- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Mechanisms for human sinoatrial node (SAN) dysfunction are poorly understood and whether human SAN excitability requires voltage-gated sodium channels (Nav) remains controversial.
Here, we report that neuronal (n)Nav blockade and selective nNav1.6 blockade during high-resolution optical mapping in explanted human hearts depress intranodal SAN conduction, which worsens
during autonomic stimulation and overdrive suppression to conduction failure. Partial cardiac (c)Nav blockade further impairs automaticity and intranodal conduction, leading to beat-to-beat
variability and reentry. Multiple nNav transcripts are higher in SAN vs atria; heterogeneous alterations of several isoforms, specifically nNav1.6, are associated with heart failure and
chronic alcohol consumption. In silico simulations of Nav distributions suggest that _I_Na is essential for SAN conduction, especially in fibrotic failing hearts. Our results reveal that not
only cNav but nNav are also integral for preventing disease-induced failure in human SAN intranodal conduction. Disease-impaired nNav may underlie patient-specific SAN dysfunctions and
should be considered to treat arrhythmias. SIMILAR CONTENT BEING VIEWED BY OTHERS INHIBITION OF G PROTEIN-GATED _K__+_ CHANNELS BY TERTIAPIN-Q RESCUES SINUS NODE DYSFUNCTION AND
ATRIOVENTRICULAR CONDUCTION IN MOUSE MODELS OF PRIMARY BRADYCARDIA Article Open access 17 June 2020 ALTERED MICRORNA AND MRNA PROFILES DURING HEART FAILURE IN THE HUMAN SINOATRIAL NODE
Article Open access 29 September 2021 DETRIMENTAL PROARRHYTHMOGENIC INTERACTION OF CA2+/CALMODULIN-DEPENDENT PROTEIN KINASE II AND NAV1.8 IN HEART FAILURE Article Open access 15 November
2021 INTRODUCTION Normal cardiac rhythm is maintained by the human sinoatrial node (SAN) complex, which is compartmentalized into multiple intranodal pacemakers and conduction pathways
(sinoatrial conduction pathways, SACPs) within an intramural three-dimensional (3D) structure1,2,3. Robust SAN pacemaking and conduction is maintained amidst a plethora of internal and
external perturbations by several ion channels, which are heterogeneously expressed within the SAN compartments3,4,5. Impairment of pacemaking or conduction between SAN compartments can
result from a variety of cardiac pathologies or extrinsic factors (e.g., antiarrhythmic drugs) and may lead to SAN dysfunction (SND), a disease that can significantly impact quality of
life6. In addition to rhythm abnormalities, SND can often facilitate deterioration to atrial fibrillation (AF)7 and heart failure (HF)8. Currently, patients with symptomatic SND rely on a
single treatment option of electrical pacemaker implantation9, which acts as a crutch to support but not heal the heart. The development of optimal alternative treatments for SND will
require in-depth knowledge of the mechanisms involved in robust human SAN rhythm regulation. However, there is a paucity of studies addressing mechanisms that contribute to automaticity and
intranodal conduction directly in the human SAN complex at the molecular, cellular, and tissue levels. Disease-induced remodeling of many of the molecular components critical to SAN function
including hyperpolarization-activated cyclic nucleotide-gated (HCN) channels and G-protein-coupled inwardly rectifying potassium channels, adenosine receptors (A1R), and L-type calcium
channels, as well as structural fibrotic remodeling can lead to SND3,10,11,12,13. However, majority of these molecular components critical to SAN function have been studied only in animal
models5, which have significantly different functional and anatomical features compared with the human SAN, especially when studying aged/diseased human SAN with SND. In particular, the
roles of voltage-gated sodium channels (Nav), which are major contributors to cardiac and neuronal excitability, remain controversial in human SAN pacemaking and conduction. Although the
presence of Nav has been reported in the SAN from multiple species14,15,16, direct evidence confirming the expression and functional role of cardiac (cNav) and/or neuronal (nNav) isoforms in
the human SAN is lacking17. Some patients with symptomatic SND are found to harbor loss-of-function mutations in the _SCN5A_ gene, which encodes the α-subunit of the cardiac isoform Nav1.5
(ref.18), suggesting that functional voltage-gated Na+ current (_I_Na) may be necessary for maintaining human SAN pacemaking and conduction. Furthermore, although antiarrhythmic drugs that
block Nav have been shown to inadvertently unmask SND in patients predisposed to the arrhythmia19,20,21, no study has determined the specific Nav isoform(s) mediating this detrimental effect
on the human SAN. Hence, there is a critical need to determine the functional contributions of Nav channels in maintaining SAN robustness, by studying their role directly in the unique 3D
human SAN complex2,3,4. Therefore, the primary goal of the current study is to determine the existence and specific role of nNav and cNav isoforms in human SAN pacemaking and intranodal
conduction, and to reveal their disease-induced alterations in explanted human hearts. Some of the comorbidities associated with the explanted human hearts used in this study include, but
are not limited to, HF, AF, hypertension (HTN), and modifying risk factors such as smoking, chronic alcohol consumption, and drug abuse (Supplementary Tables 1 and 2). We employed
high-resolution near-infrared sub-surface optical mapping, the only approach currently able to reveal intramural human SAN pacemaking and conduction. Furthermore, our functional studies are
complemented by human SAN computational simulations and molecular mapping of multiple nNav and cNav transcripts and protein expression. Here, we report that in the human SAN, nNav mainly
contribute to intranodal conduction, whereas cNav play dual roles in both pacemaking and conduction. Impairment of Nav can lead not only to depression of SAN pacemaker and conduction but
also to a perfect storm of SAN exit block, disorganized intranodal pacemakers, and SAN micro- and macro-reentry. Our data also suggest that by altering nNav in the SAN and/or atria, HF and
chronic alcohol consumption could promote a patient-specific, mechanistic substrate for tachy-brady arrhythmias and SAN conduction blocks. RESULTS ESSENTIAL ROLES OF NAVS UNDER PHYSIOLOGICAL
CONDITIONS Under control conditions, all optically mapped human hearts (non-failing _n_ = 12 and HF _n_ = 2) exhibited stable intrinsic sinus rhythm of 56 to 116 beats per minute, with a
sinus cycle length (SCL) of 729 ± 200 ms (Supplementary Table 1 and Supplementary Fig. 1). These values are comparable with “intrinsic” rates reported in vivo during autonomic blockade in
adult human subjects with/without cardiac comorbidities22,23,24. For more details on the stability and experimental protocols, please see the Methods section. All functional data presented
below are averages of all human SAN preparations (HF and non-failing) studied for each specific drug protocol. As the small sample size precludes subset analysis, heart-specific data are
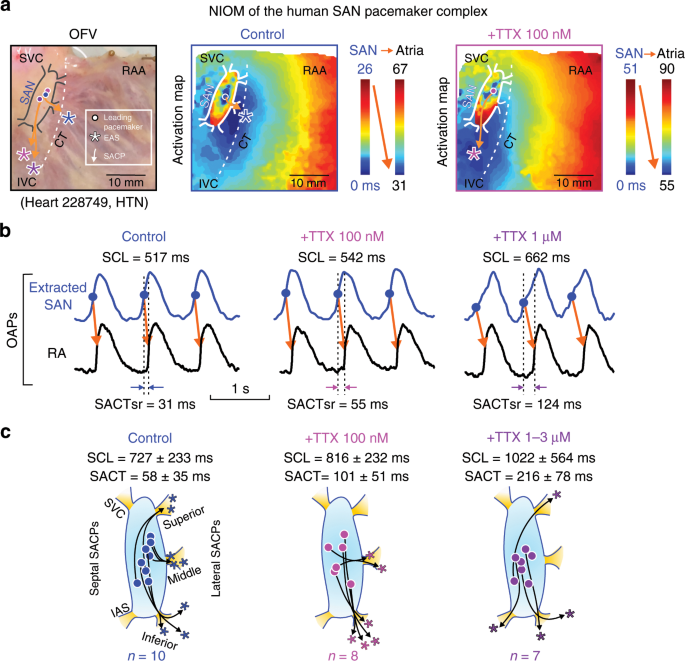
presented in Supplementary Fig. 1. Tetrodotoxin (TTX) dose-dependently prolonged SCL by 6 ± 5% (mean ± SD, _n_ = 8, _P_ < 0.05, Wilcoxon’s test) at 100 nM and 43 ± 41% (_n_ = 7, _P_ <
0.05, Wilcoxon’s test) at 1–3 µM, and caused shifts of the intranodal leading pacemaker and preferential SACP (Fig.1; Table 1). Sinoatrial conduction time at sinus rhythm (SACTsr) was
increased to ~250% (_n_ = 6, _P_ < 0.05, Wilcoxon’s test) by TTX 100 nM and to ~610% (_n_ = 6, _P_ < 0.05, _t_-test) by TTX 1–3 µM. Figure 2a, b shows the effects of TTX 1 µM on SCL
and SACTsr. Furthermore, TTX 1–3 µM significantly increased SACTsr/SCL ratio from 8% to 23% (_P_ < 0.01), suggesting that prolongation of atrial CL by cNav blockade was primarily due to
depression of SAN conduction rather than automaticity. Nav blockade also prolonged intranodal conduction (measured from the SAN leading pacemaker to the SAN border) by 31% and 47% (_P_ <
0.05, _t_-test) at TTX 100 nM and 1–3 µM, respectively (Fig. 2c, d). Decremental SAN conduction (Fig. 2c) gradually increased SACTsr and eventually caused exit block (5/7 hearts) and
complete atrial arrest (3/7 hearts) at TTX 1–3 µM. We also observed significantly higher beat-to-beat variations of SCL and SACT during TTX 1–3 µM than at control conditions or TTX 100 nM
(Fig. 2e and Supplementary Fig. 2). To eliminate the possibility of natural drift of tissue properties affecting our results during the experimental mapping period of 2 h, we verified in all
studied human SAN experiments as well as in previous experiments3 that the drift at baseline conditions in SCL was ~5% per hour, whereas drift was not observed in atrial conduction velocity
(CV), which suggests that changes in automaticity and conduction seen with low- and high-dose TTX, respectively, were significant and not due to time-dependent changes in tissue properties
(Supplementary Fig. 3). PROTECTIVE ROLES OF NAVS DURING STRESS IN THE HUMAN SAN We next investigated the contribution of Nav in protecting SAN conduction during metabolic challenge with
adenosine bolus or overdrive suppression by fast atrial pacing, which are clinically employed tests to unmask SND25,26. Adenosine bolus was injected via the coronary artery in eight SAN
preparations, which caused temporary hyperpolarization of the cardiomyocytes. At control conditions, adenosine bolus increased the maximum SCL by 30% ± 20%, whereas after 100 nM TTX, the
same dose of adenosine prolonged SCL by 72 ± 54% (_n_ = 8, _P_ < 0.05, _t_-test) (Fig. 3). Moreover, SAN exit block was observed in five of eight hearts during adenosine bolus at 100 nM
TTX, which did not occur at control conditions (Fig. 3). Based on our molecular data that showed transcripts of nNav1.6 isoform to be higher in the SAN compared with other nNav isoforms (see
below), we studied its unique role in SAN pacemaking and conduction using a specific nNav1.6 blocker 30 nM 4,9-Anhydrotetrodotoxin27 in five human SAN experiments (Fig. 3c and Supplementary
Fig. 4). These experiments revealed that selective blockade of nNav1.6 channels did not affect SCL but could depress SAN conduction and induced SAN exit block in three out of five
experiments, during adenosine bolus and overdrive suppression; these results were comparable to the effects of 100 nM TTX (Fig. 3c). Our findings suggest that nNav1.6 blockade could be one
of the contributors of the 100 nM TTX inhibitory effect on SAN conduction, thereby emphasizing its role as an important functional nNav isoform for human SAN conduction. We also evaluated
the effect of TTX and nNav1.6 blocker on atrial conduction during pacing at 500 ms; nNav1.6 did not have a significant effect on both longitudinal and transversal CV but TTX depressed
longitudinal atrial CV by 6% and 30% (_P_ < 0.01, _t_-test) at 100 nM and 1–3 µM concentrations, respectively (Table 1 and Supplementary Fig. 5). At control conditions, atrial pacing at
500 ms did not unmask SND (cSNRTi > 525 ms) in any of the hearts tested (_n_ = 14). After selectively blocking nNav with TTX 100 nM, the same rate of atrial pacing increased cSNRTi by 236
± 234% (_n_ = 7, _P_ = 0.176, Wilcoxon’s test) and unmasked SND in two hearts (HF 421856 and non-failing 642519; Supplementary Table 1) with cSNRTi > 1400 ms due to exit block (Fig. 3;
Table 1). Faster, 300 ms atrial pacing caused exit block in 3/15 experiments at control conditions, in 2/5 experiments with nNav1.6 blocker, in 3/7 experiments at TTX 100 nM, and in 5/6
hearts at TTX 1–3 µM. Figure 4 shows an example of conduction failure/arrhythmias, resulting from a perfect storm of stochastic SAN exit block, competing intranodal pacemakers, SAN micro-
and macro-reentry, seen in two hearts (non-failing 118258 with history of chronic HTN/chronic alcohol consumption and HF 930597; Supplementary Table 1). In these hearts, preferential slowing
of SAN conduction by Nav blockade, led to intranodal unidirectional blocks and initiated intranodal micro-reentry or macro-reentry (Supplementary Movies 1 and 2). The micro-reentry pivot
waves anchored to the longitudinal block region can produce both tachycardia and paradoxical bradycardia (due to exit block), despite an atrial activation pattern and ECG morphology
identical to regular sinus rhythm28. Intranodal longitudinal conduction blocks usually coincided with interstitial fibrosis strands and SAN artery as in the case of heart 118258 as shown by
histology in Fig. 4c. SAN reentrant arrhythmias were not observed at control conditions or with TTX 100 nM in any hearts, except in one (957855) experiment during nNa1.6 blockade. MRNA AND
PROTEIN PROFILES OF NAV IN HUMAN SAN AND ATRIA We next studied the relative mRNA and protein expression levels of Nav in human SAN tissues, compared with neighboring atria (Supplementary
Table 3). The distribution of Nav isoforms, including nine α-subunits, and four β-subunits, were analyzed in human SAN and surrounding atrial tissues by quantitative PCR (qPCR). With the
exception of _SCN10A_ (Nav1.8), which was detected only at low levels in some of the human RA (see Source Data file), all other isoforms were detected in human SAN as well as in RA at the
mRNA level (Fig. 5). _SCN5A_ (Nav1.5) and _SCN1B_ (Navβ1) were the most abundantly transcribed Nav channel α- and β-subunits in both human SAN and RA, with higher levels in RA than in SAN.
In contrast, mRNA transcripts of neuronal subunits _SCN1A_ (Nav1.1), _SCN2A_ (Nav1.2), _SCN8A_ (Nav1.6), and _SCN9A_ (Nav1.7) were higher in the human SAN than RA. In addition, we also
quantified the transcript levels of other major proteins involved in human SAN pacemaking and conduction including _HCN1_, _HCN4_, _GJA1_ (Cx43), _GJA5_ (Cx40), and calcium channels
_CACNA1C_ (Cav1.2), _CACNA1D_ (Cav1.3), and _CACNA1G_ (Cav3.1). _CACNA1D_ transcripts were significantly higher in the SAN vs. RA; as expected, we found higher levels of both _HCN1_ and
_HCN4_, and lower levels of _GJA1_ transcripts in SAN vs. atria, which validated the purity of SAN tissue used for these studies (Supplementary Table 3). Protein distribution patterns of
cNav1.5 and nNav1.6 in human SAN and RA were also detected by immunostaining (Fig. 6a, b). Fluorescence density analysis confirmed that protein levels of cNav1.5 channels are higher in RA
than in SAN cardiomyocytes (_P_ < 0.01, pairwise test), whereas the nNav1.6 protein expression is higher in the SAN than RA (_P_ < 0.01, pairwise test). Localization of cNav1.5 and
nNav1.6 is shown to be cardiomyocyte specific (Fig. 6c, d and Supplementary Fig. 6). Figure 6e shows that the cNav1.5 protein expression pattern detected by western blotting is consistent
with both mRNA expression and immunostaining results. These results collectively confirm the presence of both nNav and cNav isoforms in the human SAN at the molecular level. In addition, to
correlate functional results with biochemical findings the expression levels of cNav1.5 and nNav1.6 were quantified in immunostained cryo-frozen tissue sections from three hearts
functionally mapped with the nNav1.6 blocker (957855, 283273, and 670263) (Supplementary Fig. 7). Our results show that in heart 957855, which developed exit blocks with nNav1.6 blocker
(Supplementary Fig. 6), the ratio of nNav1.6 between SAN and RA was the highest compared with two other hearts, which exhibited less sensitivity to nNav1.6 blocker. These data may begin to
explain the distinct functional contribution of nNav1.6 channels and correlate these findings with protein expression levels. NNAV MRNA ALTERED BY HF AND CHRONIC ALCOHOL CONSUMPTION As every
human heart studied is characterized by cardiac disease, aging, and/or comorbidities, we investigated whether mRNA profiles of any cNav and nNav are associated with specific disease and/or
accompanying comorbidities and risk factors. To determine the contribution of these multiple factors, we screened the donor (non-failing) and HF hearts to identify those conditions
(Supplementary Table 3). There were no significant differences in Nav mRNA levels associated with HTN, AF, coronary artery disease, gender, age, or illicit drug abuse in the hearts studied
(Supplementary Tables 4 and 5). However, interestingly, history of chronic alcohol consumption was strongly associated with downregulation of multiple mRNA transcripts, both in the RA and
SAN. _SCN2A_, _SCN8A_, and _SCN3B_ transcripts were significantly downregulated in the RA and SAN of donor non-failing hearts with a history of chronic alcohol consumption (5/10 hearts)
(Fig. 7a). Smoking history (5/10 hearts) did not show as strong effects as chronic alcohol consumption, with significant changes only in _SCN4B_ in RA and SAN (Supplementary Fig. 8a).
Besides chronic alcohol consumption, HF was also associated with significant downregulation of _SCN2A_, _SCN3A_, _SCN4A_, _SCN9A_, _SCN2B_, and _SCN4B_ in RA but not in the SAN
(Supplementary Table 3). Based on our finding that history of chronic alcohol consumption can independently modify several nNav transcripts, data were further analyzed excluding hearts with
history of chronic alcohol consumption, to determine the specific effect of HF on Nav. Interestingly, four α-subunits and three β-subunits were significantly downregulated in the HF RA,
including _SCN8A_ encoding the nNav1.6 isoform, which was also significantly altered in the HF SAN (Fig. 7b). A positive correlation was also found between heart weight and _SCN11A_ and
_SCN1A_ in RA and _SCN11A_ and _SCN3B_ in the SAN (Supplementary Fig. 8b). In addition, we also tested whether HF and chronic alcohol consumption are associated with differences in other
major pacemaker ion channels in SAN but found no significant correlations (Supplementary Table 3). COMPUTATIONAL MODELING OF NAV BLOCKADE IN SAN-SACP-RA MODELS We further explored the
compartment-specific role of Nav isoforms using two-dimensional (2D) human SAN-SACP-RA computer models with and without HF characteristics (Figs. 8a, 9a), which incorporated distinctive
structural features of human SAN and SACP2,3 (Fig. 1). The 2D human SAN-SACP-RA computer models generated realistic characteristics (resting potentials, upstrokes, and action potential
durations) of action potentials in SAN and RA regions, respectively (Fig. 8b). The computer model was validated by reproducing similar changes of SCL and SACT during 1–100 µM adenosine
perfusion recorded in our previous optical mapping experiments3. Furthermore, the computer simulation revealed that impairments in SAN conduction during adenosine perfusion and _I_Na
blockade were mainly due to inhibition of conduction in the SAN-SACP compartments (Fig. 8c–f), which directly correlate with functional mapping data (Figs. 1–4). Figure 8c–e represents the
space–time plot of action potential profiles recorded from cells along the mid-line across the SAN-RA, showing continuous and stable action potential initiation and conduction at control
condition, simulated 25 µM adenosine, or 20% _I_Na blockade in SAN (4% _I_Na blockade in RA). However, consistent with results from our optical mapping study, in contrast to solely adenosine
or 20% _I_Na blockade, the combination of 25 µM adenosine and 20% _I_Na blockade led to 3:2 SAN exit block in SACP (Fig. 8f), suggesting that high levels of adenosine and HF-induced
downregulation of multiple nNav in the RA could form a substrate for conduction blocks and rhythm failure. Under simulated HF conditions, 20% fibrosis was added to the SAN and SACP regions
in the 2D human SAN computer models (Fig. 9a) and the resulting action potentials generated in SAN, SACP, and RA regions are displayed in Fig. 9b, respectively. Figure 9c shows that
dose-dependent _I_Na inhibition by itself primarily slows SAN conduction and causes exit block at ~35% _I_Na blockade, with only ~10% SCL prolongation. However, the addition of even 10 µM
adenosine lowers the threshold for SAN exit block, wherein complete exit block occurs at ~20% _I_Na blockade. Conditions simulating HF further slow SAN conduction and lower the threshold for
both exit block and SAN arrest in the presence of adenosine or _I_Na blockade (Fig. 9d). In the non-failing model, the lowest safety factor (SF) was found in the junction between SACP-RA at
control conditions. Due to atrial influence, maximum diastolic potential decreased in SACP cells from −56 mV at SAN-SACP junction to −72 mV at SACP-RA junction, which significantly
increased functional _I_Na across the SACP (Supplementary Fig. 9). Adenosine 25 µM induced hyperpolarization of ~1.5 mV across SAN-SACP-RA, which further increased functional _I_Na in SACP
junction by 21% and SF by 5.8%, thereby maintaining source-sink balance necessary for conduction. Blocking _I_Na alone by 20% decreased SF by 14% in SACP junction. Under these conditions,
adding 25 µM adenosine effects to the model produced the same hyperpolarization as adenosine alone, but as _I_Na was unavailable to compensate, a 3:2 SAN exit block was induced due to
beat-to-beat decrease in SF to 1.003 during conduction and 0.904 during exit block. These results raised the possibility that the exit blocks could be caused by Nav channels in the atria
rather than in the SAN-SACP. Hence, _I_Na was blocked separately, only in the SAN-SACP or in the RA. Results showed that when Nav channels are blocked only in the SAN-SACP component in the
model, there are depressive effects on SCL, SACT, and thresholds of SAN conduction failure, which are similar to those of the control model, where Nav channels were blocked globally
(Supplementary Fig. 10a, b). However, when Nav channels are blocked (up 40%) only in the RA, there is no SAN conduction failure clarifying that the exit blocks are mainly due to Nav channel
blockade in the SAN-SACP (Supplementary Fig. 10c). The HF model had a similar pattern for SF across the SAN-SACP-RA with the lowest safety factor also found at the SACP-RA junction; however,
a more pronounced effect on safety factor resulted from adenosine 25 µM = 26%, _I_Na block 20% = 25% and adenosine 25 µM + _I_Na block 20% = 59% (a safety factor of 0.7981). Furthermore,
the computational human SAN models were able to reveal the mechanism for only exit blocks but the absence of entrance blocks during atrial pacing in functional mapping experiments when Nav
were blocked (Figs. 3, 4). Computational analyses in control and HF human SAN models revealed that safety factor in SACP junction is direction-dependent and it is higher for entrance (~1.03)
vs. exit conduction (~0.86) at the exit block conditions, which underlie the absence of entrance blocks during slow atrial pacing and _I_Na partial block (Supplementary Fig. 11).
Importantly, as we have previously shown3, the absence of entrance blocks in SACPs during atrial pacing further inhibited excitability in the SACP and promotes occurrence of post-pacing exit
block (Supplementary Fig. 11b) and SAN reentrant arrhythmias28, which can explain current experimental observations (Figs. 3, 4). These results from computer simulations not only support
our optical mapping findings, but also suggest that the importance of _I_Na in preserving SAN conduction is dose-dependently augmented by adenosine, resulting in SAN function failure due to
exit blocks in SACPs and complete SAN automaticity arrest (Fig. 9c). DISCUSSION Here we report findings from near-infrared optical mapping that not only elucidate a previously unknown role
for Nav channels29 in the human SAN but also suggest that this functional contribution of Nav may be unique to the human SAN, compared with the previously demonstrated roles of Nav in other
small30 and large16 animal models. A major finding is that unlike cNav, nNav may predominantly contribute to SAN intranodal conduction, rather than atrial conduction. On the other hand, cNav
play important roles in both SAN pacemaking and conduction, especially during adenosine or pacing-induced stress to prevent intranodal conduction failure. Furthermore, these functional
observations are supported by higher expression of nNav (Nav1.1 and 1.6) and lower expression of cNav1.5 in human SAN cardiomyocytes vs. surrounding atrial tissue. Our data also show that
several nNav transcripts were vulnerable to selective remodeling associated with HF, cardiac hypertrophy and modifying risk factors including history of smoking and chronic alcohol
consumption. Biophysics-based computer modeling revealed compartment-specific mechanistic insights that suggest a protective role for _I_Na against rhythm failure within the human SAN
pacemaker–conduction complex, especially in HF. The human SAN is protected by multiple fail-safe mechanisms3, which are critical to ensuring adequate cardiac performance as well as
preventing SND and cardiac arrhythmias during pathophysiological conditions13. Currently, high-resolution, near-infrared optical mapping is the only mapping technique that is able to
accurately identify intranodal conduction and hence capable of determining compartment-specific backup mechanisms2,3. Utilizing these high-resolution ex-vivo examinations was critical in our
goal to uncover the unique roles of nNav and cNav within the human SAN. In the current study, blockade of both cNav and nNav was observed to directly inhibit SAN automaticity (SCL) at
physiological conditions, manifesting an essential role for Nav in maintaining human SAN pacemaking. However, SACT proportionally increased more than SCL (SACT/SCL ratio) during Nav
blockade, which suggests that SAN impairments due to Nav dysfunction may predominantly be due to depressed SAN conduction rather than automaticity. Importantly, Nav blockade increased
beat-to-beat variability in SAN intranodal conduction and SAN reentrant arrhythmias (Fig. 2). These results establish a significant role for Nav channels in maintaining stable intranodal
conduction and robust protection of human SAN rhythm, in both HF and non-failing hearts. In the light of our findings, we suggest that impaired intranodal conduction could underlie
clinically observed symptoms of familial sick sinus syndrome including sinus bradycardia/arrest in some patients carrying _SCN5A_ mutations18,31. Interestingly, although previous studies in
animal models have identified a remarkable range of varying, species-specific roles for Nav isoforms in the SAN14,15,16,30, our findings demonstrate that Nav channels may contribute very
differently to human SAN pacemaking and conduction15,16,32. Studies have found that micromolar TTX can depress heart rate in adult mouse33 and rabbit SAN preparations34, providing evidence
that cNav can contribute to SAN automaticity in some adult mammalian hearts. However, in contrast to the mouse SAN study30, where nanomolar TTX impaired SAN automaticity but did not inhibit
conduction, we found that nanomolar TTX impaired intranodal conduction (~250%) with negligible effects on SAN automaticity (~6%) and atrial conduction (~6%) at physiological conditions. Our
data suggest that unlike other species, TTX-sensitive neuronal _I_Na, particularly nNav1.6 in addition to cardiac _I_Na, may particularly be important in maintaining intranodal conduction
within the human SAN. Increased levels of adenosine have been shown to cause SND during metabolic stress and HF35,36. In the human SAN, adenosine-induced inhibition of SAN pacemaking and
conduction are mainly due to activation of A1R and outward potassium current (_I_K,Ado/ACh)3, which can significantly hyperpolarize SAN pacemaker cells37. Similarly, fast pacing or atrial
arrhythmias could also increase the activation of outward K+ currents and reduce _I_Ca,L, which inhibit the excitability of the SACPs more than intranodal pacemaker compartments and lead to
post-pacing SAN exit block in diseased hearts2,38,39. Therefore, we used both adenosine and fast atrial pacing to simulate a pathological scenario to investigate the role of Nav channels in
this context. Our findings show that Nav blockade significantly exacerbated the depressive effects of both adenosine and overdrive suppression resulting in intranodal conduction failure and
SAN arrhythmia. These findings are in keeping with previous results from isolated SAN cells, suggesting that Nav could be preferentially activated when the cell is hyperpolarized16,17, which
may be important to counteract impaired SAN-SACP excitability during pathological challenges. Our data suggest that TTX-sensitive nNav could play a distinct protective role in human SAN
conduction, especially during pathological conditions including adenosine-mediated hyperpolarization, when the balance between the source (electrical current/charge generated by the SAN and
delivered through SACP) and sink (the current/charge required to activate the neighboring atrial myocardium) might be compromised (Figs. 3, 4). Although direct experimental data confirming
SACP-specific effects of _I_Na are lacking for the human SAN, our human compartment-specific SAN-SACP-RA computer model indeed demonstrates decreased maximal diastolic potential and
increased _I_Na activation across the SACP toward the RA. This unidirectional increase in _I_Na activation within the SACP could help maintain the source-sink balance. Computer simulations
further demonstrate that adenosine-induced hyperpolarization and _I_Na blockade can produce a synergistic negative effect, causing complete SAN exit block due to conduction failure in the
SACP (Fig. 9 and Supplementary Figs. 10 and 11). Specific combinations of adenosine and _I_Na blockade reproduced the beat-to-beat conduction variability and intermittent SAN exit block
observed in our optical mapping experiments. Recent studies have shown that in addition to cNav, multiple nNav are expressed in the human heart40; however, their presence in the human SAN
has never been investigated. Nav channels are composed of one pore-forming α-subunit associated with two different β-subunits41 and can be categorized as TTX-resistant (IC50 > 1 µM)
cardiac isoforms and TTX-sensitive (IC50 < 60 nM) neuronal channels42. In this study, mRNA transcripts of both nNav and cNav isoforms were detected, with varying distributions between the
SAN and RA. In the SAN, among the TTX-resistant Navs (1.5, 1.8, and 1.9), only cNav1.5 was found at significant levels, indicating that the observed additional functional effects during
high-dose TTX could be mediated mainly by cNav1.5. Importantly, the higher protein expression of nNav1.6 compared with cNav1.5 channels in SAN vs. RA may also explain the higher sensitivity
of SAN to nanomolar TTX compared to surrounding atrial tissue. In fact, the specific nNav1.6 blocker depressed SAN function similar to the effects of 100 nM suggesting that the effect of TTX
100 nM in the human SAN studied may be partially mediated by nNav1.6; furthermore, higher ratio of nNav1.6 between SAN and RA was found in the heart that developed exit block with nNav1.6
blocker, compared with two other hearts that exhibited less sensitivity to nNav1.6 blocker. These results from the limited number of human hearts studied suggest that nNav1.6 plays an
important role in preserving human SAN conduction potentially by preventing exit blocks. However, expression of other nNav isoforms and accessory/interacting proteins should also be
investigated in future studies, to determine their mechanistic roles in SAN function. Previous studies have shown that many cardiac diseases can cause remodeling of major ion
channels10,11,12,13 including HCN1 and HCN4, A1R, and L-type calcium channels. Indeed, we found significantly lower mRNA levels of almost all nNav isoforms in HF hearts, especially nNav1.6
in the SAN (Fig. 7). Interestingly, our data from the limited number of hearts studied show that nNav transcript levels in the human SAN and RA are also associated with other modifying risk
factors including chronic alcohol consumption. As chronic alcohol consumption has been strongly associated with arrhythmias including ventricular tachycardia and AF43, our data indicate that
Nav channels must be studied in larger samples of hearts from chronic alcohol abuse patients to potentially reveal Nav-mediated arrhythmic mechanisms associated with chronic alcohol
consumption. Interestingly, as shown in Fig. 4, our optical mapping data revealed that subsequent to nNav and partial cNav blockade, hearts with history of chronic alcohol consumption or HF
were highly susceptible to SAN intranodal conduction disturbances leading to atrial beat-to-beat variability, SAN reentrant arrhythmias, and sinus arrest. From a clinical standpoint, as
adenosine’s effects on the SAN are similar to those of vagal stimulation37, our findings emphasize avoiding/limiting the use of drugs that may block TTX-sensitive _I_Na, especially when
vagal tone is high, or in HF36 and AF44 patients with high plasma levels of adenosine. Our results also identify the human SAN as a direct target for several clinical medications designed to
block Nav channels, including Class I antiarrhythmic drugs that block Nav1.5, some anesthetics, and pain medications that block nNav. In fact, flecainide, a clinically prescribed Class I
antiarrhythmic drug, has been reported to increase cSNRTi in SND patients20 and to significantly reduce heart rate during exercise in patients with normal cardiac structure and SAN
function45. The availability of Nav channels in the human SAN and RA could be an important backup mechanism to robustly protect SAN conduction and pacemaking, and prevent exit blocks and
source-sink mismatch, in the context of multiple disease-induced conduction impairments3,46. Similar to other human studies, our data are also limited by the small number of ex-vivo human
atrial preparations with varied pre-existing disease etiologies, which may not be representative of all human atria. As the data are from HF patients that have been treated long-term with
several medications, disease-independent drug-induced effects could also have affected Nav transcript levels. Few HF hearts were available for optical mapping studies due to bicaval surgical
dissection, which can impair SAN coronary artery perfusion. We were unable to confirm protein expression of all Nav isoforms transcripts detected in the mRNA studies due to the lack of
specific antibodies suitable for human tissues. Furthermore, due low sample numbers we could not directly certify the correlation between Nav-related SAN pacemaking/conduction dysfunction
and HF or alcohol consumption in optical mapping experiments. Additional studies should record _I_Na from isolated human SAN cardiomyocytes, in order to differentiate the specific
contributions of cNav and nNav to cellular activation, relative to other ion currents including _I_f and _I_Ca. These findings provide insights to clarify the role of nNav and cNav isoforms
in maintaining the robustness of the human SAN and establish Nav channels as important players essential to protect intranodal conduction and prevent rhythm failure. METHODS PATIENT GROUPS
INCLUDED IN THE STUDY All human heart tissue research were approved by The Ohio State University Institutional Review Board and in compliance with all relevant ethical regulations. Informed
consent for tissue collection was obtained from transplant patients and families of donors. Human hearts used in this study were de-identified and labeled with 6 digit random codes for
reference. Hearts with intact SAN pacemaker complexes from transplant patients (with left ventricular hypertrophy, ischemic and non-ischemic HF, AF, and comorbidities including chronic HTN
and diabetes) and human donor hearts (without history of HF and AF but with comorbidities including HTN, diabetes, and modifying risk factors including history of smoking, chronic alcohol
consumption/abuse (_n_ = 34, 19-68 y/o, details in Supplementary Tables 1 and 2) were obtained from The Ohio State University Cardiac Transplant Team or LifeLine of Ohio Organ Procurement
Organization. Chronic Alcohol Consumption is defined as either abuse ( > 7 drinks/week for women and > 14 drinks/week for men), as reported in documented medical records, or chronic
consumption of moderate drinking ( > 2 drinks/week for at least 10 years). Alcohol consumption was first collected from medical records when available and then from retrospective
interviews with family members by LifeLine of Ohio Organ Procurement Organization for rejected donor hearts and drug abuse as listed in the patient’s electronic health record. Expanded
Materials and Methods are provided in the Supplementary Material. NEAR-INFRARED OPTICAL MAPPING AND DATA ANALYSIS Conventional clinical electrode-based mapping systems are unable to record
intramural conduction and activation patterns from within the 3D human SAN in patients, yet we were able to overcome these limitations by utilizing our recently validated high-resolution
near-infrared optical mapping3. Human SAN preparations (_n_ = 14) were coronary-perfused and stained with near-infrared dye di-4-ANBDQBS3. Regions of poor coronary perfusion/ischemia were
excluded. To distinguish the function of different classes of Nav in the human SAN, two different doses of TTX (Abcam), 100 nM and 1–3 μM, were sequentially perfused through the coronary
arteries. Lower dose, 100 nM TTX, selectively blocks majority of TTX-sensitive nNav subtypes (such as Nav1.1–1.3 and Nav1.6–1.7 with IC50 ~10 nM), but not TTX-resistant cNav1.5 with IC50 ≥ 1
μM42. As the majority of nNav were already blocked, any further effects at the higher dose, TTX 1–3 μM, was attributed to blockade of TTX-resistant cNav1.5. Optical mapping was conducted
with MiCAM Ultima-L CMOS cameras with resolution up to 330 µm2 (100 × 100 pixels)3. Optical mapping identified the leading pacemaker or earliest SAN depolarization, as well as earliest
atrial activation sites, where activation exited to the atria through SACPs. SACT reports the time of activation propagation from the SAN leading pacemaker to the earliest atrial activation
site. SAN activation patterns, SCL, SACTsr, or after atrial pacing (SACTppb), and corrected direct/indirect sinus node recovery time (cSNRT_d/i_) were measured during constant perfusion of
Tyrode’s solution at control (_n_ = 10), and sequential perfusion of 100 nM (_n_ = 8) and 1–3 μM (_n_ = 7) TTX. Overdrive atrial pacing (CL = 500 ms, 400 ms, and 300 ms) and adenosine bolus
(1 mL, 10–100 μM) were used to challenge the robustness of SAN pacemaking and conduction. RA CV was measured by RA pacing at 500 ms. In addition, a specific Nav1.6 blocker
4,9-Anhydrotetrodotoxin (30 nM)27 was used in a subset of four hearts studied with the same protocols as for TTX above. Nav1.6 blocker was tested in five experiments with Heart 957855 tested
before and after washout and addition of 1 nM isoproterenol (Supplementary Fig. 1 and Supplementary Table 1). When several drug protocols were studied, the first drug studied was washed out
from SAN preparations and isoproterenol 1–10 nM may have been used to recover sinus rhythm to its baseline levels before the effect of Nav blockade was studied (Supplementary Tables 6 and
7). MOLECULAR MAPPING AND DATA ANALYSIS To molecularly map mRNA, protein expression, and distribution of nNav and cNav, pure SAN and surrounding atrial tissues were collected from 20
unmapped human hearts (non-failing = 10; failing = 10) guided by immunostaining and Masson’s trichrome stain3,4 (Supplementary Fig. 12). Total RNA and proteins were extracted separately from
the SAN and surrounding atrial tissue4,10. qPCR was conducted in duplicates with QuantStudio 3 (Applied Biosystems), SYBR green (Qiagen), and QuantiTect primer assays (Qiagen; Supplementary
Table 8). Comparative threshold cycle (Ct) was used to compare the relative abundance of mRNAs in the samples. Primary antibodies against Nav1.5 (custom-made in Dr Mohler’s lab), Nav1.6
(Alomone), Connexin-43(Cx43), Glyceraldehyde 3-phosphate dehydrogenase, and α-actinin (Sigma-Aldrich) were used to quantify corresponding proteins by western blotting and immunostaining3,4
(Supplementary Table 9). Uncropped versions of blots have been provided in Source Data file. HUMAN COMPARTMENT-SPECIFIC SAN-SACP-RA COMPUTATIONAL MODEL A biophysics-based 2D computational
model was designed based on our study of human SAN structure2 to simulate the interactions between SAN, SACP, and RA. The human SAN and SACP were modeled with the Fabbri et al. model47,
based on isolated human SAN pacemaker cell recordings48, and adapted with optical mapping data from central SAN and SACP regions, respectively (Supplementary Table 10). The RA cells were
modeled by using the original human atrial Courtemanche et al. cell model49. The ratio of _I_Na blockage in SAN vs. RA was set to 5:1. To incorporate the effects of adenosine into the
cellular models, we utilized the acetylcholine (ACh)-activated K+ current, _I_K,ACh/Ado, previously used for the atria49 and SAN models47, equating ACh concentrations with those of adenosine
based on our optical experiments. In HF SAN model (Fig. 9a), 20% reduction in _I_f current and heterogeneously seeded fibrosis with a size of 2 × 2 within SACP regions with a ratio of 1:4
to the normal SAN/SACP cells were implemented, as well as 20% and 5% reduction in the _I_Na current in SAN-SACP and atrial cells, respectively. Conduction among SAN/atrial cells was modeled
using a mono-domain equation and solved using a paralleled finite difference approach50. We used a spatial step of 0.03 mm and temporal step of 0.0025 ms in our solver. A forward Euler
method50 was used to solve the ordinary differential equations of cellular models. The conduction in the 2D tissue model attributable to intercellular electric coupling via gap junctions was
modeled through the diffusion coefficient. In this model, we considered the regional differences in gap junctional coupling between the SAN, SACP, and RA tissues by setting the diffusion
coefficient ratio to 2:14:100 in these three regions. The SF51 was calculated to measure the success of propagation at each cell and is defined as the ratio of the total charge produced to
the total charge consumed at that cell. If the ratio is less than 1, inefficient charge is produced for downstream activation and propagation will fail. STATISTICAL ANALYSIS Data are
presented as mean ± SD. All the statistical analyses were done in R 3.4.4 using packages lme4 and emmeans. The mixed models included Heart and Category (control, TTX 100 nM and TTX 1–3 μM)
as predictors. Heart was treated as random effect and Category as fixed effect. Pairwise tests between condition levels were adjusted using Tukey’s method. Quality of fit was monitored by
visual inspection of residuals. Analysis of variance model was used to compare Categories. Analysis of the ratios for TTX 100 nM and TTX 1–3 μM categories compared with control was done
using two-tailed _t_-test. Normality assumption was verified using Shapiro–Wilk test. Non-parametric data were analyzed with two-sided Wilcoxon’s test. Analysis of exit block events was done
using two-sided proportion test with Yates continuity correction. Association analysis of Nav isoform transcription level with diseases was done using mixed models in package lme4 with
heart/patient, heart weight, and indicator variables for age, gender, smoking, chronic alcohol consumption, HF, AF, or HTN. Heart/patient was considered a random effect, others as fixed
effects. _P_-values < 0.05 were considered significant. REPORTING SUMMARY Further information on research design is available in the Nature Research Reporting Summary linked to this
article. DATA AVAILABILITY Raw source data underlying all reported averages in graphs and charts, and uncropped versions of blots presented in the figures are available as a Source Data
file. The source data underlying Figs. 1c, 2d, 3c, 5, 6 immunostaining, 6 western blotting, Table 1, and Supplementary Figs. 5, 7 are found in the source data file. All data are presented
within the manuscript, the online data supplemental file, or are available upon reasonable request to the corresponding author. CODE AVAILABILITY The ionic model utilized47 is freely
available from the repository CellML (https://www.cellml.org/). Link to computer model source code, the test and readme files are provided here:
https://github.com/Charcol97/Fabbri_HumanSAN_OSU REFERENCES * Fedorov, V. V., Glukhov, A. V. & Chang, R. Conduction barriers and pathways of the sinoatrial pacemaker complex: their role
in normal rhythm and atrial arrhythmias. _Am. J. Physiol. Heart Circ. Physiol._ 302, H1773–H1783 (2012). Article CAS PubMed Google Scholar * Csepe, T. A. et al. Human sinoatrial node
structure: 3D microanatomy of sinoatrial conduction pathways. _Prog. Biophys. Mol. Biol._ 120, 164–178 (2016). Article PubMed Google Scholar * Li, N. et al. Redundant and diverse
intranodal pacemakers and conduction pathways protect the human sinoatrial node from failure. _Sci. Transl Med_. 9, eaam5607 (2017). * Li, N. et al. Molecular mapping of sinoatrial node HCN
channel expression in the human heart. _Circ. Arrhythm. Electrophysiol._ 8, 1219–1227 (2015). Article CAS PubMed PubMed Central Google Scholar * Dobrzynski, H., Boyett, M. R. &
Anderson, R. H. New insights into pacemaker activity: promoting understanding of sick sinus syndrome. _Circulation_ 115, 1921–1932 (2007). Article PubMed Google Scholar * Mangrum, J. M.
& DiMarco, J. P. The evaluation and management of bradycardia. _N. Engl. J. Med._ 342, 703–709 (2000). Article CAS PubMed Google Scholar * John, R. M. & Kumar, S. Sinus node and
atrial arrhythmias. _Circulation_ 133, 1892–1900 (2016). Article PubMed Google Scholar * Alonso, A. et al. Association of sick sinus syndrome with incident cardiovascular disease and
mortality: the Atherosclerosis Risk in Communities study and Cardiovascular Health Study. _PLoS ONE_ 9, e109662 (2014). Article ADS PubMed PubMed Central CAS Google Scholar * Epstein,
A. E. et al. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of
Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. _Circulation_ 127, e283–e352 (2013). Article PubMed Google Scholar *
Chandler, N. J. et al. Molecular architecture of the human sinus node: insights into the function of the cardiac pacemaker. _Circulation_ 119, 1562–1575 (2009). Article PubMed Google
Scholar * Lou, Q. et al. Upregulation of adenosine A1 receptors facilitates sinoatrial node dysfunction in chronic canine heart failure by exacerbating nodal conduction abnormalities
revealed by novel dual-sided intramural optical mapping. _Circulation_ 130, 315–324 (2014). Article CAS PubMed PubMed Central Google Scholar * Zicha, S., Fernandez-Velasco, M., Lonardo,
G., L’Heureux, N. & Nattel, S. Sinus node dysfunction and hyperpolarization-activated (HCN) channel subunit remodeling in a canine heart failure model. _Cardiovasc Res._ 66, 472–481
(2005). Article CAS PubMed Google Scholar * Monfredi, O. & Boyett, M. R. Sick sinus syndrome and atrial fibrillation in older persons - A view from the sinoatrial nodal myocyte. _J.
Mol. Cell Cardiol._ 83, 88–100 (2015). Article CAS PubMed Google Scholar * Baruscotti, M., Westenbroek, R., Catterall, W. A., DiFrancesco, D. & Robinson, R. B. The newborn rabbit
sino-atrial node expresses a neuronal type I-like Na+ channel. _J. Physiol._ 498, 641–648 (1997). Article CAS PubMed PubMed Central Google Scholar * Maier, S. K. et al. An unexpected
requirement for brain-type sodium channels for control of heart rate in the mouse sinoatrial node. _Proc. Natl Acad. Sci. USA_ 100, 3507–3512 (2003). Article ADS CAS PubMed PubMed
Central Google Scholar * Protas, L., Oren, R. V., Clancy, C. E. & Robinson, R. B. Age-dependent changes in Na current magnitude and TTX-sensitivity in the canine sinoatrial node. _J.
Mol. Cell Cardiol._ 48, 172–180 (2010). Article CAS PubMed Google Scholar * Verkerk, A. O., Wilders, R., van Borren, M. M. & Tan, H. L. Is sodium current present in human sinoatrial
node cells? _Int. J. Biol. Sci._ 5, 201–204 (2009). Article CAS PubMed PubMed Central Google Scholar * Benson, D. W. et al. Congenital sick sinus syndrome caused by recessive mutations
in the cardiac sodium channel gene (SCN5A). _J. Clin. Invest._ 112, 1019–1028 (2003). Article CAS PubMed PubMed Central Google Scholar * LaBarre, A. et al. Electrophysiologic effects of
disopyramide phosphate on sinus node function in patients with sinus node dysfunction. _Circulation_ 59, 226–235 (1979). Article CAS PubMed Google Scholar * Vik-Mo, H., Ohm, O. J. &
Lund-Johansen, P. Electrophysiologic effects of flecainide acetate in patients with sinus nodal dysfunction. _Am. J. Cardiol._ 50, 1090–1094 (1982). Article CAS PubMed Google Scholar *
Kim, K. O., Chung, S., Lee, K. & Cho, H. Profound bradycardia with lidocaine during anesthesia induction in a silent sick sinus syndrome patient. _J. Clin. Anesth._ 23, 227–230 (2011).
Article PubMed Google Scholar * Jose, A. D. & Taylor, R. R. Autonomic blockade by propranolol and atropine to study intrinsic myocardial function in man. _J. Clin. Invest._ 48,
2019–2031 (1969). Article CAS PubMed PubMed Central Google Scholar * Rodeheffer, R. J. et al. Exercise cardiac output is maintained with advancing age in healthy human subjects: cardiac
dilatation and increased stroke volume compensate for a diminished heart rate. _Circulation_ 69, 203–213 (1984). Article CAS PubMed Google Scholar * Jose, A. D. & Collison, D. The
normal range and determinants of the intrinsic heart rate in man. _Cardiovasc. Res._ 4, 160–167 (1970). Article CAS PubMed Google Scholar * Gomes, J. A., Kang, P. S. & El Sherif, N.
The sinus node electrogram in patients with and without sick sinus syndrome: techniques and correlation between directly measured and indirectly estimated sinoatrial conduction time.
_Circulation_ 66, 864–873 (1982). Article CAS PubMed Google Scholar * Fragakis, N. et al. The value of adenosine test in the diagnosis of sick sinus syndrome: susceptibility of sinus and
atrioventricular node to adenosine in patients with sick sinus syndrome and unexplained syncope. _Europace_ 9, 559–562 (2007). Article PubMed Google Scholar * Rosker, C. et al. The TTX
metabolite 4,9-anhydro-TTX is a highly specific blocker of the Na(v1.6) voltage-dependent sodium channel. _Am. J. Physiol. Cell Physiol._ 293, C783–C789 (2007). Article CAS PubMed Google
Scholar * Glukhov, A. V. et al. Sinoatrial node reentry in a canine chronic left ventricular infarct model: role of intranodal fibrosis and heterogeneity of refractoriness. _Circ. Arrhythm.
Electrophysiol._ 6, 984–994 (2013). Article PubMed PubMed Central Google Scholar * Lei, M., Zhang, H., Grace, A. A. & Huang, C. L. SCN5A and sinoatrial node pacemaker function.
_Cardiovasc. Res._ 74, 356–365 (2007). Article CAS PubMed Google Scholar * Lei, M. et al. Requirement of neuronal- and cardiac-type sodium channels for murine sinoatrial node pacemaking.
_J. Physiol._ 559, 835–848 (2004). Article ADS CAS PubMed PubMed Central Google Scholar * Milanesi, R., Bucchi, A. & Baruscotti, M. The genetic basis for inherited forms of
sinoatrial dysfunction and atrioventricular node dysfunction. _J. Interv. Card. Electrophysiol._ 43, 121–134 (2015). Article PubMed PubMed Central Google Scholar * Baruscotti, M.,
DiFrancesco, D. & Robinson, R. B. A TTX-sensitive inward sodium current contributes to spontaneous activity in newborn rabbit sino-atrial node cells. _J. Physiol._ 492, 21–30 (1996).
Article CAS PubMed PubMed Central Google Scholar * Lei, M. et al. Sinus node dysfunction following targeted disruption of the murine cardiac sodium channel gene Scn5a. _J. Physiol._
567, 387–400 (2005). Article CAS PubMed PubMed Central Google Scholar * Chang, S. L. et al. Heart failure modulates electropharmacological characteristics of sinoatrial nodes. _Exp.
Ther. Med._ 13, 771–779 (2017). Article CAS PubMed Google Scholar * Watt, A. H. Sick sinus syndrome: an adenosine-mediated disease. _Lancet_ 325, 786–788 (1985). Article Google Scholar
* Funaya, H. et al. Plasma adenosine levels increase in patients with chronic heart failure. _Circulation_ 95, 1363–1365 (1997). Article CAS PubMed Google Scholar * Lerman, B. B. &
Belardinelli, L. Cardiac electrophysiology of adenosine. Basic and clinical concepts. _Circulation_ 83, 1499–1509 (1991). Article CAS PubMed Google Scholar * Lou, Q. et al. Tachy-brady
arrhythmias: the critical role of adenosine-induced sino-atrial conduction block in post-tachycardia pauses. _Heart Rhythm_ 10, 110–118 (2013). Article PubMed Google Scholar * Watanabe,
E. I., Honjo, H., Boyett, M. R., Kodama, I. & Toyama, J. Inactivation of the calcium current is involved in overdrive suppression of rabbit sinoatrial node cells. _Am. J. Physiol._ 271,
H2097–H2107 (1996). CAS PubMed Google Scholar * Kaufmann, S. G. et al. Distribution and function of sodium channel subtypes in human atrial myocardium. _J. Mol. Cell Cardiol._ 61, 133–141
(2013). Article CAS PubMed PubMed Central Google Scholar * O’Malley, H. A. & Isom, L. L. Sodium channel beta subunits: emerging targets in channelopathies. _Annu. Rev. Physiol._
77, 481–504 (2015). Article PubMed PubMed Central CAS Google Scholar * Zimmer, T., Haufe, V. & Blechschmidt, S. Voltage-gated sodium channels in the mammalian heart. _Glob. Cardiol.
Sci. Pract._ 2014, 449–463 (2014). PubMed PubMed Central Google Scholar * Balbao, C. E., de Paola, A. A. & Fenelon, G. Effects of alcohol on atrial fibrillation: myths and truths.
_Ther. Adv. Cardiovasc. Dis._ 3, 53–63 (2009). Article PubMed Google Scholar * Maille, B. et al. Adenosine plasma level in patients with paroxysmal or persistent atrial fibrillation and
normal heart during ablation procedure and/or cardioversion. _Purinergic. Signal_. 15, 45–52 (2018). Article PubMed PubMed Central CAS Google Scholar * Wang, J. A., Lau, C. P., Tai, Y.
T. & Wu, B. Z. Effects of flecainide on exercise hemodynamics and electrocardiography in patients without structural heart disease. _Clin. Cardiol._ 18, 140–144 (1995). Article CAS
PubMed Google Scholar * Csepe, T. A., Kalyanasundaram, A., Hansen, B. J., Zhao, J. & Fedorov, V. V. Fibrosis: a structural modulator of sinoatrial node physiology and dysfunction.
_Front. Physiol._ 6, 37 (2015). Article PubMed PubMed Central Google Scholar * Fabbri, A., Fantini, M., Wilders, R. & Severi, S. Computational analysis of the human sinus node action
potential: model development and effects of mutations. _J. Physiol._ 595, 2365–2396 (2017). Article CAS PubMed PubMed Central Google Scholar * Verkerk, A. O. et al. Single cells
isolated from human sinoatrial node: action potentials and numerical reconstruction of pacemaker current. _Conf. Proc. IEEE Eng. Med. Biol. Soc._ 2007, 904–907 (2007). Google Scholar *
Courtemanche, M., Ramirez, R. J. & Nattel, S. Ionic mechanisms underlying human atrial action potential properties: insights from a mathematical model. _Am. J. Physiol._ 275, H301–H321
(1998). CAS PubMed Google Scholar * Zhao, J. et al. Three-dimensional integrated functional, structural, and computational mapping to define the structural “Fingerprints” of
heart-specific atrial fibrillation drivers in human heart ex vivo. _J. Am. Heart Assoc_. 6, e005922 (2017). * Shaw, R. M. & Rudy, Y. Ionic mechanisms of propagation in cardiac tissue.
Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. _Circ. Res._ 81, 727–741 (1997). Article CAS PubMed Google Scholar
Download references ACKNOWLEDGEMENTS This work was supported by NIH HL115580 and HL135109, American Heart Association Grant in Aid #16GRNT31010036 (V.V.F.), NIH T32HL134616, NIH F30HL142179
(B.J.H.), HL114940 (B.J.B.), and by funding from the Dorothy M. Davis Heart and Lung Research Institute. We thank the Lifeline of Ohio Organ Procurement Organization and the Division of
Cardiac Surgery at The OSU Wexner Medical Center for providing the explanted hearts. We thank Mr Farbod Fazlollahi, Ms Salome Kiduko, and Ms Kyra Peczkowski for their help with tissue
processing. AUTHOR INFORMATION Author notes * These authors contributed equally: Ning Li, Anuradha Kalyanasundaram, Brian J. Hansen AUTHORS AND AFFILIATIONS * Department of Physiology and
Cell Biology, The Ohio State University Wexner Medical Center, Columbus, OH, USA Ning Li, Anuradha Kalyanasundaram, Brian J. Hansen, Esthela J. Artiga, Suhaib H. Abudulwahed, Katelynn M.
Helfrich, Galina Rozenberg, Pei-Jung Wu, Stanislav Zakharkin, Sandor Gyorke, Paul ML. Janssen, Brandon J. Biesiadecki, Federica Accornero, Peter J. Mohler & Vadim V. Fedorov * Bob and
Corrine Frick Center for Heart Failure and Arrhythmia, Davis Heart and Lung Research Institute, The Ohio State University Wexner Medical Center, Columbus, OH, USA Ning Li, Anuradha
Kalyanasundaram, Brian J. Hansen, Esthela J. Artiga, Suhaib H. Abudulwahed, Katelynn M. Helfrich, Galina Rozenberg, Pei-Jung Wu, Sandor Gyorke, Paul ML. Janssen, Bryan A. Whitson, Nahush A.
Mokadam, Brandon J. Biesiadecki, Federica Accornero, John D. Hummel, Peter J. Mohler & Vadim V. Fedorov * Auckland Bioengineering Institute, The University of Auckland, Auckland, New
Zealand Roshan Sharma & Jichao Zhao * Department of Surgery, The Ohio State University Wexner Medical Center, Columbus, OH, USA Bryan A. Whitson & Nahush A. Mokadam * Department of
Internal Medicine, The Ohio State University Wexner Medical Center, Columbus, OH, USA John D. Hummel * Division of Cardiovascular Sciences, The University of Manchester, Manchester, UK
Halina Dobrzynski * Department of Anatomy, Jagiellonian University Medical College, Cracow, Poland Halina Dobrzynski Authors * Ning Li View author publications You can also search for this
author inPubMed Google Scholar * Anuradha Kalyanasundaram View author publications You can also search for this author inPubMed Google Scholar * Brian J. Hansen View author publications You
can also search for this author inPubMed Google Scholar * Esthela J. Artiga View author publications You can also search for this author inPubMed Google Scholar * Roshan Sharma View author
publications You can also search for this author inPubMed Google Scholar * Suhaib H. Abudulwahed View author publications You can also search for this author inPubMed Google Scholar *
Katelynn M. Helfrich View author publications You can also search for this author inPubMed Google Scholar * Galina Rozenberg View author publications You can also search for this author
inPubMed Google Scholar * Pei-Jung Wu View author publications You can also search for this author inPubMed Google Scholar * Stanislav Zakharkin View author publications You can also search
for this author inPubMed Google Scholar * Sandor Gyorke View author publications You can also search for this author inPubMed Google Scholar * Paul ML. Janssen View author publications You
can also search for this author inPubMed Google Scholar * Bryan A. Whitson View author publications You can also search for this author inPubMed Google Scholar * Nahush A. Mokadam View
author publications You can also search for this author inPubMed Google Scholar * Brandon J. Biesiadecki View author publications You can also search for this author inPubMed Google Scholar
* Federica Accornero View author publications You can also search for this author inPubMed Google Scholar * John D. Hummel View author publications You can also search for this author
inPubMed Google Scholar * Peter J. Mohler View author publications You can also search for this author inPubMed Google Scholar * Halina Dobrzynski View author publications You can also
search for this author inPubMed Google Scholar * Jichao Zhao View author publications You can also search for this author inPubMed Google Scholar * Vadim V. Fedorov View author publications
You can also search for this author inPubMed Google Scholar CONTRIBUTIONS This project was designed by V.V.F., with the methodology developed by N.L., B.J.H., A.K., and V.V.F. The
investigations were performed by N.L., E.J.A., B.J.H., S.H.A., K.M.H., G.R., B.W., N.A.M., and V.V.F. Simulation was performed by R.S. and J.Z. Data analysis and statistic were performed by
N.L., S.H.A., E.J.A., P.J.W., and S.Z. Supervision of the project was provided by S.G., P.M.L.J., B.J.B., F.A., P.J.M., J.D.H., H.D., and V.V.F. The original draft was written by N.L., A.K.,
and V.V.F., and revised by N.L., A.K., E.J.A., B.J.H., K.M.H., and V.V.F. CORRESPONDING AUTHOR Correspondence to Vadim V. Fedorov. ETHICS DECLARATIONS COMPETING INTERESTS The authors
declare no competing interests ADDITIONAL INFORMATION PEER REVIEW INFORMATION _Nature Communications_ thanks Gwilym Morris, and other anonymous reviewer(s) for their contribution to the peer
review of this work. Peer reviewer reports are available. PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY MOVIE 1 SUPPLEMENTARY MOVIE 2 REPORTING SUMMARY
SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation,
distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and
indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to
the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will
need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE
CITE THIS ARTICLE Li, N., Kalyanasundaram, A., Hansen, B.J. _et al._ Impaired neuronal sodium channels cause intranodal conduction failure and reentrant arrhythmias in human sinoatrial node.
_Nat Commun_ 11, 512 (2020). https://doi.org/10.1038/s41467-019-14039-8 Download citation * Received: 24 May 2019 * Accepted: 16 December 2019 * Published: 24 January 2020 * DOI:
https://doi.org/10.1038/s41467-019-14039-8 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative