- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT _DSG2_, encoding desmoglein-2, is one of the causative genes of arrhythmogenic cardiomyopathy. We previously identified a homozygous _DSG2_ p.Arg119Ter stop-gain variant in a
patient with juvenile-onset cardiomyopathy and advanced biventricular heart failure. However, the pathological significance and prevalence of the heterozygous _DSG2_ p.Arg119Ter variant
remains uncertain. Here, we identified four unrelated patients with cardiomyopathy with heterozygous _DSG2_ p.Arg119Ter variants among 808 patients with nonischemic cardiomyopathy; the
allele frequency was 0.0037, which is more than 50-fold greater than that reported in the general Japanese population. These patients were clinically diagnosed with arrhythmogenic right
ventricular cardiomyopathy (Pt-1), dilated cardiomyopathy (DCM) after ventricular septum defect closure surgery (Pt-2), DCM (Pt-3), and end-stage hypertrophic cardiomyopathy (Pt-4). The
patients also exhibited reduced left ventricular contractile function and varying clinical courses. Genetic analysis identified additional possible causative variants, _DSG2_ p.Arg292Cys in
Pt-1 and _BAG3_ p.His166SerfsTer6 in Pt-3. Immunohistochemical analysis of endomyocardial biopsy samples revealed that the expression of not only desmoglein-2 but also desmoplakin was
markedly reduced. Transmission electron microscopy revealed pale and fragmented desmosomes and widened gaps between intercalated discs in the myocardium. A microforce test using human
cardiomyocytes differentiated from induced pluripotent stem cells (iPSC-CMs) demonstrated reduced contractility in iPSC-CMs carrying a heterozygous truncating variant in _DSG2_. These data
suggest that the _DSG2_ p.Arg119Ter variant is concealed in patients with cardiomyopathy with heart failure, and desmosome impairment may be a latent exacerbating factor of contractile
dysfunction and disease progression. SIMILAR CONTENT BEING VIEWED BY OTHERS BIALLELIC LOSS OF _LDB3_ LEADS TO A LETHAL PEDIATRIC DILATED CARDIOMYOPATHY Article Open access 17 October 2022
RECESSIVE _TMOD1_ MUTATION CAUSES CHILDHOOD CARDIOMYOPATHY Article Open access 02 January 2024 NEONATAL-LETHAL DILATED CARDIOMYOPATHY DUE TO A HOMOZYGOUS _LMOD2_ DONOR SPLICE-SITE VARIANT
Article Open access 26 January 2022 INTRODUCTION Recent genetic studies using high-throughput sequencing technologies have identified various pathogenic variants and revealed an association
between genetic factors and clinical phenotypes in patients with cardiomyopathies1,2. Desmosomal genes (_PKP2_, _JUP_, _DSP_, _DSC2_, and _DSG2_) encode the structural proteins of the
desmosome, a dynamic junctional component that maintains the structural integrity of heart tissues; genetic variants in these desmosomal genes cause arrhythmogenic cardiomyopathy (AC)
characterized by lethal arrhythmia and myocardial dysfunction, predominantly in the right ventricle3,4,5,6. Desmoglein-2, encoded by _DSG2_, is a desmosomal cadherin protein that is
expressed mainly in heart tissue, and heterozygous missense or nonsense mutations in _DSG2_ have been identified in patients with AC7,8,9,10. We previously identified a patient with
juvenile-onset severe biventricular heart failure carrying a homozygous stop-gain variant of _DSG2_ (NM_001943.5(NP_001934.2):c.355 C > T, p.Arg119Ter) and demonstrated that complete
desmoglein-2 deficiency causes abnormal deposition of desmosome proteins and disruption of intercalated discs in cardiomyocytes11. Although the deleterious effect of the homozygous _DSG2_
p.Arg119Ter variant is evident, the interpretation of the effects of the heterozygous _DSG2_ p.Arg119Ter variant is conflicting (ClinVar database)12, and its pathological role remains
uncertain. In this study, we identified four unrelated cardiomyopathy patients with heterozygous _DSG2_ p.Arg119Ter variants among 808 patients with nonischemic cardiomyopathy. The clinical
diagnoses and clinical courses varied among these four patients. Genetic analysis identified additional possible causative variants, and immunohistochemical analysis of endomyocardial biopsy
samples demonstrated reduced expression of not only desmoglein-2 but also desmoplakin. Transmission electron microscopy revealed pale and fragmented desmosomes and widened gaps between
intercalated discs in the myocardium. These data suggest that the _DSG2_ p.Arg119Ter variant may be concealed in patients with cardiomyopathy with heart failure, and desmosome impairment
combined with genetic or environmental factors may promote contractile dysfunction. MATERIALS AND METHODS GENETIC ANALYSIS Genetic analysis was approved by the Ethics Committee of Osaka
University (accession number: 680,684). All investigations conformed to the Ethical Guidelines for Medical and Health Research Involving Human Subjects in Japan. Informed consent was
obtained from all patients. Genetic analyses were performed as previously described13 with slight modifications. Briefly, genomic DNA was extracted from peripheral blood, and whole-exome
sequencing was performed. The sequencing results were searched for rare pathogenic variants of 57 known causative genes of cardiomyopathy13. Rare pathogenic variants were identified by
applying the following criteria: (i) exonic or splice-site variants excluding exonic synonymous variants; (ii) variants included in the gene list; (iii) variants with a minor allele
frequency <5%; (iv-1) variants listed as disease-causing or likely disease-causing mutations in the HGMD (2021)14 or as pathogenic variants or likely pathogenic variants in ClinVar
(clinvar_20210501)12; (iv-2) nonsense, frameshift or splice-site variants; and (iv-3) homozygous variants with a minor allele frequency <0.5% or heterozygous variants with a minor allele
frequency <0.005%. Variants meeting the criteria (i), (ii), (iii) and ((iv-1) or (iv-2) or (iv-3)) are listed in Table 2. The targeted genomic regions were amplified via PCR (KOD Fx Neo,
TOYOBO), purified, and analyzed via direct Sanger sequencing or cloning (pCR bluntII-TOPO vector, Thermo) via PCR primers (Supplementary Table 1). IMMUNOHISTOCHEMICAL ANALYSIS AND
TRANSMISSION ELECTRON MICROSCOPY (EM) ANALYSIS One of the myocardial samples collected was subjected to EM evaluation; the remaining samples were subjected to light microscopy examination.
Masson’s trichrome staining and immunohistochemistry were performed on 2-µm-thick formalin-fixed and paraffin-embedded tissue sections from the right ventricular wall. We used a fully
automated immunohistochemical staining system, Bond-III (Leica Microsystems). Briefly, the samples were deparaffinized, and antigens were retrieved from the instrument. All the slides were
incubated with primary antibodies against desmoglein-2 (diluted 1:10; PROGEN 651119), desmoplakin (diluted 1:10; PROGEN 65146), plakoglobin (diluted 1:10; PROGEN 65105), and plakophilin-2
(diluted 1:10; PROGEN 651101). Antibody binding was visualized via the avidin-biotin complex method according to the manufacturer’s instructions (Vectastain ABC; Vector). Microstructural
abnormalities were assessed by sequential EM. Endomyocardial biopsy samples were fixed in 2.5% glutaraldehyde and post-fixed in 1% osmium tetroxide. The samples were dehydrated in ethanol
and embedded in Epok 812 (Ernest F. Fullam, Inc., Latham, NY, USA). Ultrathin sections were cut using an ultramicrotome, stained with lead citrate and uranyl acetate, and examined under an
electron microscope (HT7650; Hitachi Ltd., Ibaraki, Japan). Induced pluripotent stem cells (iPSCs) generated from a patient carrying the homozygous _DSG2_ p.Arg119Ter variant were
differentiated into cardiomyocytes (iPSC-CMs) and fixed for EM evaluation as previously described6. MICROFORCE TEST USING SELF-ORGANIZED TISSUE RINGS Self-organized tissue rings (SOTRs)15
were generated from iPSC-CMs carrying a heterozygous frameshift variant (c.297dupT, p.Gly100TrpfsTer105) in _DSG2_11 and from normal iPSC-CMs without a pathogenic variant in _DSG2_ as a
control (iPSC-CMs were generated from a patient with ARVC in which the genetic variant in _PKP2_ is normally corrected by genome editing6). The active force of the SOTRs was measured with a
micron-scale mechanical testing system (MicroTester G2; CellScale Biomaterials Testing) as previously described16. RESULTS PREVALENCE OF THE _DSG2_ (C.355 C > T, P.ARG119TER) VARIANT IN
NONISCHEMIC CARDIOMYOPATHIES _DSG2_ (c.355 C > T, p.Arg119Ter) is an extremely rare variant in the gnomAD database of exome and genome sequences worldwide, with a minor allele frequency
(MAF) of 0.000009297 (9/44,826 in the East Asian population, 6/1,179,852 in the European population (non-Finnish), and none in other cohorts)17. Importantly, the MAFs of _DSG2_ (c.355 C >
T, p.Arg119Ter) in the East Asian population (0.0002008) and in the Japanese cohort in the Tohoku Medical Megabank (0.000064 (7/108,574)) (jMorp database)18 were approximately 21.6-fold and
6.9-fold higher, respectively, than those in the total gnomAD database (Table 1). We screened 808 patients diagnosed with nonischemic cardiomyopathy at Osaka University Hospital from 2011
to 2023 via exome sequence analysis and identified five patients with _DSG2_ (c.355 C > T, p.Arg119Ter) variants, including one patient with a previously reported homozygous variant11 and
four patients with heterozygous variants. The MAF of _DSG2_ (c.355 C > T, p.Arg119Ter) was 0.003712871, which was more than 50-fold greater than that in the general Japanese population.
These data suggest that the _DSG2_ (c.355 C > T, p.Arg119Ter) variant is more common in East Asian populations than in other cohorts and is concentrated in patients with nonischemic
cardiomyopathy (Table 1). CLINICAL COURSES OF CARDIOMYOPATHY PATIENTS WITH HETEROZYGOUS _DSG2_ (C.355 C > T, P.ARG119TER) VARIANTS The patient with a homozygous _DSG2_ (c.355 C > T,
p.Arg119Ter) variant presented with juvenile-onset severe biventricular heart failure11, whereas patients with heterozygous variants presented various diagnoses and clinical courses. These
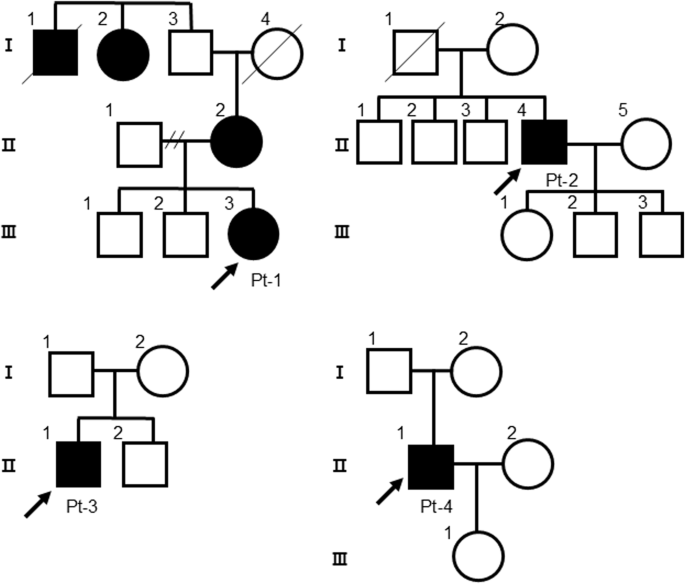
four patients (patient (Pt)-1, 2, 3, and 4) were identified within unrelated families, and only Pt-1 had a family history of cardiovascular disease or abnormal findings on cardiovascular
examination (Fig. 1). Figure 2 shows the clinical courses and time courses of the echocardiographic parameters of these patients. Pt-1 (a 19-year-old woman) was aware of congestion at 17
years of age and was diagnosed with arrhythmogenic right ventricular cardiomyopathy (ARVC) according to the TASC Force Criteria in 2010 at 19 years of age19. The mother of the proband (II-2)
carried a heterozygous _DSG2_ (c.355 C > T, p.Arg119Ter) variant and presented with an enlarged heart on a chest X-ray, but she was not diagnosed with ARVC. An echocardiogram revealed a
left ventricular ejection fraction (LVEF) of 47–57% (normal range: 66 ± 5% in women20), suggesting not only right ventricular but also left ventricular contractile dysfunction. Cardiac MRI
revealed right ventricular dilatation and reduced contraction of both the right and left ventricles but did not detect fatty degeneration. Owing to the diagnosis of ARVC and frequent
premature ventricular contractions, Pt-1 received an implantable cardioverter-defibrillator (ICD). Pt-2 (a 52-year-old man) underwent patch closure for a ventricular septum defect (VSD) at 2
years of age via open heart surgery and did not experience any cardiac symptoms until his thirties. From the age of 40 years, the left ventricular diastolic and systolic diameters
(LVDd/LVDs) gradually increased, the LVEF gradually decreased (LVDd/Ds: 64/51 mm, LVEF: 40% at 50 years of age), and nonsustained ventricular tachycardia (NSVT) was detected. Pt-2 was
diagnosed with dilated cardiomyopathy (DCM) at 52 years of age and underwent catheter ablation for symptomatic atrial fibrillation and atrial flutter. Pt-3 (a 56-year-old man) developed
fatigue and shortness of breath for the first time and was subsequently diagnosed with DCM. Echocardiographic evaluation revealed a markedly enlarged left ventricle and severe left
ventricular dysfunction (LVDd/s: 83/77 mm, LVEF: 15%). Oral and intravenous administration of inotropic drugs was required because of severe advanced heart failure. Pt-4 (a 68-year-old man)
was diagnosed with hypertrophic obstructive cardiomyopathy (HOCM) via echocardiogram in his 40 s. From the age of 60 years, the LVDd/LVDs gradually increased, and the LVEF and
intraventricular septum thickness (IVST) gradually decreased. Pt-4 was diagnosed with end-stage hypertrophic cardiomyopathy (HCM) at 68 years of age. PATHOGENIC VARIANTS IDENTIFIED IN THE
FOUR CARDIOMYOPATHY PATIENTS WITH _DSG2_ (C.355 C > T, P.ARG119TER) The pathogenic variants identified in these patients after filtering the results of whole-exome sequencing analysis
followed by Sanger sequencing analysis are shown in Table 2 and Figs. S1, S2. A variant list of all the data used for the filtering procedure is shown in Supplementary Table 2. In Pt-1, the
_DSG2_ (c.874 C > T, p.Arg292Cys) variant was identified, and Sanger sequencing after PCR cloning confirmed that _DSG2_ p.Arg119Ter and p.Arg292Cys were compound heterozygous (Fig. S1).
In Pt-2, no rare pathogenic variants other than the _DSG2_ p.Arg119Ter variant were identified. In Pt-3, a heterozygous frameshift variant (c.486_487insCCAGCCT, p.His166SerfsTer6) was
identified in _BAG3_ (NM_004281.4(NP_004272.2)), encoding BLC2-associated athanogene 3 located on the sarcomere Z-disc, which is a causative gene of DCM21 (Fig. S2). In Pt-4, no pathogenic
variants were identified in the established causal genes associated with sarcomere function (_MYH7_, _MYBPC3_, _TNNT2_, _TNNI3_, _TPM1_, _ACTC1_, _MYL2_, _MYL3_, and _CSRP3_)22, although the
clinical diagnosis of Pt-4 was HOCM. Five rare variants were identified after the filtering procedure in Pt-1 and Pt-4 (_EYA4_ p.Pro170Thr, _TTN_ p.Glu22134Lys, and _VCL_ p.Pro291Gln in
Pt-1 and _RYR2_ p.Val1810Leu and _TTN_ p.Arg22007Cys in Pt-4); however, based on previous studies and genetic databases, it was not evident whether these variants were related to the
development of ARVC in Pt-1 or HCM in Pt-4. HISTOPATHOLOGICAL EVALUATION OF THE PATIENTS’ MYOCARDIUM We previously demonstrated that the complete loss of desmoglein-2 due to the homozygous
_DSG2_ p.Arg119Ter variant causes abnormal deposition of desmosome proteins and disruption of intercalated discs in cardiomyocytes, and iPSC-CMs generated from patients recapitulate reduced
contractility, abnormal excitation, and aberrant myocardial fiber structures11. We confirmed the pathological findings in desmosomes from both the LV myocardium and iPSC-CMs generated from
the patient with the homozygous _DSG2_ p.Arg119Ter variant by transmission electron microscopy analysis (Fig. S3). To clarify how the heterozygous _DSG2_ p.Arg119Ter variant affects the
expression of desmosome proteins, serial sections of myocardium samples obtained by right ventricular myocardial biopsy on Pts 1, 2, and 3 were examined by immunostaining. Pt-4 did not
undergo myocardial biopsy. Masson’s trichrome (MTC) staining revealed massive fibrosis in Pt-1 and modest fibrosis in Pt-2 and Pt-3 (Fig. 3). Desmoglein-2 reactivity was diminished in Pt-1
as evaluated by immunostaining, suggesting that the combination of these variants (Arg119Ter and Arg292Cys) severely affected protein expression. Desmoglein-2 reactivity decreased in Pt-3
but was preserved in Pt-2. The reactivities of plakophilin-2 and plakoglobin were preserved in Pts 1, 2 and 3. Notably, desmoplakin reactivity was significantly decreased in Pt-1 and Pt-3.
Although the expression of desmosome proteins was preserved in Pt-2, their distribution in the intercalated discs exhibited abnormal deposition rather than a normal peripheral distribution.
The myocardial samples were further evaluated via transmission electron microscopy. Abnormal pale junctions characterized by repeating couplings, elongated desmosomes, and widened gaps in
intercalated discs, typically observed in ARVC patients23, were detected in the myocardium of Pt-1, Pt-2, and Pt-3 (Figs. 4A–C). Notably, fragmented and aggregated desmosomes were detected
in the myocardium of Pt-1 (Fig. 4A). Similar findings were observed in a patient with biventricular heart failure due to complete loss of desmoglein-2 caused by the homozygous _DSG2_
p.Arg119Ter variant11. REDUCED CONTRACTILITY IN IPSC-CMS CARRYING A HETEROZYGOUS TRUNCATING VARIANT IN _DSG2_ To evaluate whether the reduced expression of desmoglein-2 due to a heterozygous
truncating variant in _DSG2_ potentially affects contractility in human cardiomyocytes, we generated self-organized tissue rings (SOTRs)15 from iPSC-CMs with or without a heterozygous
frameshift variant in _DSG2_ (details in “Materials and Methods”) and measured the active force as previously described6. A microforce test on the generated SOTRs demonstrated that the
active force was significantly lower in SOTRs with the heterozygous truncating variant in _DSG2_ than in control SOTRs (Fig. 5A, B), suggesting the potential pathological role of this
variant in human cardiomyocytes. DISCUSSION Here, we identified four unrelated cardiomyopathy patients carrying heterozygous _DSG2_ p.Arg119Ter variants who exhibited various clinical
courses. The allele frequency of the _DSG2_ p.Arg119Ter variant among 808 patients with nonischemic cardiomyopathy, including one homozygous patient and four heterozygous patients, was
0.0037, which was more than 50-fold greater than that reported in the general Japanese population (0.0000064)18. Because patients diagnosed with nonischemic cardiomyopathy and eligible for
genetic analysis in our hospital require detailed cardiovascular examinations due to disease progression, the _DSG2_ p.Arg119Ter variant might be more common in patients with advanced heart
failure than in the general population. Compound heterozygous _DSG2_ p.Arg119Ter and p.Arg292Cys variants were identified in Pt-1. The homozygous _DSG2_ p.Arg292Cys variant was identified in
a patient with juvenile ARVC and sudden death24, and compound heterozygous _DSG2_ p.Arg292Cys and p.Ser194Leu variants were identified in an ARVC patient25. Genetic studies of 99 unrelated
Japanese ARVC probands revealed three homozygous and 11 heterozygous _DSG2_ p.Arg292Cys variants in 14 of 75 Japanese ARVC patients, and their family members rarely developed ARVC-related
symptoms26. These data suggest that the compound heterozygous _DSG2_ p.Arg119Ter and p.Arg292Cys variants are the most likely cause of ARVC in Pt-1. No cardiomyopathy-related pathological
variants other than _DSG2_ p.Arg119Ter were identified in Pt-2. A cohort study following 174 patients who underwent VSD closure surgery for 40 years postoperatively demonstrated that LV
systolic function was impaired but stable in 21% of patients and that LVDd/Ds and fractional shortening (FS) evaluated by echocardiography were 50 ± 6/33 ± 6 mm and 35 ± 8%, respectively,
32–44 years after the operation27. As ventricular interactions are mediated by mechanical coupling through the ventricular septum and common myocardial fibers, inadequate deformation of the
ventricular septal fibers associated with left ventricular spherization contributes to reduced contractile performance28. Pt-2 exhibited progression of LV dysfunction (LVDd/Ds: 64/51 mm, EF:
43%, FS: 19%) at 52 years of age compared with the previously reported natural history, suggesting that interventricular plate dysfunction combined with the _DSG2_ p.Arg119Ter variant might
contribute to the pathogenesis of DCM. A novel frameshift variant, _BAG3_ p.His166SerfsTer6, was identified in Pt-3. The _BAG3_ p.His166SerfsTer6 mutation was considered the causative
variant of DCM because the neighboring frameshift variant _BAG3_ p.Pro163GlnfsTer48 is defined as pathogenic17, and heterozygous frameshift or stop-gain variants of _BAG3_ have been
identified previously in patients with DCM29,30. A cohort study comprising 129 individuals with _BAG3_ variants demonstrated that disease penetrance in individuals over 40 years of age was
80% and that the LVEF and LVDd were 34.1 ± 13.0% and 64.5 ± 7.9 mm, respectively, at the first evaluation in patients diagnosed with DCM21. Echocardiographic evaluation of Pt-3 revealed more
advanced LV dysfunction (LVEF: 15%, LVDd: 83 mm) than previously reported parameters, suggesting that digenic heterozygous _DSG2_ p.Arg119Ter and _BAG3_ p.His166SerfsTer6 variants might
contribute to disease progression in Pt-3. No cardiomyopathy-related pathological variants other than the _DSG2_ p.Arg119Ter variant were identified in Pt-4, although Pt-4 was diagnosed with
HCM. As our genetic analysis was based on whole-exome sequencing, we cannot exclude the possibility of noncoding intronic and intergenic variants that are associated with known HCM-causing
genetic variants31. A cohort study evaluating genotype‒phenotype correlations in 1000 patients with HCM revealed 27 deleterious rare desmosomal variants, including _DSG2_ p.Arg119Ter, in 24
(2.4%) patients with HCM and demonstrated that deleterious rare desmosomal variants are associated with distinctive clinical features, including a higher incidence of RV involvement,
ventricular arrhythmias, and conduction block32. In HCM pathogenesis, the activation of calcium sensitivity and stress-responsive molecular pathways mediate the reprogramming of cardiac
hypertrophy22, thereby causing continuous stress on cardiomyocytes. Therefore, the continuous myocardial stress caused by HCM pathology combined with the _DSG2_ p.Arg119Ter variant may have
contributed to the transition to end-stage HCM in Pt-4. Our histopathological analysis of myocardial samples of patients demonstrated reduced reactivity for desmoglein-2 and desmoplakin in
Pt-1 and Pt-3 and abnormal deposition of desmosome proteins in Pt-2. These data are consistent with a previous finding that the signal for desmoplakin as evaluated by immunohistochemical
analysis was absent in the explanted heart of an ARVC patient carrying a null allele in desmoglein-2, possibly due to its abnormal redistribution into nonjunctional pools33. BAG3 is a
cochaperone in the protein degradation machinery and has a role related to cell-to-cell junctions; in addition, _BAG3_ knockdown decreases the levels of connexin 43 by changing the stability
of the protein34. These mechanisms may underlie the decreased expression of desmoglein-2 in the heart tissue of Pt-3. Transmission electron microscopy (TEM) further revealed abnormal pale
junctions and widened gaps in the intercalated discs in these myocardial samples. The abnormal redistribution of desmosome proteins due to desmoglein-2 deficiency despite preserved protein
expression may cause the abnormal findings observed via electron microscopy in Pt-2. These data suggest that the structural abnormalities in intercalated discs commonly observed in patients
with ARVC23 may contribute to the mechanism of heart failure progression in patients diagnosed with DCM. In Pt-1, fragmented and aggregated desmosomes, which are uncommon histopathological
features of ARVC, were observed. Similar findings were detected in a patient with biventricular heart failure who was homozygous for the _DSG2_ p.Arg119Ter variant11 and may be a
histopathological characteristic caused by complete deficiency of desmoglein-2. Genotypic analysis of 99 patients with ARVC revealed that _DSG2_ was the most frequently affected gene in the
Japanese population26. Combined with previous findings, our data highlight the importance of genetic screening for desmosomal gene variants in patients with broad cardiomyopathy with
progressive heart failure. REFERENCES * Haas, J. et al. Atlas of the clinical genetics of human dilated cardiomyopathy. _Eur. Heart J._ 36, 1123–1135 (2015). Article CAS PubMed Google
Scholar * Tobita, T. et al. Genetic basis of cardiomyopathy and the genotypes involved in prognosis and left ventricular reverse remodeling. _Sci. Rep._ 8, 1998 (2018). Article PubMed
PubMed Central Google Scholar * Al-Jassar, C., Bikker, H., Overduin, M. & Chidgey, M. Mechanistic basis of desmosome-targeted diseases. _J. Mol. Biol._ 425, 4006–4022 (2013). Article
CAS PubMed PubMed Central Google Scholar * Delmar, M. & McKenna, W. J. The cardiac desmosome and arrhythmogenic cardiomyopathies: From gene to disease. _Circ. Res_. 107, 700–714
(2010). Article CAS PubMed Google Scholar * Higo, S. Disease modeling of desmosome-related cardiomyopathy using induced pluripotent stem cell-derived cardiomyocytes. _World J. Stem
Cells_ 15, 71–82 (2023). Article PubMed PubMed Central Google Scholar * Inoue, H. et al. Modeling reduced contractility and impaired desmosome assembly due to plakophilin-2 deficiency
using isogenic iPS cell-derived cardiomyocytes. _Stem Cell Rep._ 17, 337–351 (2022). Article CAS Google Scholar * Awad, M. M., Calkins, H. & Judge, D. P. Mechanisms of disease:
molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. _Nat. Clin. Pr. Cardiovasc Med_ 5, 258–267 (2008). Article CAS Google Scholar * Awad, M. M. et al. DSG2
mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. _Am. J. Hum. Genet_ 79, 136–142 (2006). Article CAS PubMed PubMed Central Google Scholar * Pilichou,
K. et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. _Circulation_ 113, 1171–1179 (2006). Article CAS PubMed Google Scholar *
Sen-Chowdhry, S. et al. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease
expression. _Circulation_ 115, 1710–1720 (2007). Article PubMed Google Scholar * Shiba, M. et al. Phenotypic recapitulation and correction of desmoglein-2-deficient cardiomyopathy using
human induced pluripotent stem cell-derived cardiomyocytes. _Hum. Mol. Genet_ 30, 1384–1397 (2021). Article CAS PubMed PubMed Central Google Scholar * Landrum, M. J. et al. ClinVar:
improving access to variant interpretations and supporting evidence. _Nucleic Acids Res_. 46, D1062–D1067 (2018). Article CAS PubMed Google Scholar * Tabata, T. et al. Phospholamban
p.Arg14del cardiomyopathy: A Japanese case series. _Intern Med_. 61, 1987–1993 (2022). Article PubMed Google Scholar * Stenson, P. D. et al. The Human Gene Mutation Database (HGMD((R))):
optimizing its use in a clinical diagnostic or research setting. _Hum. Genet_ 139, 1197–1207 (2020). Article PubMed PubMed Central Google Scholar * Li, J. et al. Circulating re-entrant
waves promote maturation of hiPSC-derived cardiomyocytes in self-organized tissue ring. _Commun. Biol._ 3, 122 (2020). Article PubMed PubMed Central Google Scholar * Kameda, S. et al.
Modeling reduced contractility and stiffness using iPSC-derived cardiomyocytes generated from female becker muscular dystrophy carrier. _JACC Basic Transl. Sci._ 8, 599–613 (2023). Article
PubMed PubMed Central Google Scholar * Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. _Nature_ 581, 434–443 (2020). Article CAS
PubMed PubMed Central Google Scholar * Tadaka, S. et al. jMorp: Japanese multi-omics reference panel update report 2023. _Nucleic Acids Res_. 52, D622–D632 (2024). Article CAS PubMed
Google Scholar * Marcus, F. I. et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. _Circulation_ 121,
1533–1541 (2010). Article PubMed PubMed Central Google Scholar * Ohte, N. et al. JCS 2021 guideline on the clinical application of echocardiography. _Circ. J._ 86, 2045–2119 (2022).
Article PubMed Google Scholar * Dominguez, F. et al. Dilated cardiomyopathy due to BLC2-associated athanogene 3 (BAG3) mutations. _J. Am. Coll. Cardiol._ 72, 2471–2481 (2018). Article
CAS PubMed PubMed Central Google Scholar * Marian, A. J. & Braunwald, E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. _Circ.
Res_. 121, 749–770 (2017). Article CAS PubMed PubMed Central Google Scholar * Basso, C. et al. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right
ventricular cardiomyopathy: an electron microscopy investigation on endomyocardial biopsies. _Eur. Heart J._ 27, 1847–1854 (2006). Article PubMed Google Scholar * Sato, T., Nishio, H.
& Suzuki, K. Sudden death during exercise in a juvenile with arrhythmogenic right ventricular cardiomyopathy and desmoglein-2 gene substitution: a case report. _Leg. Med (Tokyo)_ 13,
298–300 (2011). Article CAS PubMed Google Scholar * Nakajima, T. et al. Compound and digenic heterozygosity in desmosome genes as a cause of arrhythmogenic right ventricular
cardiomyopathy in Japanese patients. _Circ. J._ 76, 737–743 (2012). Article CAS PubMed Google Scholar * Wada, Y., Ohno, S., Aiba, T. & Horie, M. Unique genetic background and outcome
of non-Caucasian Japanese probands with arrhythmogenic right ventricular dysplasia/cardiomyopathy. _Mol. Genet Genom. Med_ 5, 639–651 (2017). Article CAS Google Scholar * Menting, M. E.
et al. The unnatural history of the ventricular septal defect: outcome up to 40 years after surgical closure. _J. Am. Coll. Cardiol._ 65, 1941–1951 (2015). Article PubMed Google Scholar *
Schwarz, K., Singh, S., Dawson, D. & Frenneaux, M. P. Right ventricular function in left ventricular disease: pathophysiology and implications. _Heart Lung Circ._ 22, 507–511 (2013).
Article PubMed Google Scholar * Norton, N. et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy.
_Am. J. Hum. Genet_ 88, 273–282 (2011). Article CAS PubMed PubMed Central Google Scholar * Franaszczyk, M. et al. The BAG3 gene variants in Polish patients with dilated cardiomyopathy:
Four novel mutations and a genotype-phenotype correlation. _J. Transl. Med_ 12, 192 (2014). Article PubMed PubMed Central Google Scholar * Marian, A. J. Molecular genetic basis of
hypertrophic cardiomyopathy. _Circ. Res._ 128, 1533–1553 (2021). Article CAS PubMed PubMed Central Google Scholar * Wu, G. et al. Deleterious rare desmosomal variants contribute to
hypertrophic cardiomyopathy and are associated with distinctive clinical features. _Can. J. Cardiol._ 38, 41–48 (2022). Article PubMed Google Scholar * Gehmlich, K. et al. Molecular
changes in the heart of a severe case of arrhythmogenic right ventricular cardiomyopathy caused by a desmoglein-2 null allele. _Cardiovascular Pathol._ 21, 275–282 (2012). Article CAS
Google Scholar * Kirk, J. A., Cheung, J. Y. & Feldman, A. M. Therapeutic targeting of BAG3: considering its complexity in cancer and heart disease. _J. Clin. Investig._ 131, e149415
(2021). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by the Japan Agency for Medical Research and Development
(24bm0804035h0003, 24ak0101202h0002, 24ek0109760h0001, 24ek0109639h0002, 24ek0109617h0003, 23mk0101226h0002), JSPS KAKENHI (22K19526, 23K15128, 24K11239, 24K02446, 22K16138), the Next
Generation Researcher Development Project, and the Health and Labor Sciences Research Grant (23FC1050). This study was supported by the Center for Medical Innovation and Translational
Research and the Center for Medical Research and Education at the Graduate School of Medicine, Osaka University. We would like to thank Editage (www.editage.jp) for English language editing.
AUTHOR INFORMATION Author notes * These authors contributed equally: Takuya Sumida, Shou Ogawa. AUTHORS AND AFFILIATIONS * Faculty of Medicine, Osaka University, Suita, Osaka, 565-0871,
Japan Takuya Sumida * Department of Cardiovascular Medicine, Osaka University Graduate School of Medicine, Suita, Osaka, 565-0871, Japan Shou Ogawa, Shuichiro Higo, Yuki Kuramoto, Ryo Eto,
Congcong Sun, Tomoka Tabata, Yoshihiro Asano, Mikio Shiba, Yasuhiro Akazawa, Daisuke Nakamura, Takafumi Oka, Tomohito Ohtani & Yasushi Sakata * Department of Pathology, National Cerebral
and Cardiovascular Center, Suita, Osaka, 564-8565, Japan Yoshihiko Ikeda * Photonics Cell Evaluation Laboratory, Graduate School of Engineering, Osaka University, Suita, Osaka, 565-0871,
Japan Junjun Li & Li Liu * Department of Genomic Medicine, National Cerebral and Cardiovascular Center, Suita, Osaka, 564-8565, Japan Yoshihiro Asano * Cardiovascular Division, Osaka
Police Hospital, Osaka, 543-0035, Japan Mikio Shiba Authors * Takuya Sumida View author publications You can also search for this author inPubMed Google Scholar * Shou Ogawa View author
publications You can also search for this author inPubMed Google Scholar * Shuichiro Higo View author publications You can also search for this author inPubMed Google Scholar * Yuki Kuramoto
View author publications You can also search for this author inPubMed Google Scholar * Ryo Eto View author publications You can also search for this author inPubMed Google Scholar *
Yoshihiko Ikeda View author publications You can also search for this author inPubMed Google Scholar * Congcong Sun View author publications You can also search for this author inPubMed
Google Scholar * Junjun Li View author publications You can also search for this author inPubMed Google Scholar * Li Liu View author publications You can also search for this author inPubMed
Google Scholar * Tomoka Tabata View author publications You can also search for this author inPubMed Google Scholar * Yoshihiro Asano View author publications You can also search for this
author inPubMed Google Scholar * Mikio Shiba View author publications You can also search for this author inPubMed Google Scholar * Yasuhiro Akazawa View author publications You can also
search for this author inPubMed Google Scholar * Daisuke Nakamura View author publications You can also search for this author inPubMed Google Scholar * Takafumi Oka View author publications
You can also search for this author inPubMed Google Scholar * Tomohito Ohtani View author publications You can also search for this author inPubMed Google Scholar * Yasushi Sakata View
author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Shuichiro Higo. ETHICS DECLARATIONS COMPETING INTERESTS The authors
declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
SUPPLEMENTARY INFORMATION SUPPLEMANTARY_MATERIAL RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits
use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the
Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated
otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds
the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Sumida, T., Ogawa, S., Higo, S. _et al._ Four cardiomyopathy patients with a heterozygous _DSG2_ p.Arg119Ter variant. _Hum Genome Var_ 11, 47
(2024). https://doi.org/10.1038/s41439-024-00304-w Download citation * Received: 28 August 2024 * Revised: 20 November 2024 * Accepted: 21 November 2024 * Published: 20 December 2024 * DOI:
https://doi.org/10.1038/s41439-024-00304-w SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative





