- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Most patients with homozygous or compound heterozygous pathogenic ACO2 variants present with muscular hypotonia features, namely, infantile cerebellar-retinal degeneration.
Recently, two studies reported rare familial cases of ACO2 variants presenting as complex hereditary spastic paraplegia (HSP) with broad clinical spectra. Here, we report the case of a
20-year-old Japanese woman with complex HSP caused by compound heterozygous ACO2 variants, revealing a new phenotype of episodic visual loss during febrile illness. SIMILAR CONTENT BEING
VIEWED BY OTHERS HOMOZYGOUS _TFG_ GENE VARIANTS EXPANDING THE MUTATIONAL AND CLINICAL SPECTRUM OF HEREDITARY SPASTIC PARAPLEGIA 57 AND A REVIEW OF LITERATURE Article 25 March 2021 A JAPANESE
HEREDITARY SPASTIC PARAPLEGIA FAMILY WITH A RARE NONSYNONYMOUS VARIANT IN THE _SPAST_ GENE Article Open access 25 May 2021 A CLINICAL AND GENETIC STUDY OF SPG31 IN JAPAN Article 07 February
2022 The _ACO2_ gene on chromosome 22 encodes the aconitase 2 (ACO2) protein in the mitochondrial matrix; ACO2 catalyzes the stereospecific isomerization of citrate to isocitrate in the
tricarboxylic acid (TCA) cycle1. Pathogenic _ACO2_ variants were first reported in eight individuals from two Arab families, and they had infantile cerebellar-retinal degeneration (ICRD,
OMIM#614559)2. Subsequently, ~20 cases of pathogenic homozygous or compound heterozygous _ACO2_ variants have been reported, including mild cases such as isolated optic atrophy (optic
atrophy 9, OMIM#616289)2,3,4,5,6. Most patients initially present with muscular hypotonia, ataxia, seizures, progressive optic atrophy, retinal degeneration, and intellectual disabilities.
Decreased aconitase activity in fibroblastic or lymphoblastic cells suggests that impaired energy metabolism in the TCA cycle is a major cause of symptoms in patients with pathogenic _ACO2_
variants. Recently, cases from two families with pathogenic _ACO2_ variants represented by early- or late-onset spastic paraplegia with intellectual disability and broad clinical spectra
were reported4,5. Here, we describe pathogenic variants in the _ACO2_ gene presenting as complex hereditary spastic paraplegia (HSP) with a new phenotype of episodic visual loss after every
febrile infection and progressive optic atrophy. This is the third familial report and the first Asian patient with complex HSP caused by pathogenic _ACO2_ variants. The proband was born to
nonconsanguineous healthy parents at 38 weeks gestational age after unremarkable delivery. She did not have a family history of neuromuscular disorders or motor development delay. Her birth
weight was 2482 g, and her head circumference was 32 cm. Her motor development was delayed, and she could not walk independently at 1 year and 10 months because of progressive lower limb
spasticity. Physiotherapy was subsequently provided, and she started walking independently at 2 years and 6 months. Her cognitive level was moderate disability (estimated development
quotient: 50) at this time. From 3 years of age, she experienced recurrent encephalopathy-like episodes, episodic visual loss, ataxia, and altered consciousness after every febrile illness
episode. During febrile illness, she often accidentally hit her head on the wall because of her poor vision. Her visual loss recovered after defervescence, although the other symptoms
remained for several weeks. In the acute phase, magnetic resonance imaging (MRI) of the cerebrum and the retrobulbar optic nerve and ophthalmoscopy revealed no abnormalities. The following
laboratory results were normal: blood cell count; routine serum chemistry; glucose, ammonia, creatine kinase, lactate, and amino acid levels; and thyroid function. The urinary organic acid
and amino acid profiles and the cerebrospinal fluid (CSF) results for cells, glucose, protein, and lactate were within normal limits. The electroencephalogram showed diffuse slow waves and
focal spikes compatible with nonspecific encephalopathy; subsequently, antiepileptic drug therapy was initiated. However, despite treatment with the medications, episodic attacks repeatedly
occurred after every episode of fever. Her lower limb spasticity and reflexes progressed with sustained clonus and extensor plantar responses. At 18 years of age, she was admitted to our
hospital with acute psychomotor agitation after infection. Cerebral and spinal MRI, CSF analysis, metabolic screening, and ophthalmological evaluations were performed during admission.
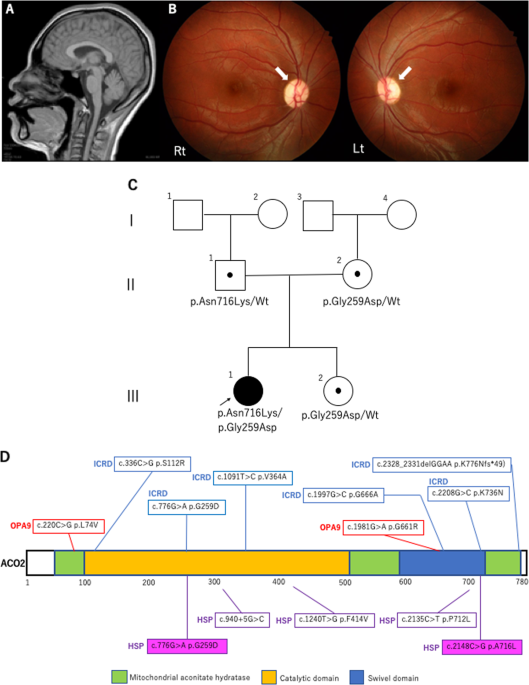
Laboratory results showed no abnormalities, whereas cerebral MRI showed mild cerebellar vermis atrophy, and ophthalmoscopy showed bilaterally pale optic discs and suspected optic atrophy
(Fig. 1A, B). These findings suggested a genetic cause for the complex HSP. Written informed consent was obtained from her parents in accordance with the Review Board and Ethics Committee of
Kyoto University, and whole-exome sequencing (WES) was performed when she was 19 years old. Trio-based WES was performed using the SuperSelect XT Human All Exon v6 (Agilent Technologies,
Santa Clara, CA). Captured libraries were sequenced using NovaSeq 6000 (Illumina, San Diego, CA). WES identified compound heterozygous missense variants in _ACO2_. The first variant was in
exon 6 (NM_001098.2: c.776 G > A, p.Gly259Asp) and was predicted to be deleterious by SIFT (score 0; http://sift.jcvi.org/) and disease-causing by MutationTaster (prob 1;
http://www.mutationtaster.org/). This variant is known to be a disease-causing mutation: rs786204828 (pathogenic)6. The second variant was located in exon 17 (NM_001098.2: c.2148 C > G,
p.Asn716Lys) and has not been reported as a pathogenic variant; it was found to have an extremely low allele frequency (1.59 × 10−6) in the Genome Aggregation Database
(http://gnomad.broadinstitute.org). This variant was predicted to be deleterious by SIFT (score 0.04) and disease-causing by MutationTaster (prob 0.999). Both variants were confirmed by
Sanger sequencing; the p.Gly259Asp and p.Asn716Lys variants were found to be maternally and paternally inherited, respectively. The unaffected younger sister had a heterozygous p.Gly259Asp
variant inherited maternally (Fig. 1C). We evaluated the pathogenicity of these two variants in accordance with the 2015 guidelines of the American College of Medical Genetics and Genomics.
The c.776 G > A, p.Gly259Asp and c.2148 C > G, p.Asn716Lys variants were classified as pathogenic and likely pathogenic, respectively. At the age of 20 years, the proband had severe
cognitive function (estimated intelligence quotient: ~30) and moderate visual impairment. She walked on her toes with spastic scissor gait and required a walking aid. Mitochondrial ACO2 is a
critical enzyme in the TCA cycle, which is the primary source of cellular metabolic energy1. Other TCA enzymopathies, such as deficiencies of alpha-ketoglutarate dehydrogenase, fumarase,
succinate dehydrogenase, and succinyl-CoA synthase, have been previously reported to cause severe encephalopathy with muscle hypotonia, developmental delay, and retinitis
pigmentosa7,8,9,10,11. The underlying pathophysiological mechanism may involve a disruption of energy metabolism and oxidative phosphorylation7. Therefore, aconitase deficiency is also
thought to disrupt cellular energy metabolism; consistent with this, a study reported mitochondrial dysfunction in the fibroblasts of an aconitase-deficient patient12. Aconitase enzymopathy
is more difficult to diagnose than other TCA enzymopathies. One reason is the poor abnormalities in metabolic screening samples. An elevation of lactate levels in the blood and CSF and of
specific organic acids in the urine, which is typically detected in other TCA enzymopathies, is not observed in aconitase enzymopathy. Another reason is the broad clinical spectrum of
_ACO2-_related disorders, ranging from isolated optic atrophy to syndromic optic atrophy, such as ICRD that involves hypotonia, retinal degeneration, severe encephalopathy, epilepsy, and
cerebellar ataxia6. Moreover, new phenotypes associated with spastic paraplegia were recently reported, including in this study. Table 1 shows the clinical manifestations of _ACO2_-related
disorders (optic atrophy 9, ICRD, and complex HSP)4,5,6,13. Most patients with _ACO2_-related disorders have optic nerve involvement, which might be a hallmark feature of aconitase
enzymopathy, but not all patients have optic atrophy4,12. Thus, biochemical testing and clinical phenotypes are insufficient for diagnosing aconitase enzymopathy, thereby indicating the
importance of WES. Figure 1D shows the structure of the ACO2 protein and reported variants in _ACO2_-related disorders, including in this study. There is no hot-spot region in any
_ACO2_-related disorder, and there seems to be poor genotype–phenotype correlation. Compared to previously reported _ACO2_-related disorders, the novel characteristic phenotype in the
present patient was episodic visual loss during febrile infection (Table 1). The phenotypes in the present patient indicated that ACO2 plays a crucial role in energy production in the optic
nerve and retina, which are highly energy-dependent structures14. Previous findings suggested that phenotype variation and severity depend on residual aconitase enzymatic activity12.
Metodiev et al. reported a very severe case involving homozygous Gly259Asp variants6. The patient presented with syndromic optic neuropathy along with encephalopathy and cerebellar atrophy
and died at 57 days because of central apnea6. The aconitase enzymatic activity in the patient’s fibroblasts was extremely low (~5%)6. However, a case report of a mild phenotype despite a
marked reduction in aconitase enzyme activity12 suggested poor genotype–phenotype correlations in _ACO2_ variants and the coexistence of genomic modifiers. HSP is not a single disease;
rather, it is a mixture of genetically heterogeneous conditions resulting in broadly overlapping clinical phenotypes. Using single-gene direct sequencing and next-generation sequencing
technologies, various HSP-related gene variants have been identified15. These genes encode proteins with diverse molecular functions, axonal transport, specific lipid metabolism, synaptic
formation, axon development, and mitochondrial function15. In addition to _ACO2_ variants, several other HSP-associated gene variants, such as those in _PGN_, _HSPD1_, _DDHD1_, _REEP1_, and
_MT-ATP6_, have been found to impair mitochondrial function16,17,18,19,20. Further reports on the causative genes of HSP would improve the understanding of the crucial role of mitochondrial
dysfunction in HSP pathogenesis. In conclusion, this case represents the third report of HSP caused by pathogenic _ACO2_ variants. Although most patients with _ACO2_-related disorders
present with muscular hypotonia features, it should be recognized that pathogenic _ACO2_ variants comprise one of the causes of complex HSP. Patients with _ACO2_-related disorders should be
evaluated for signs such as early-onset spastic paraplegia, especially those with episodic visual loss after febrile infection and progressive optic atrophy. The identification of pathogenic
_ACO2_ variants in patients with HSP could contribute to the development of specific therapies against HSP caused by mitochondrial dysfunction. HGV DATABASE The relevant data from this Data
Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.2951 and https://doi.org/10.6084/m9.figshare.hgv.2954. REFERENCES * Beinert, H. &
Kennedy, M. C. Aconitase, a two-faced protein: enzyme and iron regulatory factor. _FASEB J._ 7, 1442–1449 (1993). Article CAS Google Scholar * Spiegel, R. et al. Infantile
cerebellar-retinal degeneration associated with a mutation in mitochondrial aconitase, _ACO2_. _Am. J. Hum. Genet._ 90, 518–523 (2012). Article CAS Google Scholar * Fukada, M. et al.
Identification of novel compound heterozygous mutations in ACO2 in a patient with progressive cerebral and cerebellar atrophy. _Mol. Genet. Genom. Med._ 7, e00698 (2019). Google Scholar *
Bouwkamp, C. G. et al. _ACO2_ homozygous missense mutation associated with complicated hereditary spastic paraplegia. _Neurol. Genet_. 4, e223 (2018). Article CAS Google Scholar *
Marelli, C. et al. _ACO2_ mutations: a novel phenotype associating severe optic atrophy and spastic paraplegia. _Neurol. Genet._ 4, e225 (2018). Article CAS Google Scholar * Metodiev, M.
D. et al. Mutations in the tricarboxylic acid cycle enzyme, aconitase 2, cause either isolated or syndromic optic neuropathy with encephalopathy and cerebellar atrophy. _J. Med. Genet._ 51,
834–838 (2014). Article CAS Google Scholar * Munnich, A. Casting an eye on the Krebs cycle. _Nat. Genet_. 40, 1148–1149 (2008). Article CAS Google Scholar * Odièvre, M. H. et al. A
novel mutation in the dihydrolipoamide dehydrogenase E3 subunit gene (DLD) resulting in an atypical form of alpha-ketoglutarate dehydrogenase deficiency. _Hum. Mutat._ 25, 323–324 (2005).
Article Google Scholar * Bourgeron, T. et al. Mutation of the fumarase gene in two siblings with progressive encephalopathy and fumarase deficiency. _J. Clin. Invest._ 93, 2514–2518
(1994). Article CAS Google Scholar * Ostergaad, E. et al. Deficiency of the alpha subunit of succinate-coenzyme A ligase causes fatal infantile lactic acidosis with mitochondrial DNA
depletion. _Am. J. Hum. Genet_. 81, 383–387 (2007). Article Google Scholar * Elpeleg, O. et al. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with
encephalomyopathy and mitochondrial DNA depletion. _Am. J. Hum. Genet_. 76, 1081–1086 (2005). Article CAS Google Scholar * Sadat, R. et al. Functional cellular analyses reveal energy
metabolism defect and mitochondrial DNA depletion in a case of mitochondrial aconitase deficiency. _Mol. Genet. Metab._ 118, 28–34 (2016). Article CAS Google Scholar * Srivastava, S. et
al. Increased survival and partly preserved cognition in a patient with ACO2-related disease secondary to a novel variant. _J. Child. Neurol._ 32, 840–845 (2017). Article Google Scholar *
Barot, M., Gokulgandhi, M. R. & Mitra, A. K. Mitochondrial dysfunction in retinal diseases. _Curr. Eye Res._ 36, 1069–1077 (2011). Article CAS Google Scholar * Fink, J. K. Hereditary
spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. _Acta Neuropathol._ 126, 307–328 (2013). Article CAS Google Scholar * Atorino, L. et al. Loss of m-AAA
protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. _J. Cell Biol._ 163, 777–787 (2003). Article CAS Google
Scholar * Bross, P. et al. The Hsp60-(p.V98 I) mutation associated with hereditary spastic paraplegia SPG13 compromises chaperonin function both in vitro and in vivo. _J. Biol. Chem._ 283,
15694–15700 (2008). Article CAS Google Scholar * Bouslam, N. et al. Mapping of a new form of pure autosomal recessive spastic paraplegia (SPG28). _Ann. Neurol._ 57, 567–571 (2005).
Article CAS Google Scholar * Tesson, C. et al. Alteration of fatty-acid-metabolizing enzymes affects mitochondrial form and function in hereditary spastic paraplegia. _Am. J. Hum. Genet_
91, 1051–1064 (2012). Article CAS Google Scholar * Verny, C. et al. Hereditary spastic paraplegia-like disorder due to a mitochondrial ATP6 gene point mutation. _Mitochondrion_ 11, 70–75
(2011). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank the patient and her family members for their cooperation in this study. This work was supported by the
Japan Agency for Medical Research and Development (grant number JP19ek0109301). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Pediatrics, Graduate School of Medical Science,
Kyoto Prefectural University of Medicine, Kyoto, Japan Takenori Tozawa, Yoshihiro Taura & Tomohiro Chiyonobu * Department of Pediatrics, Ayabe City Hospital, Ayabe, Japan Takenori Tozawa
& Tamaki Ueno * Department of Neonatology, Japanese Red Cross Society Kyoto Daiichi Hospital, Kyoto, Japan Akira Nishimura * Department of Pediatrics, Tokai Central Hospital,
Kakamigahara, Japan Tamaki Ueno * Kyoto Prefectural Maizuru Rehabilitation Center for Children, Maizuru, Japan Akane Shikata * Department of Pediatrics, Kyoto University Graduate School of
Medicine, Kyoto, Japan Takeshi Yoshida * Department of Medical Ethics/Medical Genetics, Kyoto University School of Public Health, Kyoto, Japan Naoko Nakagawa, Takahito Wada & Shinji
Kosugi * Center for Medical Genetics, Keio University School of Medicine, Tokyo, Japan Tomoko Uehara & Kenjiro Kosaki * Department of Pediatrics, Keio University School of Medicine,
Tokyo, Japan Toshiki Takenouchi Authors * Takenori Tozawa View author publications You can also search for this author inPubMed Google Scholar * Akira Nishimura View author publications You
can also search for this author inPubMed Google Scholar * Tamaki Ueno View author publications You can also search for this author inPubMed Google Scholar * Akane Shikata View author
publications You can also search for this author inPubMed Google Scholar * Yoshihiro Taura View author publications You can also search for this author inPubMed Google Scholar * Takeshi
Yoshida View author publications You can also search for this author inPubMed Google Scholar * Naoko Nakagawa View author publications You can also search for this author inPubMed Google
Scholar * Takahito Wada View author publications You can also search for this author inPubMed Google Scholar * Shinji Kosugi View author publications You can also search for this author
inPubMed Google Scholar * Tomoko Uehara View author publications You can also search for this author inPubMed Google Scholar * Toshiki Takenouchi View author publications You can also search
for this author inPubMed Google Scholar * Kenjiro Kosaki View author publications You can also search for this author inPubMed Google Scholar * Tomohiro Chiyonobu View author publications
You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Takenori Tozawa. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare that they
have no conflict of interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and
reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes
were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If
material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain
permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Tozawa, T., Nishimura, A., Ueno, T. _et al._ Complex hereditary spastic paraplegia associated with episodic visual loss caused by _ACO2_ variants. _Hum Genome Var_ 8, 4 (2021).
https://doi.org/10.1038/s41439-021-00136-y Download citation * Received: 13 November 2020 * Revised: 15 December 2020 * Accepted: 15 December 2020 * Published: 26 January 2021 * DOI:
https://doi.org/10.1038/s41439-021-00136-y SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative







