- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Outcomes for cancer patients vary greatly even within the same tumor type, and characterization of molecular subtypes of cancer holds important promise for improving prognosis and
personalized treatment. This promise has motivated recent efforts to produce large amounts of multidimensional genomic (multi-omic) data, but current algorithms still face challenges in the
integrated analysis of such data. Here we present Cancer Integration via Multikernel Learning (CIMLR), a new cancer subtyping method that integrates multi-omic data to reveal molecular
subtypes of cancer. We apply CIMLR to multi-omic data from 36 cancer types and show significant improvements in both computational efficiency and ability to extract biologically meaningful
cancer subtypes. The discovered subtypes exhibit significant differences in patient survival for 27 of 36 cancer types. Our analysis reveals integrated patterns of gene expression,
methylation, point mutations, and copy number changes in multiple cancers and highlights patterns specifically associated with poor patient outcomes. SIMILAR CONTENT BEING VIEWED BY OTHERS
MLOMICS: CANCER MULTI-OMICS DATABASE FOR MACHINE LEARNING Article Open access 30 May 2025 BENCHMARKING JOINT MULTI-OMICS DIMENSIONALITY REDUCTION APPROACHES FOR THE STUDY OF CANCER Article
Open access 05 January 2021 THE CURATED CANCER CELL ATLAS PROVIDES A COMPREHENSIVE CHARACTERIZATION OF TUMORS AT SINGLE-CELL RESOLUTION Article 08 May 2025 INTRODUCTION Cancer is a
heterogeneous disease that evolves through many pathways, involving changes in the activity of multiple oncogenes and tumor suppressor genes. The basis for such changes is the vast number
and diversity of somatic alterations that produce complex molecular and cellular phenotypes, influencing each individual tumor’s behavior and response to treatment. Due to the diversity of
mutations and molecular mechanisms, outcomes vary greatly. It is therefore important to identify cancer subtypes based on common molecular features, and correlate those with outcomes. This
will lead to an improved understanding of the pathways by which cancer commonly evolves, as well as better prognosis and personalized treatment. Efforts to distinguish subtypes are
complicated by the many kinds of genomic changes that contribute to cancer. While gene expression clustering is often used to discover subtypes (e.g., the _PAM50_ subtypes1 of breast
cancer), analysis of a single data type does not typically capture the full complexity of a tumor genome and its molecular phenotypes. For example, a copy number change may be relevant only
if it causes a gene expression change; gene expression data ignores point mutations that alter the function of the gene product; and point mutations in two different genes may have the same
downstream effect, which may become apparent only when also considering methylation or gene expression. Therefore, comprehensive molecular subtyping requires integration of multiple data
types. In order to use multiple data types for subtyping, some approaches carry out separate clustering of each data type followed by manual integration of the clusters2. However, clusters
based on different data may not be clearly correlated. More rigorous methods for integration include pathway analysis on multi-omic data, followed by clustering on the inferred pathway
activities3, similarity network fusion (SNF)4, rank matrix factorization5, and Bayesian consensus clustering6. There are also several sparse clustering methods, such as iCluster+7, which
assume that only a small fraction of features are relevant. These methods are either highly dependent on feature selection, or enforce sparsity, thus neglecting potentially useful
information. A recent method, Perturbation clustering for data INtegration and disease Subtyping (PINS)8, introduces a novel strategy of identifying clusters that are stable in response to
repeated perturbation of the data. One drawback common to many of the more principled methods is that they are computationally too intensive to be routinely applied to large data sets, due
to the need for parameter selection or repeated perturbations. Moreover, they treat all data types equally, which may not be biologically appropriate. As a result, the discovered clusters
often show poor association with patient outcomes9,10. We therefore set out to develop a novel method that does not have these drawbacks. Cancer Integration via Multikernel LeaRning (CIMLR)
is based on Single-cell Interpretation via Multi-kernel LeaRning (SIMLR), an algorithm for analysis of single-cell RNA-Seq data11. CIMLR learns a measure of similarity between each pair of
samples in a multi-omic dataset by combining multiple gaussian kernels per data type, corresponding to different, complementary representations of the data. It enforces a block structure in
the resulting similarity matrix, which is then used for dimension reduction and _k_-means clustering. CIMLR is capable of incorporating complete genomes and scaling to many data types, and
does not assume equal importance for each data type. As such, it is well suited to modeling the heterogeneity of cancer data. Here, we apply CIMLR to discover integrative subtypes within 36
types of cancer. We recover known as well as novel subtypes, and show that our method outperforms current state-of-the-art tools in speed, accuracy, and prediction of patient survival. This
systematic subtype analysis, the most comprehensive to date, provides valuable insights into the biology underlying tumor variability. RESULTS SUBTYPING OF 36 CANCER TYPES USING CIMLR We
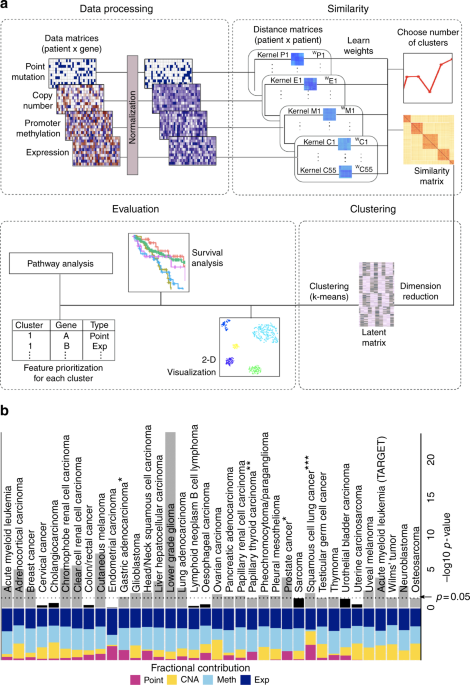
carried out a systematic subtype analysis using CIMLR (Fig. 1a) across all 32 cancer types available from TCGA, on a total of 6645 patients. Four data types were considered: point mutations,
copy number alterations, promoter CpG methylation, and gene expression. We evaluated the clusters produced by CIMLR based on (1) survival analysis, (2) silhouette (a measure of cohesion and
separation of clusters12), (3) stability of the clusters, and (4) significant differences in pathway activity between clusters (Table 1, Supplementary Table 1, Supplementary Table 2). To
demonstrate the value of multi-omic subtyping, we compared the performance of CIMLR using all four data types against analysis using only methylation or expression (Table 1, Supplementary
Data 1). We also compared CIMLR to four existing methods for integrative subtyping: iCluster+7, Bayesian consensus clustering6, PINS8, and SNF4. CIMLR outperformed all other methods on all
tested metrics (Table 1, Supplementary Data 2). In particular, the clusters obtained using CIMLR show significant differences in patient survival in 23 of 32 cancer types from TCGA (Fig.
1b), exceeding the performance of all other approaches. Additionally, we applied CIMLR to four types of pediatric cancers using data from the TARGET initiative13. Remarkably, the clusters
obtained by CIMLR present significant differences in overall survival for all four pediatric tumor types (Fig. 1b, Supplementary Table 3), exceeding the performance of other approaches
(Table 1, Supplementary Data 3, Supplementary Data 4). CIMLR learns weights for each data type instead of assigning equal importance to each. We note that the contributions of each data
type, measured as the fraction of total kernel weight contributed by kernels based on that data type, are very different between cancers (Fig. 1b). While expression and methylation each
contribute 30–50% of the kernel weight in almost all cancers, the contributions of point mutations and copy number are highly variable. We observe some association between these kernel
weights and the C/M classification of cancers14, with M-type cancers, such as endometrial and colorectal cancers having high contributions from point mutations while copy number changes
contribute more to subtyping of some C-type cancers, such as ovarian cancer. CIMLR can thus give us insight into which data types are most informative for subtyping in different cancers.
Finally, all other approaches except SNF proved impractically time-consuming and computationally intensive to run (on the order of days using 64 cores for a single configuration), while
CIMLR takes minutes to run on a laptop for each cancer type. In summary, we find that multi-omic data integration using CIMLR is the most effective method for integrative subtyping based on
technical performance, discovery of clinical and biological differences, and practical usability. BIOLOGICAL VALIDATION OF CIMLR ON LOWER-GRADE GLIOMAS Lower-grade (also called low-grade)
gliomas are a well-studied example for genomic subtyping, which is why we chose it for validation of CIMLR via reproduction of known results. Three subtypes of lower-grade gliomas have been
characterized15, based on _IDH1/2_ point mutations and chromosome 1p/19q codeletion. CIMLR finds 3 to be the best number of clusters for lower-grade gliomas, with additional peaks at 7 and
13 (Fig. 2a). The three clusters show strong separation (Fig. 2b) and correspond to the known molecular subtypes. Cluster 1 is composed almost entirely of _IDH_-wild type samples with a loss
of chromosome 10 and gain of chromosome 7. Cluster 2 (non-codel) is composed of mostly _IDH_ mutant samples with additional point mutations in _TP53_ and _ATRX_. Cluster 3 (codel) is
composed of _IDH_ mutant tumors with a chromosome 1p/19q codeletion (Fig. 2c). The _IDH_-wild type cluster has the worst overall and disease-specific survival, followed by the non-codel
cluster (Fig. 2d). A recent study2 comprising lower-grade gliomas and glioblastomas hinted at a finer classification of these tumors, finding a CIMP-low subgroup of _IDH_ mutant non-codel
tumors, with lower methylation and worse survival than the rest of the non-codel group. The codel group, on the other hand, was not divided further. To further characterize lower-grade
gliomas, we investigated the results by CIMLR for seven clusters, which are near-perfect subsets of the three major clusters (Fig. 2e). We find that the codel and non-codel groups are
divided into three subclusters each. In both groups, there are two CIMP-high subclusters and one CIMP-low subcluster (Fig. 2f). We examined the subclusters of cluster 2. Subcluster 2c is
characterized by reduced methylation (Fig. 2f), similar to the CIMP-low subgroup described previously2. The three subclusters have significantly different overall (log-rank _p_ = 0.043) and
disease-specific (log-rank _p_ = 0.012) survival (Fig. 2g). Analysis of pathway activity scores16 showed that they have significantly different activity of the PI3K, MAPK, and hypoxia
pathways (Fig. 2h). Further, subcluster 2a, which has the worst survival outcomes, is associated with more copy number changes than 2b or 2c; 68% of samples in 2a have a partial loss of 19q
(19q13.31–13.43), unlike the complete-arm loss in the codel group. 57% have a loss of 11p (Fig. 2i), including the tumor suppressor _TRIM3_, which also showed reduced expression in the same
samples. _TRIM3_ loss has been associated with increased proliferation and stem cell-like properties of glioblastomas17. Thus, CIMLR reproduces known molecular subtypes and also reveals
novel subgroups within lower-grade gliomas. This provides empirical evidence that CIMLR can discover meaningful and robust biological subtypes using multi-omic data. We therefore evaluated
the clusters found by CIMLR for all cancer types. To characterize the biological changes that lead to survival differences between clusters, we identified genetic alterations that were
enriched in specific clusters, and used gene set enrichment analysis (GSEA) and PROGENy16 to identify cancer-related biological pathways that were activated differently between clusters.
Below we present results for eight cancers where we obtain a significant difference in survival and improve over previous clustering studies. LIVER HEPATOCELLULAR CARCINOMA Hepatocellular
carcinoma is associated with several risk factors including chronic hepatitis B virus (HBV) and hepatitis C virus (HCV) infection, and alcohol consumption. iCluster+ has been used to find
three integrative subtypes18; however, there was no significant difference in patient outcomes, although some differences were seen in an external cohort that was tracked over a longer time.
CIMLR separates 359 liver hepatocellular carcinomas into eight clusters, associated with significant differences in overall (Fig. 3a) and disease-specific survival within the cohort.
