S. S. & Reis, C. A. Glycosylation in cancer: mechanisms and clinical implications. _Nat. Rev. Cancer_ 15, 540–555 (2015). Article
CAS PubMed Google Scholar * Vajaria, B. N. & Patel, P. S. Glycosylation: a hallmark of cancer? _Glycoconj. J._ 34, 147–156 (2017). Article CAS PubMed Google Scholar * Mereiter,
S., Balmaña, M., Campos, D., Gomes, J. & Reis, C. A. Glycosylation in the era of cancer-targeted therapy: where are we heading? _Cancer Cell_ 36, 6–16 (2019). Article CAS PubMed
Google Scholar * Büll, C., Stoel, M. A., den Brok, M. H. & Adema, G. J. Sialic acids sweeten a tumor’s life. _Cancer Res._ 74, 3199–3204 (2014). Article PubMed CAS Google Scholar *
Gaziel-Sovran, A. et al. miR-30b/30d regulation of GalNAc transferases enhances invasion and immunosuppression during metastasis. _Cancer Cell_ 20, 104–118 (2011). Article CAS PubMed
PubMed Central Google Scholar * Agrawal, P. et al. A systems biology approach identifies FUT8 as a driver of melanoma metastasis. _Cancer Cell_ 31, 804–819 (2017). Article CAS PubMed
PubMed Central Google Scholar * Santini, R. et al. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. _Oncogene_ 33, 4697–4608 (2014). Article CAS PubMed
PubMed Central Google Scholar * Bora-Singhal, N., Perumal, D., Nguyen, J. & Chellappan, S. Gli1-mediated regulation of Sox2 facilitates self-renewal of stem-like cells and confers
resistance to EGFR inhibitors in non-small cell lung cancer. _Neoplasia_ 17, 538–551 (2015). Article CAS PubMed PubMed Central Google Scholar * Kar, S., Sengupta, D., Deb, M., Pradhan,
N. & Patra, S. K. SOX2 function and Hedgehog signaling pathway are co-conspirators in promoting androgen independent prostate cancer. _Biochim. Biophys. Acta Mol. Basis Dis._ 1863,
253–265 (2017). Article CAS PubMed Google Scholar * Santini, R. et al. Hedgehog-GLI signaling drives self-renewal and tumorigenicity of human melanoma-initiating cells. _Stem Cells_ 30,
1808–1818 (2012). Article CAS PubMed Google Scholar * Laidler, P. et al. Characterization of glycosylation and adherent properties of melanoma cell lines. _Cancer Immunol. Immunother._
55, 112–118 (2006). Article CAS PubMed Google Scholar * Chong, Y. K. et al. ST3GAL1-associated transcriptomic program in glioblastoma tumor growth, invasion, and prognosis. _J. Natl
Cancer Inst._ 108, djv326 (2015). PubMed PubMed Central Google Scholar * Wu, X. et al. Sialyltransferase ST3GAL1 promotes cell migration, invasion, and TGF-β1-induced EMT and confers
paclitaxel resistance in ovarian cancer. _Cell Death Dis._ 9, 1102 (2018). Article PubMed PubMed Central CAS Google Scholar * Riker, A. I. et al. The gene expression profiles of primary
and metastatic melanoma yields a transition point of tumor progression and metastasis. _BMC Med. Genomics_ 1, 13 (2008). Article PubMed PubMed Central CAS Google Scholar * Kabbarah, O.
et al. Integrative genome comparison of primary and metastatic melanomas. _PLoS ONE_ 5, e10770 (2010). Article ADS PubMed PubMed Central CAS Google Scholar * Andreucci, E. et al.
Carbonic anhydrase IX inhibition affects viability of cancer cells adapted to extracellular acidosis. _J. Mol. Med._ 95, 1341–1353 (2017). Article CAS PubMed Google Scholar * Fang, X. et
al. The SOX2 response program in glioblastoma multiforme: an integrated ChIP-seq, expression microarray, and microRNA analysis. _BMC Genomics_ 12, 11 (2011). Article CAS PubMed PubMed
Central Google Scholar * Sasaki, H., Hui, C., Nakafuku, M. & Kondoh, H. A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can
respond to Shh in vitro. _Development_ 124, 1313–1322 (1997). CAS PubMed Google Scholar * Pandolfi, S., Montagnani, V., Lapucci, A. & Stecca, B. HEDGEHOG/GLI-E2F1 axis modulates iASPP
expression and function and regulates melanoma cell growth. _Cell Death Differ._ 22, 2006–2019 (2015). Article CAS PubMed PubMed Central Google Scholar * Chiang, C. H. et al. A novel
sialyltransferase inhibitor AL10 suppresses invasion and metastasis of lung cancer cells by inhibiting integrin-mediated signaling. _J. Cell Physiol._ 223, 492–499 (2010). ADS CAS PubMed
Google Scholar * Rankin, E. B. & Giaccia, A. J. The receptor tyrosine kinase AXL in cancer progression. _Cancers_ 8, E103 (2016). Article PubMed CAS Google Scholar * Restivo, G. et
al. Low neurotrophin receptor CD271 regulates phenotype switching in melanoma. _Nat. Commun._ 8, 1988 (2017). Article ADS PubMed PubMed Central CAS Google Scholar * Kovacs, E., Zorn,
J. A., Huang, Y., Barros, T. & Kuriyan, J. A structural perspective on the regulation of the epidermal growth factor receptor. _Annu. Rev. Biochem._ 84, 739–764 (2015). Article CAS
PubMed PubMed Central Google Scholar * Boone, B. et al. EGFR in melanoma: clinical significance and potential therapeutic target. _J. Cutan. Pathol._ 38, 492–502 (2011). Article PubMed
Google Scholar * Linger, R. M., Keating, A. K., Earp, H. S. & Graham, D. K. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human
cancer. _Adv. Cancer Res._ 100, 35–83 (2008). Article CAS PubMed PubMed Central Google Scholar * Sensi, M. et al. Human cutaneous melanomas lacking MITF and melanocyte differentiation
antigens express a functional Axl receptor kinase. _J. Invest. Dermatol._ 131, 2448–2457 (2011). Article CAS PubMed Google Scholar * Revach, O. Y., Sandler, O., Samuels, Y. & Geiger,
B. Cross-talk between receptor tyrosine kinases AXL and ERBB3 regulates invadopodia formation in melanoma cells. _Cancer Res._ 79, 2634–2648 (2019). Article CAS PubMed Google Scholar *
Zuo, Q. et al. AXL/AKT axis mediated-resistance to BRAF inhibitor depends on PTEN status in melanoma. _Oncogene_ 37, 3275–3289 (2018). Article CAS PubMed PubMed Central Google Scholar *
Cui, H., Lin, Y., Yue, L., Zhao, X. & Liu, J. Differential expression of the e to BRAF i acid residues in breast cancer is associated with metastatic potential. _Oncol. Rep._ 25,
1365–1371 (2011). CAS PubMed Google Scholar * Picco, G. et al. Over-expression of ST3Gal-I promotes mammary tumorigenesis. _Glycobiology_ 20, 1241–1250 (2010). Article CAS PubMed
PubMed Central Google Scholar * Girouard, S. D. et al. SOX2 contributes to melanoma cell invasion. _Lab Invest._ 92, 362–370 (2012). Article CAS PubMed Google Scholar * Laga, A. C. et
al. SOX2 and nestin expression in human melanoma: an immunohistochemical and experimental study. _Exp. Dermatol._ 20, 339–345 (2011). Article CAS PubMed PubMed Central Google Scholar *
Ravindran Menon, D. et al. CDK1 interacts with Sox2 and promotes tumor initiation in human melanoma. _Cancer Res._ 78, 6561–6574 (2018). Article PubMed Google Scholar * Jia, Q. et al. Low
levels of Sox2 are required for melanoma tumor-repopulating cell dormancy. _Theranostics_ 9, 424–435 (2019). Article CAS PubMed PubMed Central Google Scholar * Andreucci, E. et al.
SOX2 as a novel contributor of oxidative metabolism in melanoma cells. _Cell Commun. Signal._ 16, 87 (2018). Article CAS PubMed PubMed Central Google Scholar * Stecca, B. et al.
Melanomas require Hedgehog-Gli signaling regulated by interactions between Gli1 and the RAS-MEK/AKT pathways. _Proc. Natl Acad. Sci. USA_ 104, 5895–5900 (2007). Article ADS CAS PubMed
PubMed Central Google Scholar * Alexaki, V. I. et al. GLI2-mediated melanoma invasion and metastasis. _J. Natl Cancer Inst._ 102, 1148–1159 (2010). Article CAS PubMed PubMed Central
Google Scholar * Gunarta, I. K. et al. Critical role of glioma-associated oncogene homolog 1 in maintaining invasive and mesenchymal-like properties of melanoma cells. _Cancer Sci._ 108,
1602–1611 (2017). Article CAS PubMed PubMed Central Google Scholar * Zhu, C., Wei, Y. & Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions,
molecular mechanisms and clinical applications. _Mol. Cancer_ 18, 153 (2019). Article PubMed PubMed Central Google Scholar * Sasaki, T. et al. Structural basis for Gas6-Axl signalling.
_EMBO J._ 25, 80–87 (2006). Article CAS PubMed Google Scholar * Axelrod, H. & Pienta, K. J. Axl as a mediator of cellular growth and survival. _Oncotarget_ 5, 8818–8852 (2014).
Article PubMed PubMed Central Google Scholar * Boshuizen, J. et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. _Nat.
Med._ 24, 203–212 (2018). Article CAS PubMed Google Scholar * Arozarena, I. & Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. _Nat. Rev.
Cancer_ 19, 377–391 (2019). Article CAS PubMed Google Scholar * Konieczkowski, D. J. et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. _Cancer
Discov._ 4, 816–827 (2014). Article CAS PubMed PubMed Central Google Scholar * De Polo, A. et al. AXL receptor signalling suppresses p53 in melanoma through stabilization of the
MDMX-MDM2 complex. _J. Mol. Cell Biol._ 9, 154–165 (2017). Article PubMed CAS Google Scholar * Vasconcelos-Dos-Santos, A. et al. Biosynthetic machinery involved in aberrant
glycosylation: promising targets for developing of drugs against cancer. _Front. Oncol._ 5, 138 (2015). Article PubMed PubMed Central Google Scholar * Riverso, M., Montagnani, V. &
Stecca, B. KLF4 is regulated by RAS/RAF/MEK/ERK signaling through E2F1 and promotes melanoma cell growth. _Oncogene_ 36, 3322–3333 (2017). Article CAS PubMed PubMed Central Google
Scholar * Pietrobono, S. et al. Down-regulation of SOX2 underlies the inhibitory effects of the triphenylmethane gentian violet on melanoma cell self-renewal and survival. _J. Invest.
Dermatol._ 136, 2059–2069 (2016). Article CAS PubMed Google Scholar * Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. _Nature_ 566, 496–502 (2019).
Article ADS CAS PubMed PubMed Central Google Scholar * Tasdogan, A. et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. _Nature_ 577, 115–120 (2020).
Article ADS CAS PubMed Google Scholar * Pietrobono, S. et al. Targeted inhibition of Hedgehog-GLI signaling by novel acylguanidine derivatives inhibits melanoma cell growth by inducing
replication stress and mitotic catastrophe. _Cell Death Dis._ 9, 142 (2018). Article PubMed PubMed Central CAS Google Scholar * Turk, H. F. & Chapkin, R. S. Analysis of epidermal
growth factor receptor dimerization by BS³ cross-linking. _Methods Mol. Biol._ 1233, 25–34 (2015). Article PubMed PubMed Central Google Scholar * Wiśniewski, J. R., Zougman, A., Nagaraj,
N. & Mann, M. Universal sample preparation method for proteome analysis. _Nat. Methods_ 6, 359–362 (2009). Article PubMed CAS Google Scholar * Cox, J. & Mann, M. MaxQuant
enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. _Nat. Biotechnol._ 26, 1367–1372 (2008). Article CAS PubMed
Google Scholar * Cox, J. et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. _J. Proteome Res._ 10, 1794–1805 (2011). Article ADS CAS PubMed Google
Scholar * Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. _Cancer Discov._ 2, 401–404 (2012). Article PubMed
Google Scholar * Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. _Sci. Signal._ 6, 11 (2013). Article CAS Google Scholar
Download references ACKNOWLEDGEMENTS This work was supported by a grant from the Italian Association for Cancer Research (AIRC; IG-23091) and institutional funding from the Institute for
Cancer Research, Prevention and Clinical Network (ISPRO) to B.S., by CA136526 to M.E.F.Z. and by K08CA215105 to A.M.; S.P. was supported by AIRC fellowship (project n. 21168). The authors
wish to thank the Cassa di Risparmio di Padova e Rovigo (Cariparo) Holding for funding the acquisition of the LTQ-Orbitrap XL mass spectrometer. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS *
Core Research Laboratory – Institute for Cancer Research and Prevention (ISPRO), Viale Pieraccini 6, 50139, Florence, Italy Silvia Pietrobono, Giulia Anichini, Sara Pepe & Barbara
Stecca * Department of Medical Biotechnologies, University of Siena, Viale M. Bracci 16, 53100, Siena, Italy Giulia Anichini & Sara Pepe * Department of Clinical and Experimental
Medicine, University of Florence, Viale Morgagni 50, 50134, Florence, Italy Cesare Sala * Department of Biotechnology, Chemistry and Pharmacy, University of Siena, Via A. Moro 2, 53100,
Siena, Italy Fabrizio Manetti * Schulze Center for Novel Therapeutics, Division of Oncology Research, Department of Oncology, Mayo Clinic, Rochester, MN, 55905, USA Luciana L. Almada, Ryan
M. Carr, Brooke D. Paradise & Martin E. Fernandez-Zapico * Department of Radiation Oncology, Mayo Clinic, Rochester, MN, 55905, USA Jann N. Sarkaria * Department of Health Sciences
Research, Mayo Clinic, Rochester, Rochester, MN, 55905, USA Jaime I. Davila * Department of Neurosciences, Psychology, Drug Research and Child Health, University of Florence, Viale
Pieraccini 6, 50139, Florence, Italy Lorenzo Tofani * Proteomics Center, University of Padova and Azienda Ospedaliera di Padova, Via G. Oris 2B, 35129, Padova, Italy Ilaria Battisti &
Giorgio Arrigoni * Department of Biomedical Sciences, University of Padova, Via U. Bassi 58B, 35131, Padova, Italy Giorgio Arrigoni * Department of Dermatology, Mayo Clinic, Rochester, MN,
55905, USA Li Ying & Alexander Meves * Department of Molecular Pharmacology & Experimental Therapeutics, Mayo Clinic, Rochester, MN, 55905, USA Cheng Zhang & Hu Li Authors *
Silvia Pietrobono View author publications You can also search for this author inPubMed Google Scholar * Giulia Anichini View author publications You can also search for this author inPubMed
Google Scholar * Cesare Sala View author publications You can also search for this author inPubMed Google Scholar * Fabrizio Manetti View author publications You can also search for this
author inPubMed Google Scholar * Luciana L. Almada View author publications You can also search for this author inPubMed Google Scholar * Sara Pepe View author publications You can also
search for this author inPubMed Google Scholar * Ryan M. Carr View author publications You can also search for this author inPubMed Google Scholar * Brooke D. Paradise View author
publications You can also search for this author inPubMed Google Scholar * Jann N. Sarkaria View author publications You can also search for this author inPubMed Google Scholar * Jaime I.
Davila View author publications You can also search for this author inPubMed Google Scholar * Lorenzo Tofani View author publications You can also search for this author inPubMed Google
Scholar * Ilaria Battisti View author publications You can also search for this author inPubMed Google Scholar * Giorgio Arrigoni View author publications You can also search for this author
inPubMed Google Scholar * Li Ying View author publications You can also search for this author inPubMed Google Scholar * Cheng Zhang View author publications You can also search for this
author inPubMed Google Scholar * Hu Li View author publications You can also search for this author inPubMed Google Scholar * Alexander Meves View author publications You can also search for
this author inPubMed Google Scholar * Martin E. Fernandez-Zapico View author publications You can also search for this author inPubMed Google Scholar * Barbara Stecca View author
publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Experimental design: S.P. and B.S.; qPCR and western blots: S.P. and G.A.1,2; Generation of expression
vectors: S.P.; In vitro assays: S.P., G.A.1,2, and F.M.; Immunohistochemistry: S.P.1,2 and G.A.1,2; ChIP and Luciferase reporter assays: S.P.; Clinical samples acquisition and pathological
evaluation, histology, and lung metastasis evaluation: B.S.; Flow cytometry and CTC evaluation: C.S. and S.P.; Proteomic analysis: G.A.8,9 and I.B.; PDX development and tissue processing:
L.Y., B.D.P., J.N.S., and A.M.; RNA libraries, RNA-sequencing, scRNA-sequencing, and bioinformatic analysis: L.L.A., R.M.C., L.Y., C.Z., H.L., J.I.D., and M.E.F.Z.; Validation of MS and
RNA-seq data: S.P.; Statistical analysis: L.T., B.D.P., and C.Z.; Manuscript writing: S.P., M.E.F.Z., and B.S. All authors provided intellectual input, reviewed, and edited the manuscript.
CORRESPONDING AUTHOR Correspondence to Barbara Stecca. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PEER REVIEW INFORMATION
_Nature Communications_ thanks Srikumar Chellappan, Mitchell Fane and the other, anonymous, reviewer for their contribution to the peer review of this work. Peer reviewer reports are
available. PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY
INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3 REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND
PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any
medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Pietrobono, S.,
Anichini, G., Sala, C. _et al._ ST3GAL1 is a target of the SOX2-GLI1 transcriptional complex and promotes melanoma metastasis through AXL. _Nat Commun_ 11, 5865 (2020).
https://doi.org/10.1038/s41467-020-19575-2 Download citation * Received: 23 October 2019 * Accepted: 21 October 2020 * Published: 17 November 2020 * DOI:
https://doi.org/10.1038/s41467-020-19575-2 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative





