- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Hypomagnesemia 1 (HOMG1) is an extremely rare disease with autosomal recessive inheritance that is caused by mutations in the transient receptor potential melastatin 6 gene
(_TRPM6_). Here, we describe a pediatric HOMG1 case with novel compound heterozygous mutations of _TRPM6_ (c.1483 C > T [p.Gln495*] and c.2715del [p.Trp905*]) in a 2-month-old boy who
developed refractory seizures due to hypomagnesemia with secondary hypocalcemia. CASE REPORT Hypomagnesemia 1 (HOMG1: OMIM # 602014) is a rare autosomal recessive disease characterized by a
high reduction of serum magnesium levels and is often accompanied by secondary hypocalcemia. In general, HOMG1 patients develop generalized convulsions, tetany, or neuromuscular excitability
in early infancy1. The first case report of an HOMG1 patient was described by Paunier et al.2. Schlingmann et al. reported that mutations in the transient receptor potential melastatin 6
gene (_TRPM6_), which is located on chromosome 9q22, are responsible for HOMG13. The TRPM6 protein has a crucial role in magnesium metabolism in human and mammals. Magnesium absorption
predominantly occurs in the small intestine. The TRPM6 protein is an epithelial magnesium channel expressed along the brush border membrane of the small intestine. Intestinal absorption of
magnesium occurs according to paracellular simple diffusion via concentration gradient and active transcellular uptake through the magnesium transporter of the TRPM6 protein at a low
concentration of magnesium. Moreover, reabsorption of magnesium occurs in the kidney. The TRPM6 protein also plays an essential role in the apical influx of magnesium ion in the distal
convoluted tubule cells4. Impaired channel activity of the TRPM6 protein caused by _TRPM6_ mutations induces hypomagnesemia in infancy. Several mutations have been reported in families with
HOMG1. Here, we describe a pediatric case of HOMG1 with novel compound heterozygous mutations. A 2-month-old boy suffering from refractory seizures was referred to our hospital. He was born
to non-consanguineous Japanese parents at a gestational age of 39 weeks after an uneventful pregnancy and delivery. Birth weight and birth height were 3342 g (+1.1 SD) and 50.5 cm (+0.9 SD),
respectively. There were neither epileptic disorders nor electrolyte disorders in his family history. On admission, we found hypocalcemia (serum ionic calcium, 3.16 mg/dL; normal range,
4.60–5.17 mg/dL) and hypomagnesemia (serum magnesium 0.6 mg/dL; normal range, 1.8–2.4 mg/dL). Initial data of fractional excretion of magnesium (FEMg) was 0.09% (normal range, 3–5% for
normomagnesemic individuals). Serum inorganic phosphate was 6.9 mg/dL (normal range, 4.5–6.2 mg/dL), and alkaline phosphatase was 1290 U/L (normal range, 480–1620 U/L). Although serum intact
parathyroid hormone (PTH) was 80 pg/mL (normal range, 10–65 pg/ml) and 1,25-dihydroxyvitamin D3 was 85 pg/mL (normal range, 20–70), serum 25-hydroxyvitamin D3 was insufficient (4.1 ng/mL;
normal range, 10–30 ng/mL). Brain MRI and cerebrospinal fluid examination revealed no abnormalities. Replacement therapy with intravenous infusion of calcium gluconate and magnesium sulfate
was successfully performed to suppress the seizures. Calcium infusion therapy was discontinued when normal calcium levels were achieved. FEMg was elevated up to 4.93% during magnesium
intravenous infusion, and serum magnesium was 1.6 mg/dL. He was discharged after switching from intravenous magnesium replacement to oral magnesium administration (magnesium oxide, 90
mg/kg/day). The Ellsworth–Howard test disclosed a normal response of urinary cAMP excretion to PTH stimulation (urinary cAMP level: before PTH injection, 570 pmol/mL; 1 h after PTH
injection, 99,000 pmol/L). These results do not suggest the presence of pseudohypoparathyroidism. FEMg values ranged from 0.16 to 1.88% (median, 0.76%; normal range, 3–5% for normomagnesemic
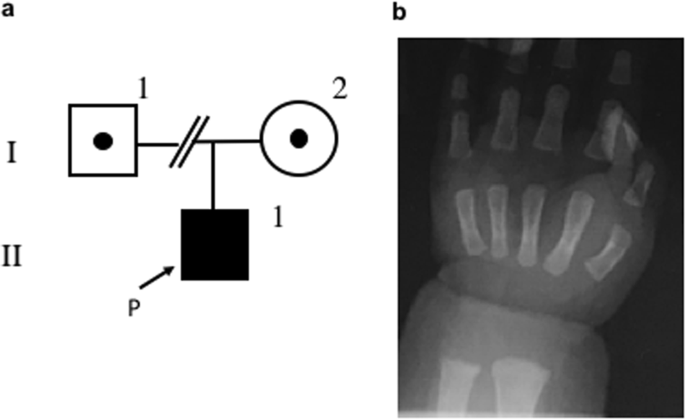
individuals). The urinary calcium/creatinine ratio ranged from 0.01 to 0.23 (median, 0.1; normal range, <0.81). A roentgenogram showed a cupping lesion in the distal ulnar (Fig. 1).
Since oral alfacalcidol (a derivative of vitamin D3) treatment was not able to maintain serum ionic calcium concentration within the normal range, we discontinued it. At an age of 1 year,
only oral magnesium treatment (magnesium oxide, 45–90 mg/kg/day) was successful in maintaining serum 25-hydroxyvitamin D3 levels within the normal range without vitamin D3 supplementation.
We concluded that nutritional vitamin D3 deficiency was not the primary reason for hypocalcemia. However, hypomagnesemia recurred with reduction of oral magnesium administration (magnesium
oxide, 20 mg/kg/day). Serum ionic calcium levels varied between 4.41 and 5.24 mg/dL, and magnesium levels were maintained between 1.3 and 1.7 mg/dL with treatment. At an age of 1 year and 8
months, he showed age-appropriate physical development and achieved developmental milestones. There were no adverse events of magnesium administration. We performed molecular genetic
analysis on the proband and his mother after obtaining written informed consent from his mother. Genomic DNA was extracted from peripheral blood samples. The father’s blood sample was not
taken because we could not contact the divorced father. Inherited hypomagnesemia-responsible genes including _TRPM6, CLCNKB, BSND, SLC12A3, CASR, KCNJ10, CLDN16, CLDN19, FXYD2, EGF, KCNA1,
CNNM2_, and _HNF1B_ were screened by next-generation sequencing analysis with targeting sequencing5,6. Finally, Sanger sequencing confirmed compound heterozygous mutations (NM_017662.5
(TRPM6_v001): c.1483C>T [p.Gln495*] and NM_017662.5 (TRPM6_v001): c.2715del [p.Trp905*]) in _TRPM6_ (Fig. 2). p.Gln495* was also detected in his mother’s genome. These two novel mutations
have not been documented in several open databases, including ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/), ExAc
(http://exac.broadinstitute.org/about), and the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php). We used the GenBank transcript ID (NM_017662.5) as a reference to
examine the pathological significance of the mutations. We have described a pediatric case with novel compound heterozygous mutations of _TRPM6_, two of which also introduced a stop codon,
namely the codon TGA/TAA. The mutations in the present case are predicted to cause premature termination of _TRPM6_ coding. Theoretically, the mutated TRPM6 proteins in the present case are
associated with loss of biological channel activity. _TRPM6_ comprises 39 exons and encodes a large protein of 2022 amino acids7. The TRPM6 protein consists of six transmembrane segments as
well as N-terminal and C-terminal domains. The p.Gln495 position of the amino acid sequence in the TRPM6 protein is located in the intracellular N-terminal domain. The p.Gln495* mutation
predicts a truncated version of the TRPM6 protein that does not contain part of the magnesium-ion gating channel. This prediction suggests a loss-of-function mutation. However, the p.Trp905*
mutation due to a frameshift nucleotide change (c.2715del) is located in the cytoplasmic loop of the TRPM6 protein. The TRPM6 protein that results from the p.Trp905*mutation is also
predicted to be non-functional. In addition, we found that the two mutations were novel mutations according to databases. HOMG1 patients primarily exhibit impairment of intestinal absorption
of magnesium3. Indeed, the initial value of FEMg on admission was extremely low in the current patient. FEMg increased to nearly the upper limit of the normal range when intravenous
infusion of magnesium was continued. These findings suggested that renal magnesium reabsorption was also impaired in the patient. However, magnesium leakage in the kidney will lead to
difficulty in achieving normal magnesium levels even if adequate supplemental treatment of magnesium is given orally in patients with HOMG1, as was observed in the present patient. The index
patient is a sporadic case of HOMG1 with a typical clinical presentation including refractory seizures. In general, magnesium greatly contributes to PTH release through cAMP elevation by
adenylate cyclase activation in the presence of magnesium in the parathyroid gland8. Therefore, secondary hypocalcemia subsequently occurs in patients with HOMG19. In that context, it is a
clinical observation of great interest that serum intact PTH was elevated in the current case, although previous HOMG1 cases have shown inappropriately low levels of serum intact PTH despite
hypocalcemia10. In the current case, the initial data of a low level of serum 25-hydroxyvitamin D3 and roentgenogram findings supported a diagnosis of calcipenic rickets induced by vitamin
D3 deficiency. Moreover, we considered that serum calcium levels were dependent on magnesium administration in the current case. With respect to vitamin D3 metabolism, magnesium plays an
essential role in the activation of 25-hydroxylase, which converts vitamin D3 to 25-hydroxyvitamin D311. We speculated that magnesium deficiency could lead to secondary dysregulation of
vitamin D3 metabolism. HGV DATABASE The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.2540 REFERENCES *
Schlingmann, K. P. et al. Novel TRPM6 mutations in 21 families with primary hypomagnesemia and secondary hypocalcemia. _J. Am. Soc. Nephrol._ 16, 3061–3069 (2005). Article Google Scholar *
Paunier, L., Radde, I. C., Kooh, S. W., Conen, P. E. & Fraser, D. Primary hypomagnesemia with secondary hypocalcemia in an infant. _Pediatrics_ 41, 385–402 (1968). CAS PubMed Google
Scholar * Schlingmann, K. P. et al. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. _Nat. Genet_ 31, 166–170 (2002).
Article CAS Google Scholar * Knoers, N. V. Inherited forms of renal hypomagnesemia: an update. _Pediatr. Nephrol._ 24, 697–705 (2009). Article Google Scholar * Horinouchi, T. et al.
Diagnostic strategy for inherited hypomagnesemia. _Clin. Exp. Nephrol._ 21, 1003–1010 (2017). Article CAS Google Scholar * Nozu, K. et al. Cryptic exon activation in SLC12A3 in Gitelman
syndrome. _J. Hum. Genet_ 62, 335–337 (2017). Article CAS Google Scholar * Schlingmann, K. P., Waldegger, S., Konrad, M., Chubanov, V. & Gudermann, T. TRPM6 and TRPM7-Gatekeeper of
human magnesium metabolism. _Biochim. Biophys. Acta_ 1772, 813–821 (2007). Article CAS Google Scholar * Grubbs, R. D. & Maguire, M. E. Magnesium as a regulatory cation: criteria and
evaluation. _Magnesium_ 6, 113–127 (1987). CAS PubMed Google Scholar * Anast, C. S., Mohs, J. M., Kaplan, S. L. & Burns, T. W. Evidence for parathyroid failure in magnesium
deficiency. _Science_ 177, 606–608 (1972). Article CAS Google Scholar * Schlingmann, K. P., Konrad, M. & Seyberth, H. W. Genetics of hereditary disorders og magnesuium homeostasis.
_Pediatr. Nephrol._ 19, 12–25 (2004). Article Google Scholar * Deng, X. et al. Magnesium, vitamin D status and mortality: results from US National Health and Nutrition Examination Survey
(NHANES) 2001 to 2006 and NHANES III. _BMC Med._ 11, 187 (2013). Article Google Scholar Download references AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Pediatrics, National
Hospital Organization Maizuru Medical Center, Kyoto, Japan Takeshi Goda, Hiroshi Komatsu & Hisakazu Nakajima * Department of Pediatrics, Kobe University Graduate School of Medicine,
Hyogo, Japan Kandai Nozu * Department of Pediatrics, North Medical Center Kyoto Prefectural University of Medicine, Kyoto, Japan Hisakazu Nakajima Authors * Takeshi Goda View author
publications You can also search for this author inPubMed Google Scholar * Hiroshi Komatsu View author publications You can also search for this author inPubMed Google Scholar * Kandai Nozu
View author publications You can also search for this author inPubMed Google Scholar * Hisakazu Nakajima View author publications You can also search for this author inPubMed Google Scholar
CORRESPONDING AUTHOR Correspondence to Hisakazu Nakajima. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare that they have no conflict of interest. ADDITIONAL INFORMATION
PUBLISHER’S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. RIGHTS AND PERMISSIONS OPEN ACCESS This article is
licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in
this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative
Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a
copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Goda, T., Komatsu, H., Nozu, K. _et al._ A pediatric
case of hypomagnesemia 1 (HOMG1) caused by novel compound heterozygous mutations in _TRPM6_. _Hum Genome Var_ 6, 13 (2019). https://doi.org/10.1038/s41439-019-0043-0 Download citation *
Received: 24 January 2019 * Revised: 31 January 2019 * Accepted: 01 February 2019 * Published: 06 March 2019 * DOI: https://doi.org/10.1038/s41439-019-0043-0 SHARE THIS ARTICLE Anyone you
share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the
Springer Nature SharedIt content-sharing initiative