Clusters 1, 2, and 3 have relatively high overall and disease-specific survival. We do not observe any common point mutations or copy number changes in cluster 1 (Fig. 3b); however, this may
be due to the low purity and higher immune infiltration of these tumors19. Cluster 2 is associated with HBV infection (60% samples) and Asian ethnicity. Although most of these tumors are
wild-type for _TP53_, they show gain and increased expression of _MDM4_, which encodes a p53 repressor, and low p53 pathway activity16 (Fig. 3c). This cluster has a universal loss on
chromosome 1p including the succinate dehydrogenase gene _SDHB_, accompanied by reduced _SDHB_ expression. Reduced SDHB blocks respiration leading to a metabolic shift toward glycolysis; the
accumulation of succinate also inhibits demethylases leading to a CIMP (high methylation) phenotype20, which we observe in this cluster (Fig. 3d). This cluster also displays losses on
chromosome 16, including the tumor suppressors _CYLD_ and _TSC2_, and the DNA repair gene _PALB2_, as well as reduced expression of all three. It is also enriched (28% samples) for mutations
in _AXIN1_, a tumor suppressor gene that regulates the Wnt signaling pathway. GSEA shows that this cluster is enriched for tumors with reduced expression of genes for oxidative
phosphorylation and the G1/S checkpoint. Cluster 3 is enriched for mutations in _CTNNB1_ (beta-catenin). While _CTNNB1_ mutations are also common in other clusters, the tumors in cluster 3
also display high expression of _GLUL_ (Glutamine synthase), a well-characterized target of beta-catenin21, suggesting that beta-catenin activation leads to glutamine synthesis and cellular
proliferation in these tumors. Patients in cluster 6 are more likely to be female (Fisher’s test _p_ = 0.001), non-drinkers, and do not have HBV or HCV infection. This cluster is enriched
for mutations in the tumor suppressor _BAP1_, which is involved in chromatin remodeling as well as double-strand break repair (42% samples). 63% of samples also share a loss of _BAP1_ on 3p,
along with reduced expression. These tumors have high DNA methylation, a phenotype previously associated with _BAP1_ mutations in renal cancers22, and frequently lack the 8p loss/8q gain
that is seen in other clusters. In addition, they show strongly reduced expression of genes for normal hepatocyte functions, such as bile acid metabolism, fatty acid metabolism, xenobiotic
metabolism, and coagulation. Clusters 4, 7, and 8 are associated with _TP53_ point mutations, as well as losses on 13q (_RB1_) and 17p (_MAP2K4, TP53_). However, clusters 7 and 8 have
significantly worse overall survival than cluster 4 (log-rank _p_ = 0.045 and _p_ = 0.036 respectively). Both show increased expression of Myc and E2F target genes, as well as genes involved
in mTORC1 signaling and the mitotic spindle. In addition, cluster 8 shows reduced expression of genes involved in normal hepatocyte function (as seen in cluster 6), higher immune
infiltration and macrovascular invasion. p53 and PI3K pathway activities16 are significantly associated with the clusters (_p_ < 10−12 for both, Kruskal–Wallis test), with cluster 8
showing the lowest p53 activity and highest PI3K activity. LUNG ADENOCARCINOMA Lung adenocarcinoma, often caused by smoking, is the leading cause of cancer death globally. Previous studies
identified transcriptional23 and histological24 subtypes, as well as six integrated clusters9, which, however, showed no significant association with patient survival. CIMLR separates 188
lung adenocarcinomas into eight clusters, significantly associated with overall and disease-specific survival (Fig. 3e). Clusters 1–3 are predominantly wild-type for _TP53_, whereas the
remaining clusters (4–8) are associated with _TP53_ mutations (Fig. 3f). In general, the _TP53_ mutant clusters are associated with worse survival outcomes; the exception is cluster 4, which
has significantly better overall and disease-specific survival outcomes than the other _TP53_-mutant clusters, comparable to clusters 1–3 (Fig. 3h). Cluster 1 is characterized by loss of
19p, including the tumor suppressor _STK11_; this is associated with reduced _STK11_ expression. It is enriched for point mutations in _STK11_ and _KEAP1_, as well as high expression of
_CCND3_ (cyclin D3), the transcriptional regulator _MUC1_, the Wnt pathway activator _PYGO2_ and the p53 inhibitor _MDM4_. In addition, it shows low DNA methylation (Fig. 3g), high
expression of genes for fatty acid metabolism and peroxisome function, and low expression of genes involved in apoptosis and the G2/M checkpoint. Cluster 3, like cluster 1, has low
methylation, and is associated with _STK11_ loss and point mutations. In addition, it is enriched for point mutations in _ATM_ and _KRAS_. It has a gain on 14q and losses on 1p, 21q (_BTG3,
PRMT2, HMGN1_), and 15q (_FAN1_), as well as reduced expression of those genes. This cluster is associated with high expression of the oncogene _KIT_ and the chromatin modifiers _CHD7_ and
_SUDS3_, as well as high expression of genes involved in membrane fusion and budding, and the unfolded protein response. Among the five _TP53_-mutated clusters, cluster 4, which has the best
outcomes, has a loss on chromosome 15, as well as low expression of genes involved in DNA repair and oxidative phosphorylation. Interestingly, tumors in this cluster are enriched for
splice-site mutations in _TP53_ (20% of _TP53_ mutations in this cluster), and mutations in exon 4 of _TP53_ (25%), whereas the other clusters are dominated by missense and nonsense
mutations in exons 5–10. However, neither exon 4 nor splice site mutations, nor both combined, were significantly associated with survival in this dataset. Cluster 6 is a small cluster of 14
samples, associated with high DNA methylation, _KRAS_ mutations and increased expression of the chromatin remodeling factor _SATB2_. Finally, cluster 8 shows the worst overall survival; it
is associated with males, a high rate of point mutations, and low methylation. In addition to _TP53_ point mutations, it has a loss of 19p (_MAP2K7, STK11_; >50% samples also have reduced
expression of both these genes), high expression of the RNA methyltransferase _NSUN2_, and high expression of genes for the mitotic spindle, Myc targets, E2F targets, and mTORC1 signaling.
HEAD AND NECK SQUAMOUS CELL CARCINOMA Head and neck squamous cell carcinomas (HNSCCs) are very heterogeneous in etiology and phenotype. They are stratified by tumor site, stage and
histology, and human papilloma virus (HPV) has been associated with better patient outcomes25. We separate 495 HNSCCs into eight subtypes, which are significantly associated with overall
(Fig. 4a) and disease-specific survival. Tumors in clusters 1 and 2 are predominantly HPV+ and _TP53_ wild-type (Fig. 4f). They are found mostly in the tonsils and base of tongue (Fig. 4e),
and share a loss on 11q. These HPV+ clusters have significantly higher overall (log-rank _p_ = 6.1 × 10−3) and disease-specific (log-rank _p_ = 2.1 × 10−3) survival than the remaining
clusters. However, cluster 2 has significantly worse disease-specific survival than cluster 1 (Fig. 4b), and differs in gene expression. While cluster 1 is associated with high expression of
59 genes including the oncogenes _DEK_ and _PIK3CA_, cluster 2 shows elevated _NFKB2_ expression, and reduced expression of _CDH1_ and _MAP2K4_. GSEA shows that tumors in cluster 2 also
show reduced expression of genes involved in PI3K/AKT/mTOR signaling. Consistent with these features, cluster 2 has significantly higher NFkB pathway activity than cluster 1, whereas cluster
1 has significantly higher activity of the PI3K pathway16 (Fig. 4c). Finally, 62% of the samples in cluster 2 have loss of chromosome 3p, compared to 27% of cluster 1. 3p loss, occurring
jointly with HPV status, has been associated with worse prognosis26. The remaining six clusters are HPV-negative and tend to have point mutations in _TP53_. We do not find significant
survival differences within this group, although they differ in molecular features. Cluster 4 has high DNA methylation (Fig. 4d) and is enriched for females and nonsmokers. This cluster
lacks the common 3q gain but is enriched for point mutations in _CASP8_, _FAT1_, _HRAS_, _HUWE1_ and the histone methyltransferase _KMT2B_. Clusters 5, 6, 7 and 8 all have high genomic
instability. However, cluster 5 is associated with loss of function of the histone methyltransferase NSD1; 68% of the samples have a point mutation in _NSD1_ while an additional 6% have
homozygous deletion of this gene. Tumors in this cluster are hypomethylated, a pattern previously associated with _NSD1_ loss27, and have losses on 13q and 9p. Cluster 8, which has the
highest genomic instability, is enriched for a gain on 7q (including _SMURF1_; also increased in expression) and a loss on 4q, and high expression of 35 genes including _PIK3CA_ and the
transcriptional regulator _YEATS2_, as well as low expression of the ubiquitin-conjugating enzyme _UBE2D3_, a phenotype linked to cell cycle progression, reduced apoptosis, and telomere
stability28. CLEAR CELL RENAL CELL CARCINOMA Clear cell renal cell carcinomas are the most common kidney cancers. Common genetic alterations include mutations in _VHL_ and _PBRM1_, 3p loss
and 5q gain. Using 260 clear cell renal cell carcinoma samples, CIMLR finds two cluster number peaks, at 4 and 10. The four major clusters show significant differences in overall (Fig. 5a)
and disease-specific survival. Clusters 1 and 2 have the best survival outcomes; cluster 2 shows higher genomic instability, particularly a gain on chromosome 7 (Fig. 5b). Cluster 3 has
significantly worse survival outcomes than clusters 1 and 2 (log-rank _p_ = 0.022), and is characterized by a loss on chromosome 14, including the tumor suppressor _WDR20_, which also shows
reduced expression; this gene suppresses growth and apoptosis in renal cancer cell lines29. Cluster 4 is a small cluster with significantly worse overall and disease-specific survival than
the other three clusters. These tumors have only one point mutation each in coding regions (mostly in _VHL_) (Fig. 5c), high hypoxia pathway activity16 (Fig. 5d), low expression of the
chromatin modifier _SETD2_, and high expression of the helicase _DDX11_, which is overexpressed in multiple cancers and associated with proliferation and survival in melanomas30. On
examining the split into 10 clusters, we found that several of these were subsets of the four major clusters. Interestingly, a subset of cluster 1, characterized by fewer copy number
alterations, shows significantly worse overall (Fig. 5e) and disease-specific (log-rank _p_ = 0.013) survival than the rest of cluster 1. We also identified a subcluster within cluster 3
which shows significantly better overall (Fig. 5f), and disease-specific (log-rank _p_ = 0.004) survival than the rest of cluster 3. This low-CNA group lacks a loss on chromosome 9
(including _NOTCH1_ and the tumor suppressor _TSC1_) which is present in the rest of the cluster. Instead, it has reduced expression of several genes involved in DNA repair (_CCNK, MLH3,
MTA1, APEX1_). CUTANEOUS MELANOMA Cutaneous melanoma is particularly difficult to subtype since it frequently has a high mutational burden. These tumors have been classified on the basis of
common mutations; however, this classification is not predictive of patient outcomes10. Instead, CIMLR separates 262 cutaneous melanomas into four clusters significantly associated with
overall and disease-specific survival, and a second-best split at 10. Clusters 1, 2, and 3 are not significantly different in terms of survival; however, cluster 4 has significantly worse
overall and disease-specific survival than all other clusters (Fig. 5g). Cluster 1 is characterized by relatively low purity and high immune cell infiltration (Fig. 5h). Cluster 2 has high
expression of genes involved in mTORC1 signaling and DNA synthesis. While the outcomes for these patients are similar, on examining the split into 10 clusters, we identify a subcluster (2a)
that has significantly worse disease-specific survival than the rest of cluster 2, and is in fact comparable to cluster 4 (Fig. 5i). This subcluster has a distinctive expression pattern,
which does not appear to be driven by copy number. This includes high expression of genes for autophagy, organelle fusion and protein transport, and low expression of genes involved in the
G2/M checkpoint, splicing, DNA repair, RNA metabolism, and chromatin remodeling. Cluster 3 is differentiated by a loss of 47 genes on chromosome 6q and by reduced expression of genes
involved in oxidative phosphorylation. Finally, cluster 4 is distinguished by a low point mutation burden (~80 coding mutations per tumor) (Fig. 5j), as well as high expression of three
genes (_BTBD9, CDYL, TFAP2A_) and high methylation at 100 promoters. BREAST CANCER Breast cancers are frequently classified by intrinsic subtypes1 or by ER, PR, and HER2 receptors. Another
classification, IntClust31, comprises 10 clusters based on copy number and expression. CIMLR separates 663 breast tumors into 13 clusters, which are significantly different in overall (Fig.
6a) and disease-specific survival. Ten of these are predominantly ER+ while three are predominantly triple-negative (Fig. 6c and d). There are significant differences in survival within each
group and we examine them separately. Among the predominantly ER+ clusters, Clusters 1, 2, and 3 share a loss on 11q that includes _SDHD, ATM, ARHGEF12_, and _EI24_. Cluster 1 has the best
survival outcomes and is enriched for point mutations in _GATA3_ (71% samples). On the other hand, clusters 2 and 3 are enriched for HER2+ tumors and have gains on 17q and 20, as well as a
loss on 17p, including _RPA1_, which encodes an ssDNA-stabilizing protein. In addition, Cluster 3 has a gain on 16p, which is shared by clusters 4 and 5, and is enriched for patients with
African ancestry. Cluster 12 is a small cluster of 11 patients that display global DNA hypermethylation (Fig. 6b) and high expression of genes for telomere maintenance. Cluster 11 has
significantly worse survival outcomes than the other predominantly ER+ clusters (except clusters 4 and 12 which have small sample size). This cluster is differentiated from the other ER+
clusters by methylation (it shows significant hypermethylation of 128 promoters and hypomethylation of 186 promoters) and by higher MAPK pathway activity16. It has low expression of
_NEURL4_; this gene encodes a regulator of centrosome organization and its depletion results in mitotic abnormalities in human cell lines32. This cluster also has high expression of _TAF2_,
encoding a transcriptional regulator associated with dedifferentiation and proliferation in ovarian cancer33. Three clusters—7, 8, and 13—are dominated by triple-negative tumors. All three
are characterized by _TP53_ mutations and similar patterns of copy number changes, expression and methylation, and clusters 8 and 13 are enriched for patients with African ancestry. However,
cluster 13 has significantly worse survival outcomes than clusters 7 and 8 (log-rank _p_ = 0.031 and _p_ = 0.045, respectively, for disease-specific survival). This cluster is
differentiated from clusters 7 and 8 by elevated expression of 230 genes and reduced expression of 442 genes including the tumor suppressors _APC, CREB1, NCOR1_, and _NUP98_. In addition, it
has significantly higher VEGF activity than clusters 7 and 816, suggesting higher angiogenesis. It is notable that the six ER+ tumors in this cluster share the expression changes described
above. PROSTATE CANCER CIMLR finds three clusters in a dataset of 490 primary prostate tumors. For this cancer, we do not consider overall survival as very few patients died during the
10-year follow-up period; instead, we observe significant differences in progression-free interval (Fig. 6e) and disease-free interval. Clusters 1 and 2 differ primarily in expression, as
well as methylation of 13 promoters. Cluster 3 has significantly worse outcomes than both clusters 1 and 2. It is characterized by high genomic instability (Fig. 6f), including loss of the
tumor suppressor _TRIM35_ on chromosome 8, reduced expression of the tumor suppressor _RHOBTB2_, and high promoter methylation (Fig. 6g). It also has higher activity of the VEGF pathway and
lower activity of the apoptotic Trail pathway (Fig. 6h). 63% of the samples in this cluster have p53 mutation and/or loss, and the cluster has lower p53 pathway activity than the others16.
Apart from the detailed analysis for eight cancers presented above, we provide clustering results for all cancers (Supplementary Data 7, Supplementary Data 8), and summarize the main
features of the clusters for each cancer (Supplementary Note 2, Supplementary Figs. 1–36). CIMLR VALIDATION ON UNSEEN DATA For five cancers, we were able to find sufficient additional
multi-omic data to validate our biological findings. For lower-grade glioma, clear cell renal cell carcinoma, cutaneous melanoma and breast cancer, we obtained new unseen samples recently
released by TCGA34. For prostate cancer, we used a non-TCGA dataset35. For each cancer, we classified the tumors in the new dataset into high-risk and low-risk groups based on the original
clusters, using genomic features that differed significantly between clusters. We then assessed whether the survival differences discovered in the original dataset were reproduced in the
test data (Table 2). For example, for lower-grade gliomas, we classified 226 tumors into the three major clusters found by CIMLR and validated that Cluster 1 has lower survival than the rest
of the population. We then selected the tumors predicted to belong to cluster 2 and classified them into high-risk (subcluster 2a) and low-risk (subclusters 2b + 2c) groups. Our novel
finding that tumors of subcluster 2a have worse overall survival outcomes than the rest of this cluster was validated in this dataset. For clear cell renal cell carcinoma, we classified 138
samples as high (cluster 4), intermediate (cluster 3) or low-risk (cluster 1 + cluster 2). Only two samples in the validation set were classified into the high-risk group (cluster 4).
However, samples classified as intermediate risk (cluster 3) had significantly worse overall survival than samples classified as low-risk. Similarly, we validated worse survival outcomes for
cluster 4 in cutaneous melanoma, cluster 11 for non-triple negative breast cancers, and cluster 3 for prostate cancer, in their respective external datasets. This analysis demonstrates that
the survival differences discovered by CIMLR are reproducible and potentially clinically useful. Further, in order to ask whether multi-omic subtyping results in prognostic value beyond
clinical variables commonly employed to predict survival, we also evaluated the prognostic value of the CIMLR clusters using Cox proportional hazard regression in both the discovery and
validation sets. We found that CIMLR clusters were associated with significant hazard ratios and high concordance index (CI)36 values. We also note that CI values were similar in each of the
matched discovery and validation sets. Moreover, in 11 cancers in the discovery sets, as well as 3 of the external validation sets, CIMLR clusters were associated with significant hazard
even after adjusting for common clinical variables (Supplementary Note 1, Supplementary Data 5, Supplementary Data 6). These results provide strong evidence that multi-omic subtyping using
CIMLR offers significant prognostic value beyond that of commonly used clinical features. DISCUSSION The importance of integrative cancer subtyping has been recognized for several years, and
multiple algorithms have been developed to exploit the growing amount of available multidimensional data4,5,6,7,8. CIMLR addresses many of the weaknesses of current integrative subtyping
algorithms, outperforming all tested methods in terms of cluster separation and stability. Furthermore, most of the alternative algorithms proved impractically time-consuming and
computationally intensive to run on the considerable volume of data analyzed in this study. As the amount of genomic data is growing rapidly and more types of data are becoming available
(such as gene fusions, structural variants, proteomes, miRNA, and ATAC-Seq), efficient methods are essential. Of the available methods, CIMLR is not only superior in terms of performance but
is also capable of practically scaling to large-scale analyses with many more data types. We therefore anticipate significant use of this method in the future. The subtyping achieved by
CIMLR demonstrates both biological and clinical relevance. The discovered clusters exhibit significant differences in the activity of oncogenic and tumor suppressor pathways, and show
significant differences in patient survival in 27 of 36 cancer types. The discovered subtypes provide valuable biological insights and are more predictive of survival than other commonly
used classifications. For example, for thymomas the CIMLR subtypes perform better at predicting survival than histological classification (Supplementary Note 2), while the CIMLR subtypes of
cutaneous melanoma are much better at predicting survival than classification based on BRAF, RAS, and NF1 mutations. For head and neck squamous cell carcinomas, we separate HPV+ tumors into
two groups with significantly different survival outcomes and pathway activity; a previous subtyping attempt using gene expression did not predict survival37. Similarly, in clear cell renal
carcinomas, where chromosome 14 loss has been associated with poor prognosis38, we not only find a cluster enriched for chromosome 14 loss but show that this is divided into two subclusters
only one of which is associated with poor prognosis. In breast cancer, we separate triple-negative cancers for the first time into three clusters, one of which is considerably more
aggressive than the others and is associated with reduced expression of several tumor suppressor genes. Finally, we validate several of the survival differences discovered by CIMLR in
external datasets, showing that CIMLR discovers molecular subtypes associated with robust, reproducible clinical outcomes. Our results demonstrate the value of machine learning-based
multi-omic subtyping in cancer, and the need for more effective and practically usable algorithms. As more data becomes available, the predictive power of CIMLR and related approaches will
continue to increase. We expect that subtyping will be useful in stratifying patients for prediction of outcomes and drug response to improve personalized treatment. In addition, our work
can be used as a resource for future studies aimed at understanding the biology and evolution of these cancers. METHODS DATA PREPROCESSING We considered all the 32 cancer types studied by
TCGA and collected, for each of them, multi-omic data comprising somatic point mutations (as TCGA Mutation Annotation Format files and converted to binary values, 0 to report absence of a
mutation in a gene and 1 to report its presence), copy number alterations (log2 ratios between tumor and normal tissue), methylation (beta-values, i.e. continuous values between 0 and 1),
and expression (_z_-scores normalized to normal tissue or to tumors with diploid genomes). For the TARGET data, we considered four pediatric tumors: acute myeloid leukemia, Wilms tumor,
neuroblastoma, and osteosarcoma. For each of them we collected multi-omic data comprising copy number alterations (log2 ratios between tumor and normal tissue), methylation, and RNA
expression. Moreover, we removed extreme values for both copy number log2 ratios and expression _z_-scores by setting values greater than 10 to 10 and values lower than −10 to −10. We refer
to TCGA guidelines for a detailed description of the data obtained from the consortium at the following Website: https://wiki.nci.nih.gov/display/TCGA. All the considered data were within
the Open Access Data Tier. Each data type was modeled as a matrix _N_ × _M_, where _N_ represents the samples, i.e., the patients, and _M_ a set of genes. Each data matrix was normalized so
that values ranged between 0 and 1. CIMLR We extended the original implementation of SIMLR11 to use multi-omic data. The version of SIMLR adopted here is the default version rather than the
large-scale version which leaves out the similarity enhancement by diffusion step. The original method11 takes as input a dataset where rows are samples and columns are genes, and constructs
a set of Gaussian kernels for the dataset by fitting multiple hyperparameters. Gaussian kernels are defined as follows: $$K\left( {x_i,x_j} \right) = \frac{1}{{{\it{\epsilon }}_{ij}\sqrt
{2\pi } }}{\mathrm{exp}}\left( { - \frac{{\left\Vert {x_i - x_j} \right\Vert_2^2}}{{2{\it{\epsilon }}_{ij}^2}}} \right)$$ (1) where _x__i_ and _x__j_ denote the _i_th and _j_th rows (i.e.,
samples) of the input data and \({\it{\epsilon }}_{ij}^2\) is the variance. For CIMLR, we represented each of the data types as a patient × gene matrix. We then performed the above procedure
for each data type independently, to obtain a set of 55 gaussian kernels with different variance per data type. The number of 55 kernels per data type was empirically derived (Supplementary
Table 4, Supplementary Fig. 37). Then, we solved the same optimization problem described in SIMLR11, but considering the Gaussian kernels for all the data types together to build one
patient × patient similarity matrix. This optimization problem is defined as follows: $$\begin{array}{l}{\mathrm{minimize}}_{S,L,w} - \mathop{\sum}\limits_{i,j,l} {w_lK_l} \left( {x_i,x_j}
\right)S_{ij} + \beta\left\Vert {S} \right\Vert_{\mathrm{F}}^2 + \gamma \,tr\left( {L^T\left( {I_N - S} \right)L} \right) + \rho \mathop {\sum}\limits_l {w_l} {\mathrm{log}}\,w_l\\
{{{\rm{subject}}\,{\rm{to}}}}\,L^TL = I_C,\mathop{\sum}\limits_l {w_l = 1} ,w_l \ge 0,\mathop {\sum}\limits_j {S_{ij} = 1} ,{\mathrm {and}}\,S_{ij} \ge 0.\end{array}$$ (2) Here, _N_ is the
number of patients, _C_ is the number of clusters, _i_ is the row (sample) index, _j_ is the column (gene) index, and _l_ is the kernel index which ranges from 1 to (55 × number of data
types). In the optimization framework, we solve for _S_, i.e., the _N_ × _N_ similarities matrix; moreover, _w__l_ represents the weight of each Gaussian kernel, _I__N_ and _I__C_ are _N_ ×
_N_ and _C_ × _C_ identity matrices, _β_ and _γ_ are non-negative tuning parameters, \(\left\| S \right\|_F\) is the Frobenius norm of _S,_ and _L_ is an auxiliary low-dimensional matrix
enforcing the low rank constraint on _S_. NUMBER OF CLUSTERS We also extended the method to estimate the best number of clusters presented in SIMLR11 based on separation cost to multi-omics.
For a given value of _C_, we aim at finding an indication matrix _Z_(_R_) = _XR_, with _X_ being the matrix of the top eigenvectors of the similarity Laplacian and _R_ a rotation matrix.
Let: $$[M(R)]_i = {\mathrm {max}}_j\left[ {Z\left(R \right)} \right]_{i,j}$$ (3) Then, we can define the following cost function to be minimized: $$J\left(R\right) = \mathop
{\sum}\limits_{i,j} {\frac{{[Z(R)]_{i,j}^2}}{{[M(R)]_i^2}}}$$ (4) The best number of clusters is the one for which we obtain the largest drop in the value of _J(R)_ over the set of values we
consider for _C_. We considered 2–15 clusters for the cancer types where we had at least 150 samples, or a maximum of _N_/10 clusters (where _N_ is the number of samples) for smaller
datasets. SURVIVAL ANALYSIS We used four outcome metrics provided by TCGA: overall survival (OS), disease-specific survival (DSS), progression-free interval (PFI) and disease-free interval
(DFI), over a time interval of 10 years. For OS, we censored data points corresponding to patients who died within 30 days or were over the age of 80 at the beginning of the observation
period. For TARGET, we only considered OS data, censored in the same way as for TCGA. Clusters with only one sample were removed prior to survival analysis. Associations between subtypes and
outcome were then calculated by Kaplan–Meier analysis using a log-rank test. Cox regression analysis was performed to estimate hazard ratios associated with individual clusters and to test
whether significant associations between clusters and survival outcomes remained after adjusting for common clinical features. Univariate Cox regression was used to select significant
(two-sided Wald test _p_ < 0.1) clinical features which were then included along with CIMLR clusters in a multivariate Cox regression model. Patient age, gender, race, ethnicity, tumor
stage, and grade were taken into account where data was available. For prostate cancer, Gleason score was taken into account. Five cancers with an insufficient number of events to fit the
Cox regression model were excluded from this analysis. Survival analysis was carried out using the survival 2.41–3R package. SIGNIFICANT FEATURE SELECTION Molecular features significantly
enriched in each cluster were selected as follows. For each cluster, we carried out a hypergeometric test for enrichment of point mutations in each gene. We selected point mutations with an
FDR-adjusted _p_-value of less than 0.05. To select genes significantly enriched for copy number alterations, we obtained GISTIC thresholded copy number data for each sample from TCGA. We
considered a value ≥1 to represent gain of the gene and ≤−1 to be loss of the gene. For each cluster, we used a hypergeometric test to assess whether the cluster was significantly enriched
for either loss or gain of the gene, and selected genes with an FDR-adjusted _p_-value less than 0.05. For additional stringency and to select the features that were most representative of
an individual cluster, we further selected only those genes that were altered in at least 2/3 of the samples in the cluster and <1/3 of the samples in at least one other cluster. To
select expression changes that were significantly enriched within a cluster, we considered a gene to be over-expressed when the _z_-score was ≥1, and under-expressed if the _z_-score was
≤−1. For each cluster, we selected enriched genes using the same criteria as for copy number. For methylation, we considered a gene to be highly methylated when the beta-value was ≥0.75 and
unmethylated when the beta-value was ≤0.25. For each cluster, we selected genes enriched for high or low methylation using the same criteria as for copy number. CLASSIFICATION OF UNSEEN DATA
To classify previously unseen samples into the CIMLR clusters, we used random forest classifiers. Features were ranked on the basis of the hypergeometric test described above and the
threshold for selecting the most significant features was tuned to obtain high (>80%) out-of-bag classification accuracy on the discovery set. We used the ranger version 0.9.0 and caret
version 6.0-79 R packages to train random forests and classify unseen samples. For all cancers other than prostate cancer, all four input data types were used for classification. For
prostate cancer, only expression and copy number data were available for the validation set. PATHWAY ANALYSIS AND IMMUNE CELL INFILTRATION GSEA was performed on each cluster using the method
of Segal et al.39. Gene sets (GO, Cancer Hallmarks, KEGG, Reactome) were obtained from mSigDB40. PROGENy pathway activity scores for 11 signaling pathways in TCGA patients were obtained
from Schubert et al.16. Estimates of tumor immune infiltration were obtained from Li et al.19. All statistical analyses were carried out in R version 3.3.3. CODE AVAILABILITY CIMLR is
available for download at https://github.com/danro9685/CIMLR. Both R and Matlab implementations are available. The Matlab version was used in this paper. DATA AVAILABILITY The authors
confirm that all relevant data generated in this study are included in the article and/or its supplementary information files. REFERENCES * Parker, J. S. et al. Supervised risk predictor of
breast cancer based on intrinsic subtypes. _J. Clin. Oncol._ 27, 1160–1167 (2009). Article Google Scholar * Ceccarelli, M. et al. Molecular profiling reveals biologically discrete subsets
and pathways of progression in diffuse glioma. _Cell_ 164, 550–563 (2016). Article CAS Google Scholar * Vaske, C. J. et al. Inference of patient-specific pathway activities from
multi-dimensional cancer genomics data using PARADIGM. _Bioinformatics_ 26, i237–i245 (2010). Article CAS Google Scholar * Wang, B. et al. Similarity network fusion for aggregating data
types on a genomic scale. _Nat. Methods_ 11, 333–337 (2014). Article CAS Google Scholar * Le Van, T. et al. Simultaneous discovery of cancer subtypes and subtype features by molecular
data integration. _Bioinformatics_ 32, i445–i454 (2016). Article Google Scholar * Lock, E. F. & Dunson, D. B. Bayesian consensus clustering. _Bioinformatics_ 29, 2610–2616 (2013).
Article CAS Google Scholar * Mo, Q. et al. Pattern discovery and cancer gene identification in integrated cancer genomic data. _Proc. Natl Acad. Sci. USA_ 110, 4245–4250 (2013). Article
ADS CAS Google Scholar * Nguyen, T., Tagett, R., Diaz, D. & Draghici, S. A novel approach for data integration and disease subtyping. _Genome Res._ 27, 2025–2039 (2017). Article CAS
Google Scholar * Collisson, E. A. et al. Comprehensive molecular profiling of lung adenocarcinoma. _Nature_ 511, 543–550 (2014). Article ADS CAS Google Scholar * Akbani, R. et al.
Genomic classification of cutaneous melanoma. _Cell_ 161, 1681–1696 (2015). Article Google Scholar * Wang, B., Zhu, J., Pierson, E., Ramazzotti, D. & Batzoglou, S. Visualization and
analysis of single-cell RNA-seq data by kernel-based similarity learning. _Nat. Methods_ 14, 414–416 (2017). Article CAS Google Scholar * Rousseeuw, P. J. Silhouettes: A graphical aid to
the interpretation and validation of cluster analysis. _J. Comput. Appl. Math._ 20, 53–65 (1987). Article Google Scholar * Ma, X. et al. Pan-cancer genome and transcriptome analyses of
1,699 paediatric leukaemias and solid tumours. _Nature_ 555, 371–376 (2018). Article ADS CAS Google Scholar * Ciriello, G. et al. Emerging landscape of oncogenic signatures across human
cancers. _Nat. Genet._ 45, 1127–1133 (2013). Article CAS Google Scholar * The Cancer Genome Atlas Research Network. Comprehensive, integrative genomic analysis of diffuse lower-grade
gliomas. _New Engl. J. Med._ 372, 2481–2498 (2015). Article Google Scholar * Schubert, M. et al. Perturbation-response genes reveal signaling footprints in cancer gene expression. _Nat.
Commun._ https://doi.org/10.1038/s41467-017-02391-6 (2018). Article PubMed PubMed Central Google Scholar * Chen, G. et al. Human brat ortholog TRIM3 is a tumor suppressor that regulates
asymmetric cell division in glioblastoma. _Cancer Res._ 74, 4536–4548 (2014). Article CAS Google Scholar * Ally, A. et al. Comprehensive and integrative genomic characterization of
hepatocellular carcinoma. _Cell_ 169, 1327–1341.e23 (2017). Article Google Scholar * Li, B. et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. _Genome
Biol_. https://doi.org/10.1186/s13059-016-1028-7 (2016). Article PubMed PubMed Central Google Scholar * Saxena, N. et al. SDHB-deficient cancers: the role of mutations that impair iron
sulfur cluster delivery. _J. Natl. Cancer Inst._ 108, djv287 (2016). Article Google Scholar * Cadoret, A. et al. New targets of β-catenin signaling in the liver are involved in the
glutamine metabolism. _Oncogene_ 21, 8293–8301 (2002). Article CAS Google Scholar * Chen, Y.-C., Gotea, V., Margolin, G. & Elnitski, L. Significant associations between driver gene
mutations and DNA methylation alterations across many cancer types. _PLoS Comput. Biol._ 13, e1005840 (2017). Article Google Scholar * Hayes, D. N. et al. Gene expression profiling reveals
reproducible human lung adenocarcinoma subtypes in multiple independent patient cohorts. _J. Clin. Oncol._ 24, 5079–5090 (2006). Article CAS Google Scholar * Travis, W. D. et al. The
2015 World Health Organization classification of lung tumors. _J. Thorac. Oncol._ 10, 1243–1260 (2015). Article Google Scholar * The Cancer Genome Atlas Network. Comprehensive genomic
characterization of head and neck squamous cell carcinomas. _Nature_ 517, 576–582 (2015). Article ADS Google Scholar * Gross, A. M. et al. Multi-tiered genomic analysis of head and neck
cancer ties TP53 mutation to 3p loss. _Nat. Genet._ 46, 939–943 (2014). Article CAS Google Scholar * Lee, S.-T. & Wiemels, J. L. Genome-wide CpG island methylation and intergenic
demethylation propensities vary among different tumor sites. _Nucleic Acids Res._ 44, 1105–1117 (2016). Article CAS Google Scholar * Yang, H. et al. Downregulation of
ubiquitin-conjugating enzyme UBE2D3 promotes telomere maintenance and radioresistance of Eca-109 human esophageal carcinoma cells. _J. Cancer_ 7, 1152–1162 (2016). Article CAS Google
Scholar * Takahashi, M. et al. Downregulation of _WDR20_ due to loss of 14q is involved in the malignant transformation of clear cell renal cell carcinoma. _Cancer Sci._ 107, 417–423
(2016). Article CAS Google Scholar * Bhattacharya, C., Wang, X. & Becker, D. The DEAD/DEAH box helicase, DDX11, is essential for the survival of advanced melanomas. _Mol. Cancer_ 11,
82 (2012). Article CAS Google Scholar * METABRIC Group et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. _Nature_ 486, 346–352 (2012).
Article Google Scholar * Li, J. et al. Neurl4, a novel daughter centriole protein, prevents formation of ectopic microtubule organizing centres. _EMBO Rep._ 13, 547–553 (2012). Article
CAS Google Scholar * Ribeiro, J. R., Lovasco, L. A., Vanderhyden, B. C. & Freiman, R. N. Targeting TBP-associated factors in ovarian cancer._Front. Oncol._
https://doi.org/10.3389/fonc.2014.00045 (2014). Article PubMed PubMed Central Google Scholar * Hoadley, K. A. et al. Cell-of-origin patterns dominate the molecular classification of
10,000 tumors from 33 types of cancer. _Cell_ 173, 291–304.e6 (2018). Article CAS Google Scholar * Taylor, B. S. et al. Integrative genomic profiling of human prostate cancer. _Cancer
Cell._ 18, 11–22 (2010). Article CAS Google Scholar * Gerds, T. A., Kattan, M. W., Schumacher, M. & Yu, C. Estimating a time-dependent concordance index for survival prediction models
with covariate dependent censoring. _Stat. Med._ 32, 2173–2184 (2013). Article MathSciNet Google Scholar * Zhang, Y. et al. Subtypes of HPV-positive head and neck cancers are associated
with HPV characteristics, copy number alterations, PIK3CA mutation, and pathway signatures. _Clin. Cancer Res._ 22, 4735–4745 (2016). Article CAS Google Scholar * Monzon, F. A. et al.
Chromosome 14q loss defines a molecular subtype of clear-cell renal cell carcinoma associated with poor prognosis. _Mod. Pathol._ 24, 1470–1479 (2011). Article CAS Google Scholar * Segal,
E., Friedman, N., Koller, D. & Regev, A. A module map showing conditional activity of expression modules in cancer. _Nat. Genet._ 36, 1090–1098 (2004). Article CAS Google Scholar *
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. _Proc. Natl Acad. Sci. USA_ 102, 15545–15550 (2005). Article
ADS CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank Dr. Noah Spies for useful discussions. This work was supported by an R01 grant to A.S. and S.B. (NIH/NCI) and gift
funding from the BRCA Foundation. A.L. is supported by a Young Investigator Award from the BRCA Foundation. The results published here are based in part upon data generated by the TCGA
Research Network (http://cancergenome.nih.gov/) and by the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) initiative, phs000218, managed by the NCI.
Information about TARGET can be found at http://ocg.cancer.gov/programs/target. AUTHOR INFORMATION Author notes * Serafim Batzoglou Present address: Illumina Mission Bay, 499 Illinois
Street, Suite 210, San Francisco, CA, 94158-250, USA * These authors contributed equally: Daniele Ramazzotti, Avantika Lal. AUTHORS AND AFFILIATIONS * Department of Pathology, Stanford
University, Stanford, CA, 94305, USA Daniele Ramazzotti, Avantika Lal & Arend Sidow * Department of Computer Science, Stanford University, Stanford, CA, 94305, USA Daniele Ramazzotti, Bo
Wang & Serafim Batzoglou * Department of Genetics, Stanford University, Stanford, CA, 94305, USA Arend Sidow Authors * Daniele Ramazzotti View author publications You can also search
for this author inPubMed Google Scholar * Avantika Lal View author publications You can also search for this author inPubMed Google Scholar * Bo Wang View author publications You can also
search for this author inPubMed Google Scholar * Serafim Batzoglou View author publications You can also search for this author inPubMed Google Scholar * Arend Sidow View author publications
You can also search for this author inPubMed Google Scholar CONTRIBUTIONS S.B., B.W., and D.R. designed CIMLR based on SIMLR. B.W. and D.R. implemented the software in MATLAB with inputs
from A.L. D.R. and A.L. processed TCGA data and analyzed the results. A.L. performed cluster annotation, pathway analysis, and external validation. A.L., D.R., and A.S. designed the overall
study and drafted the manuscript. All authors read and approved the final manuscript. CORRESPONDING AUTHOR Correspondence to Arend Sidow. ETHICS DECLARATIONS COMPETING INTERESTS The authors
declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. ELECTRONIC SUPPLEMENTARY MATERIAL SUPPLEMENTARY INFORMATION DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3
SUPPLEMENTARY DATA 4 SUPPLEMENTARY DATA 5 SUPPLEMENTARY DATA 6 SUPPLEMENTARY DATA 7 SUPPLEMENTARY DATA 8 REPORTING SUMMARY RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a
Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit
to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are
included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ramazzotti, D., Lal, A., Wang, B. _et al._ Multi-omic tumor data
reveal diversity of molecular mechanisms that correlate with survival. _Nat Commun_ 9, 4453 (2018). https://doi.org/10.1038/s41467-018-06921-8 Download citation * Received: 01 June 2018 *
Accepted: 27 September 2018 * Published: 26 October 2018 * DOI: https://doi.org/10.1038/s41467-018-06921-8 SHARE THIS ARTICLE Anyone you share the following link with will be able to read
this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative







