- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Macrophages are crucial members of the innate immune response and important regulators. The differentiation and activation of macrophages require the timely regulation of gene
expression, which depends on the interaction of a variety of factors, including transcription factors and epigenetic modifications. Epigenetic changes also give macrophages the ability to
switch rapidly between cellular programs, indicating the ability of epigenetic mechanisms to affect phenotype plasticity. In this review, we focus on key epigenetic events associated with
macrophage fate, highlighting events related to the maintenance of tissue homeostasis, responses to different stimuli and the formation of innate immune memory. Further understanding of the
epigenetic regulation of macrophages will be helpful for maintaining tissue integrity, preventing chronic inflammatory diseases and developing therapies to enhance host defense. SIMILAR
CONTENT BEING VIEWED BY OTHERS DOES TISSUE IMPRINTING RESTRICT MACROPHAGE PLASTICITY? Article 18 January 2021 REPRESSION OF CTSG, ELANE AND PRTN3-MEDIATED HISTONE H3 PROTEOLYTIC CLEAVAGE
PROMOTES MONOCYTE-TO-MACROPHAGE DIFFERENTIATION Article 20 May 2021 HISTONE DEMETHYLASE KDM5B LICENSES MACROPHAGE-MEDIATED INFLAMMATORY RESPONSES BY REPRESSING _NFKBIA_ TRANSCRIPTION Article
13 March 2023 INTRODUCTION Macrophages are essential phagocytes of the innate immune system and are present in all organs and tissues of the body as resident cells or the differentiation
products of recruited blood monocytes.1 Macrophages play a critical role in both the maintenance of tissue homeostasis and the regulation of inflammation, completing necessary
tissue-specific functions as well as protecting the organism from infection.2,3 In addition, macrophages are major producers of cytokines, which are implicated in the pathogenesis of
inflammatory disease, and are strategically placed to orchestrate both innate immune responses and adaptive immune responses.4,5 Macrophages display high heterogeneity, not only in their
inherent terminal differentiation pathway but also in their different responses to different environmental stimuli. Both tissue-resident macrophages in homeostasis and activated macrophages
under stimuli are driven by specific transcriptional changes and are controlled by complex cellular mechanisms. Among them, epigenetics now arises as a key controller of macrophage activity.
EPIGENETIC MODIFICATIONS Epigenetics regulates the expression of DNA-encoded information and determines the specific “identity” of a cell while the genetic code is not altered. The
chromatin conformation controls transcription factor binding and gene expression through DNA accessibility. Epigenetic markers have traditionally been considered stable and heritable.6
However, it has been revealed that epigenetic chromatin markers are dynamically regulated with changes in development or environmental signals.7 These results have led to additional
information regarding epigenetics, including a transient change in chromatin in response to external stimuli that control gene expression. Currently, most of the epigenetic mechanisms are
related to the protein−DNA interactions that affect gene expression, whether through the recruitment of protein complexes involved in nucleosome modification and remodeling or by chemical
modification of DNA bases, such as 5-methylcytosine. Additionally, there are other epigenetic mechanisms, such as the production of small noncoding RNAs, that can regulate mRNA in
transcription through base pairing, resulting in mRNA degradation or translational inhibition. Overall, epigenetic changes can be divided into three categories: (1) posttranscriptional
histone modifications, (2) DNA methylation, and (3) noncoding RNA. POSTTRANSCRIPTIONAL HISTONE MODIFICATIONS ASSOCIATED WITH CHROMATIN STRUCTURE AND FUNCTION In eukaryotes, approximately 147
bases of DNA wrap around a histone octamer to form a nucleosome.8 The octamer, composed of two groups of H2A, H2B, H3 and H4 proteins, is the fundamental unit of chromatin, and histone H1
connects the two nucleosomes. When nucleosomes are organized into tightly arranged bundles (heterochromatin), transcription is inhibited by restricting the entry of transcriptional
machinery. In contrast, when the chromatin is relaxed (euchromatin), the nucleosomes are arranged like a string of beads, predominantly associated with active transcription. Nucleosome-free
regions are known as open chromatin, which contain a variety of functional sequences, such as promoters and enhancers, and thus play a vital role in gene regulation.9 Promoters are short
sequences of approximately 100 bases proximal to the transcription start sites at the 5′ ends of genes. A common feature of promoters is that RNA polymerase II binds to promoters of inactive
genes in advance. When stimulated, promoters can recruit more polymerase II to initiate transcription.10 Enhancers are distal transcriptional regulatory elements that are defined as genomic
DNA elements, ranging from several to hundreds and, in rare cases, even thousands of bases in length, and contain short transcription factor (TF) recognition sequences or binding sites. In
these regulatory regions, the accessibility of DNA by protein TFs and other transcriptional mechanisms is critical for gene expression. The interplay and long-range communication of
enhancers and target gene promoters mediated by DNA looping tightly regulate gene expression. Disordered N-terminal histone tails extending from nucleosomes are substrates for a variety of
posttranscriptional enzymatic modifications. Amid a vast repertoire of histone modifications, acetylation/deacetylation and methylation/demethylation of lysine residues belong to the most
broadly studied and extensively characterized macrophage-related epigenetic modifications of posttranslational histone modification. Determined by the opposing activities of enzymatic
writers and erasers, the presence of histone modifications on the chromatin is reversible. Histone acetyltransferases promote acetylation, which is related to transcriptional activity, such
as H3K9ac and H3K27ac, whereas histone deacetylases (HDACs), the erasers, catalyze the removal of acetyl groups from histone tails, which is linked to transcriptional inhibition.11 Histone
acetylation markers are specifically recognized and bound by bromodomains, a structural motif present in specialized chromatin reader proteins that reads the histone acetylation code, and
then transcriptional mechanisms are initiated.12 Many histone acetyltransferase complexes contain a component with a bromodomain that anchors histone acetyltransferase complexes on the
already acetylated chromatin, enabling them to propagate acetyl labels to adjacent nucleosomes, hence spreading histone acetylation. Similarly, the SWI/SNF complex, the first described
ATP-dependent chromatin remodeling complex, targets the acetylated histone tails through the bromodomain subunit, which is critical for recruitment.13 Another group of histone acetyl
readers, known as the bromo- and extraterminal (BET) protein family members, can recruit TFs and chromatin remodeling complexes to regulate gene expression, and they have been demonstrated
to play a key role in the regulation of inflammatory gene expression in macrophages.14 Similar to histone acetylation, the subsequent methylation and demethylation of histones are promoted
by histone methyltransferases and histone demethylases, respectively. Specifically, every site-specific mark of lysine within the tail of H3 is written by a group of specific enzymes called
histone lysine methyltransferases (KMTs) and erased by other enzymes classified as histone lysine demethylases. Unlike acetylation, methylation modification of histone can induce
transcriptional activation or inhibition, depending on the location and number of methyl groups within the histone tail. Regulatory elements on repressed genes, such as methylation at the
H3K9 and H3K27 loci, often lead to “gene silence” and induce the heterochromatin state in chromatin regions. Conversely, methylation at H3K4 and H3K36 is associated with the assembly of
transcriptionally permissive chromatin structures and active transcription at many loci.15 Furthermore, the extent of individual lysine methylation, including the mono- (m1), di- (m2), or
trimethylation (m3) of histone lysine residues, is differentially distributed in chromatin and plays different roles in gene regulation. Transcriptional activity is correlated positively
with the trimethylation of H3 histone lysine 4 (H3K4m3) at gene promoters.16 On the other hand, the monomethylation of this residue (H3K4m1) is a typical feature of enhancers.17 DNA
METHYLATION IN THE REGULATION OF GENE EXPRESSION In addition to histones, methyl modification of DNA is a unique mechanism in epigenetic regulation. In mammals, most DNA methylation occurs
in the 5′-cytosine-phosphate-guanine-3′ dinucleotide (CpG) dinucleotide clusters named CpG islands, which are found in the promoters of approximately 40% of the genes, and greater than 70%
of CpG sites are methylated in the DNA of somatic cells.18 DNA methylation is mainly related to transcriptional inhibition, characterized by 5-methylcytosine immediately adjacent to guanine
residues, and involves the transfer of a methyl group to the cytosine ring of DNA through DNA methyltransferases (DNMTs).19 Mechanically, 5-methylcytosine binding proteins induce the
recruitment of repressor complexes to methylated promoter segments and cause transcriptional silencing.18 In particular, DNMT3A and DNMT3B are responsible for establishing de novo
methylation markers, while DNMT1 is the maintenance methyltransferase responsible for maintaining these markers, as these markers at CpG dinucleotides must be reestablished after DNA
replication in each cell division.20 DNMT1 can also participate in the regulation of histone modifications that result in a depletion of di- (H3K9me2) and trimethylation (H3K9me3) at H3K9
and a concomitant increase in H3K9 acetylation (H3K9ac).21 Recently, the methylation of non-CpG moieties has been found to occur widely on genomic DNA.22 The abundant non-CpG methylations of
DNA directly affect the binding of TFs through the methylation of TF binding sites, which provide a mechanism for regulating gene expression.22 NONCODING RNA IN POSTTRANSCRIPTIONAL
REGULATION Finally, endogenously expressed noncoding RNAs, including long noncoding RNAs (lncRNAs) and microRNAs (miRNAs), play critical roles in the posttranscriptional regulation of gene
expression by acting as competing RNAs, although they do not directly affect chromatin structure. lncRNAs are a large group of nonprotein-coding transcripts with a length of more than 200
nucleotides. XIST, one of the best characterized lncRNAs, was found to drive X-chromosome inactivation in 1991.23 Thereafter, thousands of lncRNAs have been found in different cells and to
be involved in a variety of diseases. Although the sources of these lncRNAs are different, their functional mechanisms are similar. A number of lncRNAs can directly interact with
chromatin-modifying enzymes and remodeling complexes to guide these epigenetic catalysts to specific parts of the chromatin.24 In addition, some lncRNAs can form RNA−protein complexes with
transcription factors and affect the location and activity of transcription factors binding to them, thereby regulating gene expression.25 Specific lncRNAs play a part in the programming of
silencing or activating histone modifications. For example, HOTAIR, a 2.2 kilobase lncRNA in the HOXC locus, is involved in silencing chromatin mediated by polycomb repressive complex 2
(PRC2, comprised of SUZ12, EED, and the lysine methyltransferase EZH2) and can recruit PRC2 to its target gene and methylate H3K27 via EZH2.26 Furthermore, HOTAIR has been found to be a
scaffold that targets genes in at least two different histone modification complexes.27 The 5′ domain of HOTAIR binds to PRC2, and the 3′ domain of HOTAIR binds to the CoREST/REST repressor
complexes, which contain LSD1 (KDM1/BHC110), a demethylase that mediates the enzymatic demethylation of H3K4me2. The ability to link two distinct complexes makes it possible to assemble PRC2
and LSD1 and coordinate the complexes targeting chromatin to achieve the methylation of H3K27 and the demethylation of H3K4.27 In addition, another typical example is the lncRNA ANRIL, an
antisense RNA transcript overlapping the INK4b/ARF/INK4a tumor suppressor locus, which participates in the _cis_ recruitment of both PRC1 and PRC2 to the target gene for silencing.28 Another
class of noncoding RNAs, miRNAs, which are single-stranded RNAs containing 20−24 bases. It is estimated that more than 60% of all protein-coding genes are directly regulated by miRNAs. They
can specifically bind to the 3′ or 5′ untranslated regions of mRNA, mainly as posttranscriptional inhibitors, by targeting the 3′ untranslated regions of the RNA to stimulate its
degradation and translation repression.29 In addition, a specific miRNA may bind to and regulate several targets, sometimes as part of the same signaling pathway, adding multiple regulatory
levels. Therefore, under pathophysiological stimulation, miRNAs can fine-tune gene expression patterns. CROSSTALK MECHANISMS Based on existing research, mechanisms of crosstalk among the
different epigenetic regulation systems have been revealed. They interact with each other rather than working independently. For example, histone lysine is the target of both methylation and
acetylation, such as H3K9 and H3K27. At least two enzymatic steps are required to shift from the inhibited methylated state to the activated acetylated state (and vice versa). Therefore, it
is believed that the state between the opposite nucleosome-modifying activities is altered by sequence-specific DNA-binding TFs that recruit specific chromatin remodeling agents and
modifying factors to individual loci, and crosstalk between them occurs, leading to their combinatorial effects in transcriptional control.30 Histone and DNA modification, together with
lncRNAs and miRNAs, are collectively defined as epigenetic mechanisms. EPIGENETIC MECHANISMS OF MACROPHAGES IN TISSUE HOMEOSTASIS ORIGIN OF MACROPHAGES In 1968, it was presumed that
macrophages were derived only from blood monocytes, and this concept prevailed for approximately half a century.31 Accumulating evidence, however, has demonstrated that the different
macrophages do not necessarily have the same origins. With few notable exceptions, the tissue macrophages do not arise from blood monocytes under homeostasis and even in some kinds of
inflammation; those tissue macrophages are seeded from embryonic precursors of the yolk sac macrophages and fetal monocytes prior to birth and maintain themselves throughout adulthood by
self-renewal and participate in tissue remodeling. Erythro-myeloid progenitors, derived from the yolk sac, have been found to be the embryonic precursors of yolk sac macrophages and fetal
monocytes before the emergence of hematopoietic stem cells (HSCs); thus, they are a common origin for tissue macrophages.32 Two sequential waves of erythro-myeloid progenitors were
identified to generate macrophages. An early myeloid-restricted hematogenic wave originates in the yolk sac, generates primitive macrophages without monocytic intermediates and is
independent of the transcription factor c-Myb.33 These cells either directly generate primitive yolk sac macrophages as the sole origin of tissue-resident macrophages, such as microglia, or
migrate to the fetal liver and give rise to fetal monocytes that proliferate in the embryonic tissues and differentiate into distinct tissue macrophages.34 Alternatively, a late
c-Myb-dependent wave that commences in the fetal liver to generate multiple hematopoietic lineages, including monocytic intermediates, persists throughout adult life.35 Fetal monocytes
differentiate into macrophages when recruited to fetal tissues, replacing the main components of the existing yolk-sac-derived macrophages gradually with the exception of microglia.35 With
the growth of host tissues, these primitive macrophages differentiate through proliferating in their respective tissue macrophage compartments in most tissues, except the intestine, the
heart, and the skin. Therefore, a model of macrophage origination has been established, which is distinct from the model that depends only on blood monocytes. ROLE OF THE LINEAGE-DETERMINING
FACTOR PU.1 AND ENHANCERS IN MACROPHAGES Tissue-resident macrophages are an extremely heterogeneous population, which is a necessary outcome of lineage- and tissue-specific functions during
development and adulthood, and are integral to maintenance of tissue homeostasis. Independent of each other, tissue macrophage compartments evolve locally surrounded by their organ
microenvironment, and therefore, each population of tissue macrophages is closely related to its immediate surroundings. Hence, these cells acquire additional functions and activities
tailored to maintain the steady state in local tissues through various functions, including the modification of the phagocytosis mechanism, general or specific niche nutritional factors, and
morphological specificity. Normal tissue homeostasis is regulated in a tissue-specific manner by distinct populations of tissue macrophages, ranging from pulmonary surfactant clearance to
neuron pruning and the establishment of intestinal homeostasis. Epigenetic characterization shows great plasticity in the epigenetic programs of macrophages, and more than 12,000 enhancers
can be reprogrammed when mouse differentiated macrophages are transplanted into a new microenvironment.36 Enhancers are fundamental and precise determinants of gene expression and play a key
role in how distinct signals establish cell identity and regulatory potentiality at the genomic level. The number of enhancers identified in murine macrophages exceed that of promoters;
thus, interactions between DNA and TFs at enhancers are more likely to occur than at promoters.37 Importantly, there is strong enrichment for sequences containing transcription factor
binding sites that recognize and recruit different patterns of TFs on macrophage enhancers, corresponding to a significant enrichment of DNA recognition motif combinations.38 In turn, the
binding of TFs to DNA determines the selection of new enhancers. In addition to tissue-specific regulation, which is very predictable, to be more precise, the epigenetic regulation of
macrophages is coordinated by lineage- and tissue-specific transcription factors, which are determined by the built-in programming of myeloid development as well as signals from the tissue
environment. Lineage-determining transcription factors (LDTFs), also referred to as pioneer factors or master regulators, can actively open up the local chromatin and directly combine it
with other factors.39 Multiple lines of evidence suggest that ETS family member PU.1 contributes to the basal activation state and H3K4me3 of many promoters, acts as a crucial LDTF that
occupies most macrophage enhancers where it is essential in maintaining H3K4me1, and contributes to select a large number of the cell-specific enhancer-like elements, suggesting that PU.1 is
both required and sufficient to function in genomic regions as an enhancer.40 Moreover, PU.1 is considered to be a pioneer TF in initiating chromatin accessibility, allowing the binding of
additional TFs, and PU.1 is bound at the same level in both unstimulated and stimulated cells. Additional TFs commonly found in macrophages, including C/EBP family members, IRF, NF-κB and
AP-1 factors, exhibit collaborative interactions with macrophage-specific enhancers selected by PU.1 (Fig. 1).41 Studies have validated that the collaborative and hierarchical relationship
of LDTFs and signal-dependent transcription factors (SDTFs) exist at pre-existing enhancers.41 Mutations in PU.1 motifs leading to the inhibition of PU.1 binding cause the deletion of the
binding of C/EBPα in collaboration. Conversely, mutations in C/EBP motifs also result in a corresponding reduction in nearby PU.1 binding. Moreover, mutations in LDTF motifs can abolish the
signal-dependent binding of NF-κB, whereas mutations in NF-κB motifs rarely affect the binding of PU.1 or C/EBPα. Therefore, the hierarchical model of regulatory functions in macrophages has
been described, where a relatively small group of LDTFs compete with nucleosomes to bind DNA in a cell-specific manner, among which PU.1 is a necessary LDTF of macrophages to maintain
nucleosome depletion at macrophage-specific enhancers. By initiating nucleosome remodeling and histone modification deposition that are associated with _cis_-active regulatory elements, the
binding of PU.1 is proposed to “prime” DNA, and thus, the common enhancer repertoire is activated differentially by H3K4 monomethylation at a large number of genomic regions.40
Simultaneously, or subsequently, the binding of secondary SDTF establishes tissue-specific enhancers and regulates gene expression.42 Cooccurrence motifs associated with these TFs can be
traced back to the fact that enhancers provide integration sites of TFs regulated at the genome level by internal and external environment signals. Accordingly, synergistic binding of TFs
can promote the ability to overcome a nucleosomal barrier and initiate chromatin regulatory events. In this way, tissue-specific TFs and tissue-programmed epigenetics of distinct tissue
environments control the gene expression of resident macrophages, regulating their functions and affecting the environment itself. While some lineage-specific enhancers are “open” and
labeled by H3K4me1 in HSCs, it seems that a considerable number of de novo establishments of enhancers occur during hematopoiesis. For instance, many macrophage-specific enhancers are
established in granulocyte-macrophage progenitors (e.g., H3K4me1) but are blocked in HSCs, such as the ones that drive the expression of the gene encoding CD11b, which is the integrin and
common myeloid marker.43 Particularly, NF-κB also shows the ability to select “latent” or “de novo” enhancers in cooperation with PU.1 to bind to genomic locations lacking prior
characteristics associated with active enhancers.44 In fact, a promoter can be affected by a variety of different combinations of enhancers with different SDTF motifs that are intrinsically
related to various signaling pathways. This feature of enhancers constitutes great flexibility in tuning gene expression according to the specific needs that macrophages encounter in a
context-dependent manner. Generally, active enhancers contain both H3K4me1 marks and H3K27ac marks, while enhancers in the poised state are marked by H3K4me1 and in the absence of the
activating acetylation marker H3K27ac.45 These poised enhancers shared by many tissue macrophages are not active but might indicate the potential to respond to local challenges and activate
prospective gene-expression programs.46 The research on promoters (H3K4me3+), poised enhancers (H3K4me1+ and H3K27ac−) and activated enhancers (H3K4me1+ and H3K27ac+) has suggested that,
independently of development, signals from the local microenvironment play a leading role in the formation of tissue-specific regulatory regions and in the control of gene activity in
macrophages.36 SIGNIFICANT EFFECTS OF MACROPHAGES IN SPECIFIC TISSUES The existence of multiple macrophage progenitors begs the question of whether the ontogenesis of macrophages determines
their functional characteristics. In fact, distinct macrophage populations display unique transcriptional characteristics and epigenetic marks that are specific to their tissue of residence
(Fig. 2). Microglia, the macrophages of the central nervous system (CNS), are resident macrophages in the brain and spinal cord that are entirely derived from the yolk sac during
embryogenesis, potentially and specifically due to their privacy behind the blood−brain barrier.34 Microglia play diverse roles in the healthy brain, from forming developing neuronal
circuits to shaping learning-related plasticity.47 In healthy brains, microglia are highly active and constantly observe the microenvironment with extremely motile processes and protrusions.
The transcriptomic and epigenetic phenotypes of microglia are relatively well conserved.48 Snall1 is a microglial unique TF, and its enhancers are open and active only in microglia.36
Therefore, Snall1 controls the transcriptional regulation that maintains microglial identity and physiological properties as a critical factor for CNS homeostasis.49 Recently, the functions
of HDAC1 and HDAC2 in microglia have been found to be time- and context-dependent in vivo.50 Prenatal ablation of HDAC1 and HDAC2 results in spontaneous microglial damage due to abundant
H3K9ac and H3K27ac deposits on their respective promoters, but the lack of HDAC1 and HDAC2 enhances the phagocytic function of microglial amyloid proteins during neurodegeneration.50 In
addition, miR-124, a brain-specific microRNA expressed in microglia but not in peripheral macrophages, has been detected to be a crucial regulator for maintaining the quiescent state of
microglia in the CNS.51 In terms of mechanism, miR-124 can reduce the expression of PU.1 and its downstream target, the M-CSF receptor; suppress cell proliferation; and enhance the
differentiation of primary macrophages into adult microglia via the C/EBP-α–PU.1 pathway.51 Blood−brain barrier breakdown is associated with brain pathologies and induces prominent monocyte
infiltration into the CNS. The monocytes give rise to brain macrophages and can hardly be discerned from resident embryo-derived microglia.52 Microglia and monocyte-derived cells have
distinct functions, as revealed by the study of murine multiple sclerosis and the experimental autoimmune encephalomyelitis model.53 During encephalomyelitis, microglia appear dedicated to
the clearance of debris, whereas monocyte-derived macrophages are highly phagocytic and inflammatory, with the expression of proinflammatory genes. Specifically, monocyte-derived macrophages
are unable to generate long-lived microglia and do not permanently contribute to the brain-resident macrophage compartment, which is thus composed of embryo-derived microglial cells before
and after the challenge.54 The intestine contains the largest pool of macrophages among the tissues.55 As the extreme opposite of CNS microglia, embryo-derived macrophages are found in the
intestine only shortly after birth. While embryonic precursors seed the intestinal mucosa and demonstrate extensive proliferation in the neonatal period, these cells do not appear in the
adult intestine.56 Instead, they are replaced entirely around the time of weaning by the blood inflammatory monocytes that migrate to inflamed tissues, differentiate locally into mature
tissue macrophage populations in healthy intestinal lamina propria and contribute to the maintenance of gut homeostasis. Intestinal macrophages are enriched for RUNX family motifs, and RUNX3
is highly expressed in these cells.36 The monocyte infiltration is regulated mainly by the microbiota, which leads to low-grade inflammation. In fact, intestinal macrophages spend their
existence bathed in cytokine IL-10 and maintain anti-inflammatory responses that mute any inflammatory response to the gut flora and their products.57 This progress can eliminate the
pathogen, avoid collateral damage caused by the oversecretion of proinflammatory cytokines and restore tissue integrity as a result. Alveolar macrophages (AMFs) located in the alveolar
cavity develop from fetal liver monocytes depending on CSF2 (also known as GM-CSF) through the induction of peroxisome proliferator-activated receptor-γ (PPAR-γ).58 Before birth,
embryo-derived primitive macrophages and fetal monocytes are colonized in the developing lung.59 The initial signs of AMF differentiation appear around the saccular stage of lung
development, and it is assumed that fetal monocytes, but not embryo-derived macrophages, are the main precursors of AMFs. With minimal contribution from circulating hematopoietic precursors,
AMFs self-maintain locally. After birth, AMFs play an important role in the scavenging of lung surfactant and pulmonary homeostasis. Transcriptional inhibitors Bach1 and Bach2 are required
for functional maturation; Bach2 in particular is a major contributor to this repression.60 The lncRNA MEG3-4 has been identified as a tissue-specific regulator of inflammatory responses in
alveolar macrophages during bacterial infection through the transcriptional regulation of immune response genes.61 It has been confirmed that the lncRNA MEG3-4 binds to the microRNA miR-138
in a competitive manner with mRNA encoding the proinflammatory cytokine IL-1β, thereby increasing the abundance of IL-1β and enhancing the inflammatory response to bacterial infection in
alveolar macrophages in mice.61 The spleen-macrophage chamber actually contains several different compartments, including the marginal zone (MZ), red pulp (RP), and white pulp (WP) subtypes,
which have independent and noncomplementary functions that are linked to the phagocytic capacity of specific macrophage subpopulations.62 Macrophages located near the splenic MZ, the
channel through which the bloodstream passes, seem to be uniquely committed to the stationary clearance of apoptotic cells and the selective engulfment of dying cells.63 In the MZ, the
nuclear receptor LXRA is essential for macrophage differentiation, as proven in LXRA-deficient mice, which have defective generation of the MZ and metallophilic macrophages.64 In contrast,
macrophages in the RP specifically remove aging or damaged red cells and recover the released iron. It has been discovered that the transcription factor Spi-C, a PU.1-related transcription
factor, selectively dominates the development of red pulp macrophages.65 Due to the lack of Spi-C, a deficiency of macrophages in the red pulp impairs the clearance of senescent red blood
cells and the maintenance iron homeostasis, and selective splenic iron overload occurs in mice with this deficiency.65 As for peritoneal macrophages, there are at least two distinct
macrophage subtypes in the abdominal cavity of adult mice.66 In healthy mice, “large” peritoneal macrophages account for the principal part of the components of peritoneal cavity macrophages
but disappear rapidly after stimulation. In addition, “small” peritoneal macrophages, as a source of controversy, predominate in the peritoneal cavity after stimulation. The phagocytosis of
these macrophages eliminates apoptotic cells. Evidence suggests that retinoic acid is the tissue-derived signal that induces the localization and functional polarization of peritoneal
macrophages in a tissue-specific manner through the reversible induction of GATA6, a specific TF for peritoneal macrophages associated with the establishment of the tissue-specific
transcriptional and epigenetic landscape.67 Moreover, previous research has shown that DNA methyltransferase DNMT3A maintains a high expression of HDAC9 in a DNA-methylation-dependent manner
in naïve peritoneal macrophages and epigenetically prepares these cells to activate TBK1-IRF3 signaling fully and produce interferon I after virus infection.68 As mentioned above,
macrophages promote tissue homeostasis under physiological conditions in distinct manners. In a stable state, tissue macrophages have intrinsic and potential anti-inflammatory functions.
Tissue-resident macrophages derived from the yolk sac, fetal monocytes and adult monocytes all exhibit inhibitory effects despite differences in ontogeny. Immunosuppression may be the key
function of macrophages in tissue homeostasis. Nevertheless, macrophages in homeostasis are primed to respond rapidly and robustly to subsequent challenges, maintaining low levels of
constitutive IFN-β and downstream Janus kinase (JAK)–STAT signaling.69 Two almost simultaneous studies introduced the importance of commensal microbiota in the production of low levels of
IFN.70,71 It is particularly noteworthy that the microbiota mimics the regulatory components of host protein networks. For example, influenza A virus carries a sequence that resembles H3K4
and can block interactions with readers of H3K4me3, thereby suppressing the positive function of this epigenetic marker.72 Another mechanism that primes macrophages is the maintenance of low
levels of negative H3K9me3 marks at IFN response gene loci.15 EPIGENETIC VARIATIONS IN DIFFERENT TISSUE-RESIDENT MACROPHAGES As mentioned above, analyses of enhancer landscapes have
revealed that some enhancer-like regions in various tissue-resident macrophage populations are shared and that the combination of PU.1 is required for the development of macrophages in
almost all tissues. The enhancer of Spi1, which controls the expression of PU.1, has H3K4me2 and H3K27ac marks in all macrophage populations.18 In contrast, there are epigenetic variations
in macrophage populations in different tissues. For example, the Rarb gene, induced by retinoic acid, is labeled by H3K4me2 in all macrophage populations, but H3K27ac only presents in the
peritoneal macrophage population.42 The fact that the enhancer region of the Rarb gene is poised in other tissue-resident macrophages but active in peritoneal macrophages is consistent with
another finding that peritoneal macrophages are controlled by locally produced retinoic acid, and the Rarb gene itself is retinoic acid inducible.73 This evidence suggests that the enhancers
leading to Rarb expression are selected by a series of common macrophage lineage-determining factors in other macrophages and peritoneal macrophages. However, these enhancers can become
active only in the peritoneal cavity because of the sufficient concentrations of retinoic acid in the tissue environment. EPIGENETIC REGULATION OF MACROPHAGE ACTIVATION IN HOST DEFENSE
DIFFERENT POLARIZATION STATES OF ACTIVE MACROPHAGES In addition to maintaining tissue homeostasis induced by homeostatic signals, macrophages are best known for their role as immune guards
at the forefront of tissue defense. In response to various external stimuli and diverse signals, macrophages can obtain heterogeneous activation states and customize specific functions for
specific microenvironments. The accurate and specific regulation of macrophage activation to eliminate the pathogenic insult and repair damaged tissue, in turn, is crucial for restoring
tissue homeostasis. As shown previously, tissue-specific phenotypes of macrophages, controlled by lineage-specific master regulators, can be generated by hard-wired, irreversible
differentiation processes. Alternatively, phenotypes can be reversible and induced as needed based on a functional polarization program. From the previous literature, two extreme states of
activated macrophages are M1-like macrophages (in response to inflammatory stimuli such as LPS and IFN-γ) and M2-like macrophages (in response to cytokines such as IL-4 and IL-13).74 In
addition, the growth factors GM-CSF and M-CSF, traditionally used to differentiate monocytes into dendritic cells or macrophages, have the ability alone to induce M1- and M2-like phenotypic
changes, respectively.75 In fact, the phenotypes of macrophages can switch between different functional states, and the stability of M1, M2 and other phenotypes is unclear. M1 and M2 are
more accurate when describing macrophages in vitro, which are induced by specific stimuli. However, in vivo macrophages have heterogeneous phenotypes and multiple polarization states, and
thus, they can hardly be simply binned into an M1 or M2 pool. The description of the activation states of these macrophages is currently contentious and confusing. Nonetheless, it is still
useful to conceptualize these states as they are still related to the binary M1/M2 classification, and the consensus on the definition offers a reductionist tool to describe extremes of
their function and may facilitate the study of macrophages. M1 macrophages, also known as classically activated macrophages, are inflammatory macrophages characterized by efficient antigen
presentation, high bactericidal activity, and the production of proinflammatory cytokines and reactive oxygen and nitrogen species.76 In contrast, M2 macrophages, often referred to as
alternatively activated macrophages, have immune-regulatory functions by releasing anti-inflammatory cytokines and decreasing the production of proinflammatory cytokines and show less
efficient antigen presentation. In addition, M2 macrophages are predominantly regulatory macrophages involved in tissue remodeling, tumor progression, wound healing, angiogenesis,
anti-helminth responses and allergic reactions. When early warning signals are triggered, a defined feature of M1 activation is monocyte recruitment from the blood.77 Nevertheless, M2
macrophages accumulate independently of monocytes, and in situ proliferation occurs rapidly to induce the accumulation of macrophages under the control of IL-4.78 MONOCYTE−MACROPHAGE
TRANSITION UNDER STIMULATION Monocyte-derived macrophages exist in some tissues, such as the dermis and intestine, where monocytes are considered an intermediate developmental stage between
bone marrow precursors and tissue macrophages.79 In addition, blood monocytes migrate to inflammatory tissues and differentiate into monocyte-derived macrophages that can restore tissue
integrity and eliminate the pathogen. In vitro, monocyte cultures imitate inflammatory macrophages in vivo and are used clinically as a crucial tool in basic research. Epigenetically, active
DNA demethylation is involved in the entire process of monocyte differentiation into macrophages as a major example of the role of targeted demethylation in cell differentiation.80 DNA
demethylation affects a small group of specific genes that regulate the actin cytoskeleton and phagocytosis and is therefore very important for the structure and function of macrophages.80
Notably, these regions become nucleosome-free and obtain active enhanced markers through enrichment in the binding sites of transcription factors (such as AP-1, RFX1 and KLF4), which can
open chromatin, and demethylation catalyzed by 10−11 translocation proteins occurs rapidly.81 A recent study revealed that during macrophage development, the ATP-dependent chromatin
remodeling SWI/SNF complex (also termed BAF), utilizing two alternative ATP-dependent enzymes, brahma-related gene 1 (BRG1) and brahma (BRM), activates the expression of DNA repair enzymes
by recognizing and replacing epigenetically marked nucleosomes, together with EP300 and HDAC1, constituting a functional unit.82 Along the monocyte−macrophage differentiation axis,
EP300-HDAC-SWI/SNF functional crosstalk defines the chromatin structure and transcriptional activity of DNA repair enzyme promoters.82 Recently, high-throughput epigenome analysis has
revealed that three miRNAs (miR-34, miR-146 and miR-221), which play essential roles in actin cytoskeletal reorganization, are upregulated and overexpressed in the process of
monocyte-to-macrophage differentiation.80 In contrast, other miRNAs have been found to be downregulated during monocyte-to-macrophage differentiation. miR-198, which is capable of reducing
cyclin T1 protein expression through translational inhibition, for instance, is strongly downregulated during this process.83 The decrease in miR-198 level during macrophage differentiation
is beneficial for cyclin T1 protein expression, which has been shown to be important in the regulation of macrophage gene expression.84 INITIAL INFLAMMATORY ACTIVATION BY TLRS Toll-like
receptor ligands, such as LPS, and Th1 cytokines, including IFN-γ, elicit M1 activation alone or in combination and can affect epigenetic processes and lead to epigenetic modifications (Fig.
3).85 Toll-like receptors (TLRs), especially TLR4, are key sensors in the M1 response that triggers cascades of signals to activate inflammatory processes through MAPK, NF-κB and IRF gene
networks, which have downstream genes that encode inflammatory cytokines, such as CXCL10, IL-1β, IL-6, IL-12 p40 and TNF. LPS affects diverse sets of epigenetic factors that determine the
modifications of chromatin structure and gene-expression programs, such as DNA methylation, chromatin-associated complexes, and histone modifications. Until now, however, histone
modifications in response to LPS have been studied more extensively. The first evidence on the link between LPS stimulation and epigenetic regulation in inflammatory genes dates back to
1999, as LPS stimulation induces IL-12 p40 production in murine primary activated macrophages by rapid and specific nucleosome translocation at the promoter region.86 A subsequent study
revealed that it is a TLR-4-dependent event and depends on histone H3 and H4 acetylation.87 Prior to the activation of differentiated macrophages, the locus that encodes inflammatory genes
appears to be in a relatively “open” chromatin environment, and the epigenetic landscapes are established during macrophage differentiation as stated earlier. As mentioned above, macrophage
master TFs, such as PU.1 and C/EBP family members, bind to and open the enhancers of these genes and thus “prime” them. Enhancers in this state are marked by PU.1, H3K4me1, and open
chromatin.40 In addition, there are gene-specific repressive mechanisms that restrain inflammatory cytokine gene transcription in the absence of TLR signaling. These two opposite effects
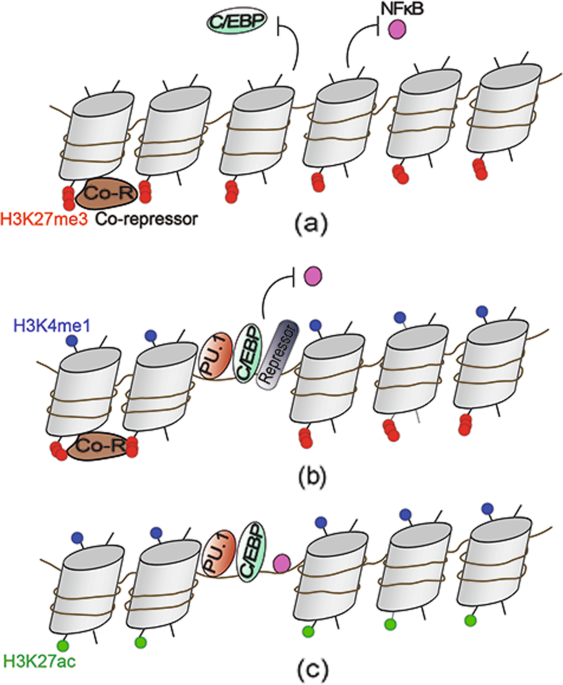
keep macrophages in a “poised” state. One of the repressive mechanisms is that negative histone marks H3K9me3, H3K27me3, and H4K20me3 are present in the inflammatory gene loci.88,89,90
Another mechanism is the occupancy of gene loci by repressors such as nuclear receptors that recruit corepressor complexes that contain sequence-specific transcriptional repressor B cell
leukemia 6 (Bcl6).91 In addition, the occlusive positioning of nucleosomes limits the chromatin accessibility of genes such as IL-12b.10 Furthermore, these epigenetic “brakes” associated
with transcriptional silencing are released after the stimulation of macrophages by TLR ligands. The corepressors are removed from gene loci, and the concomitant reduction of negative
histone marks trimethylations by using demethylases. JMJD3, which specifically demethylates trimethylated lysine 27 (H3K27me3), could be induced by NF-κB rapidly under LPS stimulation and
recruited to the promoter regions of more than 70% of LPS-induced genes in macrophages and regulate gene transcriptional elongation associated with the H3K27 demethylase KIAA1718.92,93 The
induction of inflammatory cytokines by acute-phase protein serum amyloid A depends on JMJD3 activity.94 Only a subset of proinflammatory cytokines such as IL-12b and CCL5, however, shows a
strong dependence on JMJD3 demethylase activity to achieve full activation.92 After removal of negative histone marks of inflammatory genes, positive histone marks, such as H3K4me3 and
H3K27Ac, increase. The histone marks H3K4me3 are enriched in the promoter region of the M1 marker gene CXCL10 by histone methyltransferase myeloid lymphoid leukemia (MLL), which is shown to
increase upon LPS- and IFN-γ-induced M1 activation of macrophages.95 Moreover, different kinds of MLLs, such as MLL1, MLL2/4, and MLL3, are critical for the remodeling of the enhancer
landscape of macrophage activation.44 In addition, a new study suggests that the methyltransferase Dot1l increases the production of IL-6 and TNF-α by promoting H3K79me2/3 and H3K79me2 at
their promoters, respectively.96 HDACs are strongly involved in M1 activation and play a prominent role in the regulation of immunological pathways. The expression of almost all classes of
HDACs is affected upon LPS stimulation, and the sequential regulation of HDAC controls gene-specific expression (Fig. 3). As a typical example, cyclooxygenase-2, a crucial enzyme involved in
the inflammatory reaction, is rapidly induced in macrophages upon LPS stimulation, and a number of HDACs are involved in the process.97 It has been shown that LPS inhibits the expression of
HDAC4, 5 and 7 at first and then upregulates the acetyltransferase complex, leading to cyclooxygenase-2 gene activation.98 During _L. pneumophila_ infection of pulmonary epithelial cells, a
similar mechanism has been described for IL-8 induction, accompanied by the initial decrease in HDAC1 and HDAC5.99 Other studies have shown that HDAC6 and HDAC7 are involved in the
expression of proinflammatory genes in macrophages stimulated by LPS.100,101 In fact, a specific subtype of HDAC7 (HDAC7-U) promotes the expression of TLR-induced inflammatory gene subsets,
such as EDN1, IL-12p40, and IL-6.101 The inhibition of HDAC6 activity significantly limits LPS-induced macrophage activation and proinflammatory cytokines, regulated by the effects of HDAC6
on cell adhesion and microtubule acetylation.100 In addition, an integrated genomic approach shows that, in response to LPS, HDAC3-deficient macrophages are unable to activate almost half of
the inflammatory gene-expression program.102 In addition to the histone modification associated with inflammatory genes directly, some mediators target TFs. SIRT1, a specific type of HDAC,
suppresses macrophage activation through TFs such as p65, LXR, and IRF8.103 In LPS-activated macrophages, SIRT1 expression is downregulated, and the expression level of these TFs increased.
In addition to the direct combination of pro-inflammatory genes and the regulation of their transcription, regulators aimed at the suppression mechanisms are also required. As a
methyltransferase, KMT6 (also known as EZH2) works on H3K27 as a key subunit of polycomb repressive complex 2.104 KMT6 directly targets suppressor of cytokine signaling 3 (SOCS3) to promote
H3K27me3 and inhibits the expression of SOCS3, leading to uncontrolled TLR-induced NF-κB activation and increased proinflammatory gene expression, therefore playing important roles in
regulating the inflammatory responses in macrophages. Moreover, the negative marker H3M27me3 generated by KMT6 persists after termination of the stimulus, and these inhibited genes no longer
respond to the glucocorticoids IL-4 and M-CSF.105 In addition, the repression of M2 gene expression is also related to the downregulation of transcription factor MAF and MAF-binding
enhancers, coordinated with the loss of LDTF binding and chromatin accessibility.106 Recently, it has been revealed that HDAC2 interacts with the c-Jun promoter and plays a key role in
silencing c-Jun expression specifically, the activity of which is important for inhibiting the transcription of a series of inflammatory pathway genes, thereby enhancing proinflammatory gene
expression indirectly.107 Moreover, HDAC9, interacting and colocating with many transcriptional repressors or corepressors, including NCOR13, during macrophage differentiation, represses
these nuclear receptors by forming multiprotein complexes and thus induces a proinflammatory M1 phenotype.108 In addition to histone modification, chromatin remodeling occurs during M1
activation. Chromatin remodeling occurs through the ATP-dependent chromatin remodeling complex SWI/SNF, which is preferentially targeted to enhancers and serves as a histone acetylation
sensor interacting with acetyltransferase P300 to modulate H3K27ac.109 DNA methylation affected by M1 activation is poorly investigated compared with histone modification and chromatin
remodeling. A link between DNA methylation and LPS stimulation has been found for SOCS1, a negative regulator of cytokine signals.110 DNMT1-mediated hypermethylation of SOCS1 results in a
loss of SOCS1 expression and enhances the release of LPS-induced proinflammatory cytokines such as TNF and IL-6 in macrophages.110 DNMT1 also induces the hypermethylation of the critical
regulators Notch1, PU.1, and KIF4; participates in increasing the dimethylation (H3K9me2) and trimethylation (H3K9me3) of H3K9 in these genes; and skews their polarization towards M1
macrophages.111 Noncoding RNAs are also involved in M1 differentiation and the modulation of proinflammatory polarization. For example, a higher expression of proinflammatory miR-155 and a
lower expression of miR-125b favor M1 macrophages.112 In contrast, mir-99a inhibits the phenotype and function of M1 macrophages by targeting TNF-α.113 M2 ALTERNATIVE ACTIVATION Different
from classical activation, the macrophages to be alternatively activated do not require a primed state, but appropriate inducers are needed, such as IL-4 and IL-13. Transcriptional
activation induced during M2 polarization is commonly associated with histone demethylase (Fig. 3). The increased expression of JMJD3, mediated by STAT6, contributes to transcriptional
activation of M2 marker genes. Specifically, JMJD3 induces H3K27 demethylation at the transcription factor IRF4 locus, which is essential for M2 macrophage development and thus facilitates
its expression.114 Accordingly, the JMJD3-IRF4 axis is necessary for M2 polarization in macrophages. Another study showed that αKG produced by glutamine lysis controls JMJD3-dependent
regulation, suggesting the correlation of epigenetic and metabolic reprogramming for M2 macrophages.115 JMJD3 has been indicated to be a critical modifying enzyme in M2 polarization through
JMJD3−/− bone marrow chimeras, while the role in M1 activation is dispensable.114 The dual roles of JMJD3 in M1 and M2 macrophages are not inevitably conflicting, suggesting the need to
remove the inhibitory H3K27me3 marker to respond to many environmental queues. Moreover, M2 polarization is also due to SMYD3 activity, another H3K4 methyltransferase.95 SMYD3 is
preferentially expressed in M2 macrophages and can increase H3R4me3 at the M2-related promoter site. With respect to deacetylases, the enzymes have both positive and negative effects. For
instance, SIRT2 is an NAD+-dependent deacetylase that reduces NF-κB acetylation and increases M2-associated anti-inflammatory responses.116 In contrast, HDAC3 acts as an epigenomic brake on
IL-4-induced alternative activation by deacetylating putative enhancers and thereby repressing IL-4-regulated genes characterized by M2 activation.117 Recently, IL-4-activated STAT6 was
found to act as a transcriptional repressor in an HDAC3-dependent manner.118 The repressed inflammatory enhancers are associated with reduced LDTF and p300 binding followed by a reduction in
enhancer RNA expression, H3K27 acetylation, and chromatin accessibility. Therefore, it can be inferred that HDAC3 plays an important role in M2 activation. Moreover, even without external
stimulation, macrophages lacking HDAC3 exhibit an M2-like phenotype and are highly responsive to IL-4.117 DNA methylation and noncoding RNAs are also involved in macrophage alternative
activation. DNMT3B modulates DNA methylation at the promoter of PPAR1, which is a key transcriptional factor that regulates macrophage polarization.119 Additionally, the expression of
certain miRNAs, such as miR-142-5p and miR-511, promotes M2 polarization by negatively regulating genes involved in inflammatory signaling.120,121 To answer the question of whether the
IL-4-activated macrophages are homogeneous, a recent study revealed that repeated stimulation with IL-4 induces strong phenotypic changes in macrophages through PPAR-γ, which has a
significant ligand-insensitive, genome-bound fraction.122 After the first IL-4 exposure and subsequent STAT6 activation, PPAR-γ establishes a permissive chromatin environment through the
recruitment of coactivator P300 and architectural protein RAD21, and this altered epigenome endows transcriptional memory by promoting the binding of STAT6 and RNA polymerase II. Therefore,
the robust production of enhancer and mRNAs are induced upon IL-4 restimulation, and the expression of a hidden gene program, such as extracellular matrix gene network, reaches the threshold
of activation only after the second stimulus. THE ROLE OF EPIGENETIC MODIFICATION IN MACROPHAGES DURING DISTINCT DISEASE STATES Changes in macrophages can cause a broad spectrum of
maladaptive immunity and inflammation that are causative factors of disease and thus represent key therapeutic targets.4 Moreover, the significant role of epigenetic pathways in macrophages
in disease states indicates that epigenetic modifications of macrophages can be used for the diagnosis or therapies of indifferent diseases. ATHEROSCLEROSIS Atherosclerosis is a chronic
inflammatory disorder as a result of a vascular injury caused by endothelial dysfunction. Macrophages are crucial immune cells in the progression of atherosclerosis and determining the
clinical outcome through transmission inflammatory responses, foam cell formation and the final development of necrotic core.123 In the initiation of atherosclerosis, endothelial cells are
activated by the aggregation of low density lipoprotein (LDL) and the modification of it (such as oxidation to oxLDL) in the arterial wall. Monocytes are attracted to adhere and migrate to
the vessel wall and then differentiate into macrophages and may become lipid-loaded foam cells. Apoptotic foam cells can form the core of necrosis, and thus, the differentiation of monocytes
into macrophages is a key step in the formation of atherosclerosis. Abundant studies have focused on the epigenetic changes of macrophages involved in the pathogenesis of atherosclerosis,
especially histone modification and drugs targeting atherosclerosis based on HDACs. It has been demonstrated that HDAC inhibitors have beneficial antiatherogenic effects as they partially
inhibit M1 activation, reduce proinflammatory cytokine expression and blunt apoptosis; thus, they do not increase the formation of foam cells in primary macrophages. Valproate, a
broad-spectrum HDAC inhibitor, has been shown to inhibit atherosclerosis in animal models.124 Nonspecific HDAC inhibitors, however, have contraindications that prevent their direct usage in
the treatment of atherosclerosis. Therefore, specific HDAC enzymes deserve further study. HDAC3 has been detected to be a new potential target for atherosclerosis therapy, as deletion of
HDAC3 can promote M2 activation while inhibiting M1 polarization.123 These two characteristics are considered to be the advantages of atherosclerosis therapy. Moreover, high expression of
HDAC9 is consistent with an increased risk of atherosclerosis.125 HDAC9 upregulation in macrophages during atherosclerosis represses cholesterol efflux and M2 polarization, and in
HDAC9-deficient mice, the phenotype of macrophages switches from the proinflammatory M1 to the anti-inflammatory M2 state via PPAR-γ.108 miRNAs are closely related to atherosclerosis in
various aspects, including the regulation of macrophages during atherosclerosis. miR-155 plays a key role in macrophages, supporting and improving inflammation-induced atherosclerosis by
directly repressing Bcl6.126 Moreover, it has been shown that miR-146a is involved in the pathogenesis of atherosclerosis, which negatively regulates macrophage maturation and inhibits
inflammatory activation by reducing the expression of CD86 and CD80.127 OBESITY AND TYPE 2 DIABETES Obesity and type 2 diabetes (T2D) are rapidly growing diseases and are major risk factors
for the development of cardiovascular diseases. Obesity generally tends to classically activate M1 adipose tissue macrophages and decreases alternatively activated M2 macrophages. The
different activation states of M1 and M2 contribute to obesity-induced insulin resistance and inflammation. In recent years, research has focused on the epigenetic mechanisms by which
macrophages regulate their activation as crucial controllers of inflammation in T2D and therefore provide new insights into therapeutic interventions. Hyperlipidemia and hyperglycemia in
diabetes patients could cause epigenetic changes and promote the formation of an inflammatory macrophage phenotype. In macrophages isolated from hyperlipidemia and a T2D mouse model of
hindlimb ischemia, inflammatory genes are hypomethylated, and M2 genes are hypermethylated.128 In obesity patients, DNMT3B and DNMT1 expression are enhanced, leading to DNA methylation of
the promoter of PPAR1, which may induce M2-associated gene suppression and contribute to an inflammatory macrophage phenotype.129 Therefore, the deletion of DNMT1 by using
5-aza-2′-deoxycytidine in pharmacology could promote alternative activation and inhibit macrophage inflammation. Furthermore, several lncRNAs are altered by T2D. One example is the lncRNA
E330013P06, which is upregulated in macrophages isolated from T2D mice and the monocytes of T2D patients. The lncRNA E330013P06 may play an important role in macrophage dysfunction and
related gene regulation because its overexpression in macrophages enhances the expression of inflammatory genes and increases inflammatory responses to specific signals.130 DIABETIC WOUND
HEALING Wound healing is a well-coordinated dynamic process involving diverse cells, including three main stages of coagulation and inflammation, tissue formation and remodeling. Macrophages
are one of the critical participants in wound healing. Since the occurrence of injury, the functional changes of macrophages at the wound site persist. The major M1 subtype occurs mainly in
the early inflammatory phase and is responsible only for the phagocytosis of bacteria, neutrophils, and tissue debris and the release of cytokines such as TNF-α and IL-6.131 As the
repair/remodeling phase progresses, phenotypic transformation occurs in macrophages. During the late stage of repair, macrophages provide an M2 gene-expression profile predominantly,
resulting in an increase in the number of M2 (healing-promoting) macrophages and the release of growth factors such as TGF-β, IL-10 and IGF.131 Delayed wound healing is a serious
diabetes-related complication that often leads to nontraumatic limb amputations. Given the role of epigenetic mechanisms in T2D, attention has recently been paid to epigenetic changes in
diabetic wounds, and it has been revealed that a large number of epigenetic changes in diabetes lead to delayed wound healing. Alterations in histone methylation have been suggested to be a
damaging factor for diabetic wound healing. The expression of IL-1β is increased in macrophages in diabetic wounds, and the abnormal expression of IL-1β impairs wound healing. After
stimulation with LPS, the expression of IL-1β is increased in macrophages isolated from wounds of the T2D mouse model, with increased H3K4 methylation and decreased H3K27 methylation,
suggesting that the expression of IL-1β is regulated by an epigenetic mechanism.132 It has recently been demonstrated that hyperglycemia leads to changes in the microRNA signature in wound
healing, and they have been found to play a role in the dysregulated inflammation of diabetic wounds. For instance, miR-146a has been demonstrated to be downregulated in diabetic wounds and
is unable to regulate its proinflammatory target gene expression, such as the NF-κB p65 subunit, leading to wound-healing impairment.133 SEPSIS Sepsis is a worldwide disease with high
morbidity and mortality rates, especially in intensive care units. Sepsis occurs when the body reacts to infectious and noninfectious stimuli that cause a nonresolving inflammatory response
and cytokine release and then induce the injury of tissues and organs, leading to multiorgan failure, shock, and death.134 During sepsis, white blood cells and platelets migrate to the
infection site, resulting in platelet aggregation, endothelial damage, and increased microvascular permeability. Blood flow also decreases, which may introduce ischemia-reperfusion injury.
These physiological processes can induce systemic inflammatory response syndrome, leading to multiple organ dysfunction syndrome. It is now well established that macrophages and other innate
immune cells are activated profoundly during sepsis, playing a key role in the pathogenesis of this disease. In particular, dysregulated and profound activation macrophages can influence
immune function and can directly affect the prognosis of sepsis. Macrophage epigenetics plays an important role in the immune response associated with sepsis. For instance, the
inhibition/modulation of HDACs can serve as a therapeutic approach for sepsis via modulating the epigenetic pathway. In particular, HDAC6 inhibitor has a substantial advantage in the
treatment of sepsis.135 The selective inhibition can significantly reduce levels of proinflammatory mediators, inhibit macrophage apoptosis, promote bacterial clearance, increase immune cell
phagocytosis, and improve survival in a lethal murine model of sepsis. The expression of microRNA-Let7A (let-7a) in patients with sepsis caused by gram-negative bacilli has been shown to be
significantly downregulated.136 Let-7a has been confirmed to regulate the Toll-mediated inflammatory response in sepsis, thus providing a potential target for sepsis treatment. Recently, it
has been demonstrated that lncRNA NEAT1 is upregulated in patients with sepsis and that overexpressed NEAT1 plays a key role in acute kidney injury induced by sepsis.137 Moreover, it has
been found that the lncRNAs NEAT1/Let-7a and Let-7a/TLR4 have direct combinations. In the pathogenesis of sepsis, increased lncRNA NEAT1 binds to Let-7a competitively, and TLR4 is released
from Let-7a, which is activated and stimulates downstream signals, leading to severe inflammatory responses.138 INNATE IMMUNE MEMORY IN MACROPHAGES Innate immune memory of macrophages is an
important mechanism in response to environmental stimuli and affects subsequent immune responses and can be divided into tolerance and training. Recently, it has also been demonstrated that
immune memory also occurs in tissue-resident macrophages in vivo, such as microglia.139 The molecular mechanisms responsible for memory-like activity in macrophages have not yet been
elucidated. However, epigenetic regulations most likely induce these changes (Fig. 4). TOLERANCE Acute inflammatory activation of macrophages induced by TLR and related receptors must be
tightly regulated and transient to avoid tissue damage.140 The activation state is inherently unstable and is followed by a state of tolerance. Even in the continued presence of the agonist,
the response to LPS is self-limiting. In murine macrophages, the TLR-induced response in vitro fell into two categories of LPS tolerance: the selective and transient silencing of
proinflammatory genes and the priming of the second-class genes of M2 activation.141 Following the first exposure to LPS, anti-inflammatory genes in macrophages become modified to induce the
second stimulation faster and stronger and thereby increase the efficiency of innate host defense. The consistent gene reprogramming that macrophages undergo is the result of complex
regulatory mechanisms. For the molecular mechanism, LPS tolerance is mainly controlled by epigenetic regulation, including nucleosome remodeling, the reduced recruitment of transcription
factors and chromatin remodeling complexes, and histone modification.142 Nucleosome remodeling complexes are essential in this process. Initial LPS stimulation of naïve macrophages is needed
for the silencing of proinflammatory genes and the priming of anti-inflammatory genes in tolerant macrophages. LPS-induced gene products in naïve macrophages differentially modify chromatin
at promoters of pro- and anti-inflammatory genes to silence the former and prime the latter through a second LPS stimulation. For example, BRG1 is recruited to the promoters of secondary
response genes by the products of primary response genes, which explains why the induction of anti-inflammatory genes is qualitatively and quantitatively different in naïve and tolerant
macrophages. Therefore, the SWI/SNF complexes, not for rapidly induced primary response genes, are required for the activation of primary response genes induced with delayed kinetics and
secondary response. A Mi-2bβ complex, acting antagonistically to limit the induction of these gene classes, is selectively recruited along with the SWI/SNF complexes.143 The NF-κB-associated
inhibitory mechanisms are essential in the tolerance process. NF-κB can recruit the NCOR-HDAC3-P50 repressive complex into targeted genes.144 Moreover, NF-κB can also recruit histone
methyltransferase G9a to promoters to induce H3K9 methylation and induce binding of the heterochromatin protein 1.145 Together, these mechanisms lead to epigenetic silencing. Noncoding RNAs,
especially specific miRNAs, can regulate macrophage tolerization. For example, miRNA-146a has been shown to play a central role in TLR signaling tolerance following a primary stimulus with
MyD88-dependent TLR pathways.146 Additionally, miRNA-221 and miRNA-222 are also found as regulators of the functional reprogramming of macrophages during LPS tolerization by regulating
BRG1.147 TRAINED IMMUNITY The traditional view that only the adaptive immune system can build immunological memory has been challenged by a growing number of discoveries. In organisms
lacking adaptive immunity, such as invertebrates, the innate immune system can mount long-term memory for resistance to reinfection.148 Studies that exposed macrophages to bacterial and
fungal pathogens expand this observation, where the exposures enhance their subsequent response to the following stimulation with unrelated pathogens or PAMPs.149 Exposure of microglia to
inflammatory stimuli can cause a long-lasting change or memory; when the microglia encounter subsequent inflammatory stimuli, they produce higher inflammatory responses.150 It has been
demonstrated that trained immunity is accompanied by epigenetic reprogramming, especially histone modification.151 In response to stimulation, H3K4me1 levels and the binding of TFs increase
at a subset of enhancers, named “latent enhancers”, and these enhancers do not return to a latent state when stimulation ceases, suggesting the establishment of an epigenetic memory that
regulates cell responses to subsequent stimuli.46 For the promoter, after training in macrophages, an increased level of H3K4me3 can be observed at the promoter of genes associated with
innate immunity, such as the adaptor molecule Myd88 and the proinflammatory cytokines TNF-α, IL-6, and IL-18, and it is a basis of robust transcriptional responses during trained immune
responses.149 A low concentration of oxLDL treatment of monocytes can induce a long-lasting proatherogenic macrophage phenotype as “training”, enhancing H3K4me3 marks at promoter regions of
these inflammatory genes, characterized by increased pro-inflammatory cytokine production.152 The molecular basis for this process has been found recently. Trained immune genes through
β-glucan and its receptor Dectin-1 are able to engage in chromosomal contacts with a subset of lncRNAs, and the upstream master lncRNA of the inflammatory chemokine locus can form
chromosomal contacts with the ELR + CXCL chemokines (IL-8, CXCL1, CXCL2, and CXCL3) and _cis_-directs the WDR5/MLL1 complex across the CXCL chemokine promoters, facilitating their H3K4me3
epigenetic priming before their transcriptional activation.153,154 Innate immunity training plays an important role in disease prevention, among which research on Bacillus Calmette-Guérin
(BCG) is a typical example. BCG can be used to train macrophages ex vivo and thereby provide cross-protection.155 Considering the relatively short lifespan, the ability of macrophages to
transmit memory phenotypes to offspring and provide sustained protection remains unclear. In fact, the efficacy of generating long-term innate immune memory is essential to enhance organism
immunity. Therefore, immune memory has also been studied for insights into HSCs, long-lived cells that are self-renewing and capable of producing multipotent and lineage-committed
hematopoietic progenitors that give rise to all cells of the mammalian blood system, including macrophages.156 HSCs educated with BCG produce epigenetically modified macrophages, which are
better than HSCs educated without BCG in preventing _M. tuberculosis_ infection.157 Macrophages can be trained or tolerized for specific inflammatory stimuli, depending on the type of
trigger. In trained immunity, the initial hit induces long-lasting histone marks. In the case of the second hit, H3K27ac and H3K4me3 have already primed the genes, and expression is
enhanced. In contrast, in tolerance, the first stimulation leads to the activation of macrophages, but the removal of the stimulus results in the loss of the activating marks. In addition,
suppressive nucleosome remodeling complexes and histone enzymes are involved in the process. CONCLUSION Macrophages, heterogeneous cells whose surroundings regulate their phenotypes and
functions, have central roles in danger detection, inflammation and host defense. Tissue-resident macrophages are specific to the environment and play a critical role in the maintenance of
tissue homeostasis to maintain physiological functions. Under different transcriptional profiles and various stimulations, M1/M2 macrophages have almost opposite functions and different
transcriptional profiles, but both of these cell types have unique abilities to destroy pathogens or repair inflammation-mediated damage; therefore, these cells are also necessary for
restoring homeostasis. It is becoming increasingly clear that epigenetic modifiers have the ability to determine the fate of macrophages, whether in the health tissues or at the site of
tissue injury. Therefore, the application of the epigenetic paradigm in macrophage studies is of great value. The dynamic regulation of epigenetic patterns in macrophages provides
opportunities to alter disease-related epigenetic status. Hence, it can be assumed that understanding the epigenetic regulation of macrophages will substantially contribute to medicine in
terms of diagnostic and therapeutic mechanisms and modalities. Overall, work on the epigenetic regulation of macrophages has laid the foundation for the development of human medicine, and
thus, this issue deserves more extensive and thorough study. REFERENCES * Hume, D. A. Differentiation and heterogeneity in the mononuclear phagocyte system. _Mucosal Immunol._ 1, 432–441
(2008). Article CAS PubMed Google Scholar * Kim, Y. B. et al. Programming of macrophages by UV-irradiated apoptotic cancer cells inhibits cancer progression and lung metastasis. _Cell
Mol. Immunol._ (2019). [Epub ahead of print]. * Ginhoux, F. & Guilliams, M. Tissue-resident macrophage ontogeny and homeostasis. _Immunity_ 44, 439–449 (2016). Article CAS PubMed
Google Scholar * Wynn, T. A., Chawla, A. & Pollard, J. W. Macrophage biology in development, homeostasis and disease. _Nature_ 496, 445–455 (2013). Article CAS PubMed PubMed Central
Google Scholar * Chu, Z. et al. Primed macrophages directly and specifically reject allografts. _Cell Mol. Immunol._ (2019). [Epub ahead of print]. * Karl, E., Tim, O., Bryan, M. T.,
Gwen, C. & Robin, C. A. Transient inhibition of histone deacetylation alters the structural and functional imprint at fission yeast centromeres. _Cell_ 91, 1021–1032 (1997). Article
Google Scholar * Davidson, E. J. & Yang, I. V. Role of epigenetics in the development of childhood asthma. _Curr. Opin. Allergy Clin. Immunol._ 18, 132–138 (2018). Article CAS PubMed
PubMed Central Google Scholar * Keating, S. T. & El-Osta, A. Epigenetics and metabolism. _Circ. Res._ 116, 715–736 (2015). Article CAS PubMed Google Scholar * David, S. G. &
William, T. G. Nuclease hypersensitive sites in chromatin. _Ann. Rev. Biochem._ 57, 159–197 (1988). Article Google Scholar * Ramirez-Carrozzi, V. R. et al. A unifying model for the
selective regulation of inducible transcription by CpG islands and nucleosome remodeling. _Cell_ 138, 114–128 (2009). Article CAS PubMed PubMed Central Google Scholar * Sterner, D. E.
& Berger, S. L. Acetylation of histones and transcription-related factors. _Microbiol Mol. Biol. Rev._ 64, 435–459 (2000). Article CAS PubMed PubMed Central Google Scholar *
Filippakopoulos, P. & Knapp, S. Targeting bromodomains: epigenetic readers of lysine acetylation. _Nat. Rev. Drug Discov._ 13, 337–356 (2014). Article CAS PubMed Google Scholar *
Tyagi, M., Imam, N., Verma, K. & Patel, A. K. Chromatin remodelers: we are the drivers!! _Nucleus_ 7, 388–404 (2016). Article CAS PubMed PubMed Central Google Scholar * Fu, J.,
Nguyen, T. H., Maltby, S., Eyers, F., Foster, P. S., Yang, M. Bromodomain and extra terminal (BET) inhibitor suppresses macrophage-driven steroid-resistant exacerbations of airway
hyper-responsiveness and inflammation. _PLoS ONE_ 11, e0163392 (2016). Article PubMed PubMed Central CAS Google Scholar * Ivashkiv, L. B. Epigenetic regulation of macrophage
polarization and function. _Trends Immunol._ 34, 216–223 (2013). Article CAS PubMed Google Scholar * Bernstein, B. E. et al. Genomic maps and comparative analysis of histone
modifications in human and mouse. _Cell_ 120, 169–181 (2005). Article CAS PubMed Google Scholar * Barski, A. et al. High-resolution profiling of histone methylations in the human genome.
_Cell_ 129, 823–837 (2007). Article CAS PubMed Google Scholar * Patel, D. J. A structural perspective on readout of epigenetic histone and DNA methylation marks. _Cold Spring Harb.
Perspect. Biol._ 8, a018754 (2016). Article PubMed PubMed Central CAS Google Scholar * Holliday, R. & Pugh, J. E. DNA modification mechanisms and gene activity during development.
_Science_ 187, 226–232 (1975). Article CAS PubMed Google Scholar * Manzo, M. et al. Isoform-specific localization of DNMT3A regulates DNA methylation fidelity at bivalent CpG islands.
_EMBO J._ 36, 3421–3434 (2017). Article CAS PubMed PubMed Central Google Scholar * Gilbert, N. et al. DNA methylation affects nuclear organization, histone modifications, and linker
histone binding but not chromatin compaction. _J. Cell Biol._ 177, 401–411 (2007). Article CAS PubMed PubMed Central Google Scholar * Jin, J. et al. The effects of cytosine methylation
on general transcription factors. _Sci. Rep._ 6, 29119 (2016). Article PubMed PubMed Central Google Scholar * Graeme, D. P., Graham, F. K., Steven, A. S., Sohaila, R. & Neil, B. A
gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. _Nature_ 349, 38–44 (1991). Article Google Scholar * Han, P. & Chang,
C. P. Long non-coding RNA and chromatin remodeling. _RNA Biol._ 12, 1094–1098 (2015). Article PubMed PubMed Central Google Scholar * Li, X., Wu, Z., Fu, X. & Han, W. lncRNAs:
insights into their function and mechanics in underlying disorders. _Mutat. Res. Rev. Mutat. Res._ 762, 1–21 (2014). Article CAS PubMed Google Scholar * Rinn, J. L. et al. Functional
demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. _Cell_ 129, 1311–1323 (2007). Article CAS PubMed PubMed Central Google Scholar * Tsai, M. C. et
al. Long noncoding RNA as modular scaffold of histone modification complexes. _Science_ 329, 689–693 (2010). Article CAS PubMed PubMed Central Google Scholar * Eric, P., Ingrid, L.
& Delphine, Hr Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding
RNA whose expression coclusters with ARF. _Cancer Res._ 67, 3963–3969 (2007). Article CAS Google Scholar * Bartel, D. P. MicroRNAs: target recognition and regulatory functions. _Cell_
136, 215–233 (2009). Article CAS PubMed PubMed Central Google Scholar * Suganuma, T. & Workman, J. L. Signals and combinatorial functions of histone modifications. _Annu. Rev.
Biochem._ 80, 473–499 (2011). Article CAS PubMed Google Scholar * Van Furth, R. & Cohn, Z. The origin and kinetics of mononuclear phagocytes. _J. Exp. Med._ 128, 415–435 (1968).
Article PubMed PubMed Central Google Scholar * Gomez Perdiguero, E. et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. _Nature_ 518, 547–551
(2015). Article CAS PubMed Google Scholar * Schulz, C. et al. A lineage of myeloid cells independent of myb and hematopoietic stem cells. _Science_ 335, 86–90 (2012). Article CAS
Google Scholar * Kierdorf, K. et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. _Nat. Neurosci._ 16, 273–280 (2013). Article CAS PubMed Google
Scholar * Hoeffel, G. et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. _Immunity_ 42, 665–678 (2015). Article CAS PubMed
PubMed Central Google Scholar * Lavin, Y. et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. _Cell_ 159, 1312–1326 (2014). Article CAS
PubMed PubMed Central Google Scholar * Kumaki, Y. et al. Analysis and synthesis of high-amplitude Cis-elements in the mammalian circadian clock. _Proc. Natl. Acad. Sci. USA_ 105,
14946–14951 (2008). Article CAS PubMed PubMed Central Google Scholar * Romanoski, C. E., Link, V. M., Heinz, S. & Glass, C. K. Exploiting genomics and natural genetic variation to
decode macrophage enhancers. _Trends Immunol._ 36, 507–518 (2015). Article CAS PubMed PubMed Central Google Scholar * Zaret, K. S. & Carroll, J. S. Pioneer transcription factors:
establishing competence for gene expression. _Genes Dev._ 25, 2227–2241 (2011). Article CAS PubMed PubMed Central Google Scholar * Heinz, S. et al. Simple combinations of
lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. _Mol. Cell_ 38, 576–589 (2010). Article CAS PubMed PubMed Central
Google Scholar * Heinz, S. et al. Effect of natural genetic variation on enhancer selection and function. _Nature_ 503, 487–492 (2013). Article CAS PubMed PubMed Central Google Scholar
* Gosselin, D. et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. _Cell_ 159, 1327–1340 (2014). Article CAS PubMed PubMed
Central Google Scholar * Amit, I., Winter, D. R. & Jung, S. The role of the local environment and epigenetics in shaping macrophage identity and their effect on tissue homeostasis.
_Nat. Immunol._ 17, 18–25 (2016). Article CAS PubMed Google Scholar * Kaikkonen, M. U. et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer
transcription. _Mol. Cell_ 51, 310–325 (2013). Article CAS PubMed PubMed Central Google Scholar * Creyghton, M. P. et al. Histone H3K27ac separates active from poised enhancers and
predicts developmental state. _Proc. Natl. Acad. Sci. USA_ 107, 21931–21936 (2010). Article CAS PubMed PubMed Central Google Scholar * Ostuni, R. et al. Latent enhancers activated by
stimulation in differentiated cells. _Cell_ 152, 157–171 (2013). Article CAS PubMed Google Scholar * Salter, M. W. & Stevens, B. Microglia emerge as central players in brain disease.
_Nat. Med._ 23, 1018–1027 (2017). Article CAS PubMed Google Scholar * Gosselin, D. et al. An environment-dependent transcriptional network specifies human microglia identity. _Science_
356, 1248–1259 (2017). Article CAS Google Scholar * Buttgereit, A. et al. Sall1 is a transcriptional regulator defining microglia identity and function. _Nat. Immunol._ 17, 1397–1406
(2016). Article CAS PubMed Google Scholar * Datta, M. et al. Histone deacetylases 1 and 2 regulate microglia function during development, homeostasis, and neurodegeneration in a
context-dependent manner. _Immunity_ 48, 514–529.e516 (2018). Article CAS PubMed Google Scholar * Ponomarev, E. D., Veremeyko, T., Barteneva, N., Krichevsky, A. M. & Weiner, H. L.
MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-α-PU.1 pathway. _Nat. Med._ 17, 64–70 (2011). Article CAS PubMed Google Scholar *
Varol, C., Mildner, A. & Jung, S. Macrophages: development and tissue specialization. _Annu Rev. Immunol._ 33, 643–675 (2015). Article CAS PubMed Google Scholar * Mildner, A. et al.
CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. _Brain_ 132, 2487–2500 (2009). Article PubMed Google Scholar * Ajami, B., Bennett,
J. L., Krieger, C., McNagny, K. M. & Rossi, F. M. V. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. _Nat. Neurosci._ 14, 1142–1149
(2011). Article CAS PubMed Google Scholar * Calum, C. B. & Allan, M. M. Macrophages in intestinal homeostasis and inflammation. _Immunol. Rev._ 260, 102–117 (2014). Article CAS
Google Scholar * Bain, C. C. et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. _Nat. Immunol._ 15, 929–937 (2014).
Article CAS PubMed PubMed Central Google Scholar * Simon, J. M. et al. Alterations to chromatin in intestinal macrophages link IL-10 deficiency to inappropriate inflammatory responses.
_Eur. J. Immunol._ 46, 1912–1925 (2016). Article CAS PubMed PubMed Central Google Scholar * Schneider, C. et al. Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is
critical for the differentiation of fetal monocytes into alveolar macrophages. _Nat. Immunol._ 15, 1026–1037 (2014). Article CAS PubMed Google Scholar * Guilliams, M. et al. Alveolar
macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. _J. Exp. Med_. 210, 1977–1992 (2013). Article CAS PubMed PubMed
Central Google Scholar * Ebina-Shibuya, R. et al. The double knockout of Bach1 and Bach2 in mice reveals shared compensatory mechanisms in regulating alveolar macrophage function and lung
surfactant homeostasis. _J. Biochem._ 160, 333–344 (2016). Article CAS PubMed Google Scholar * Li, R., Fang L., Pu, Q., Bu, H., Zhu, P. MEG3-4 is a miRNA decoy that regulates IL-1β
abundance to initiate and then limit inflammation to prevent sepsis during lung infection. _Science Signal, abbreviated in Sci Signal_ 11, eaao2387 (2018). Article PubMed PubMed Central
CAS Google Scholar * Mebius, R. E. & Kraal, G. Structure and function of the spleen. _Nat. Rev. Immunol._ 5, 606–616 (2005). Article CAS PubMed Google Scholar * Miyake, Y. et al.
Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell-associated antigens. _J. Clin. Invest._ 117, 2268–2278 (2007). Article CAS PubMed
PubMed Central Google Scholar * N, A. G. et al. The nuclear receptor LXRalpha controls the functional specialization of splenic macrophages. _Nat. Immunol._ 14, 831–839 (2013). Article
CAS Google Scholar * Kohyama, M. et al. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. _Nature_ 457, 318–321 (2009). Article CAS PubMed Google
Scholar * Bain, C. C. & Jenkins, S. J. The biology of serous cavity macrophages. _Cell Immunol._ 330, 126–135 (2018). Article CAS PubMed Google Scholar * Rosas, M. et al. The
transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. _Science_ 344, 645–648 (2014). Article CAS PubMed PubMed Central Google Scholar * Li, X. et al.
Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. _Nat. Immunol._ 17, 806–815 (2016). Article CAS PubMed Google
Scholar * Gough, D. J., Messina, N. L., Clarke, C. J. P., Johnstone, R. W. & Levy, D. E. Constitutive Type I interferon modulates homeostatic balance through tonic signaling. _Immunity_
36, 166–174 (2012). Article CAS PubMed PubMed Central Google Scholar * Abt, M. C. et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. _Immunity_
37, 158–170 (2012). Article CAS PubMed PubMed Central Google Scholar * Ganal, S. C. et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive
signals from commensal microbiota. _Immunity_ 37, 171–186 (2012). Article CAS PubMed Google Scholar * Marazzi, I. et al. Suppression of the antiviral response by an influenza histone
mimic. _Nature_ 483, 428–433 (2012). Article CAS PubMed PubMed Central Google Scholar * Okabe, Y. & Medzhitov, R. Tissue-specific signals control reversible program of localization
and functional polarization of macrophages. _Cell_ 157, 832–844 (2014). Article CAS PubMed PubMed Central Google Scholar * Murray, P. J. & Wynn, T. A. Protective and pathogenic
functions of macrophage subsets. _Nat. Rev. Immunol._ 11, 723–737 (2011). Article CAS PubMed PubMed Central Google Scholar * Fleetwood, A. J., Lawrence, T., Hamilton, J. A. & Cook,
A. D. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities:
implications for CSF blockade in inflammation. _J. Immunol._ 178, 5245–5252 (2007). Article CAS PubMed Google Scholar * Kim, S.-J. et al. Macrophages are the primary effector cells in
IL-7-induced arthritis. _Cell Mol. Immunol._ (2019). [Epub ahead of print]. * Soehnlein, O. & Lindbom, L. Phagocyte partnership during the onset and resolution of inflammation. _Nat.
Rev. Immunol._ 10, 427–439 (2010). Article CAS PubMed Google Scholar * Jenkins, S. J. et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2
inflammation. _Science_ 332, 1284–1288 (2011). Article CAS PubMed PubMed Central Google Scholar * Ginhoux, F. & Jung, S. Monocytes and macrophages: developmental pathways and
tissue homeostasis. _Nat. Rev. Immunol._ 14, 392–404 (2014). Article CAS PubMed Google Scholar * Wallner, S. et al. Epigenetic dynamics of monocyte-to-macrophage differentiation.
_Epigenetics Chromatin_ 9, 33 (2016). Article PubMed PubMed Central CAS Google Scholar * Pastor, W. A., Aravind, L. & Rao, A. TETonic shift: biological roles of TET proteins in DNA
demethylation and transcription. _Nat. Rev. Mol. Cell Biol._ 14, 341–356 (2013). Article CAS PubMed PubMed Central Google Scholar * Pietrzak, J., Ploszaj, T., Pulaski, L. &
Robaszkiewicz, A. EP300-HDAC1-SWI/SNF functional unit defines transcription of some DNA repair enzymes during differentiation of human macrophages. _Biochim. Biophys. Acta Gene Regul. Mech._
1862, 198–208 (2019). Article CAS PubMed Google Scholar * Hope, T. J., Sung T.-L. & Rice, A. P. miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism
of action appears to involve repression of cyclin T1. _PLoS Pathog_. 5, e1000263 (2009). * Yu, W., Wang, Y., Shaw, C. A., Qin, X. F. & Rice, A. P. Induction of the HIV-1 Tat co-factor
cyclin T1 during monocyte differentiation is required for the regulated expression of a large portion of cellular mRNAs. _Retrovirology_ 3, 32 (2006). Article PubMed PubMed Central CAS
Google Scholar * Murray, P. J. et al. Macrophage activation and polarization: nomenclature and experimental guidelines. _Immunity_ 41, 14–20 (2014). Article CAS PubMed PubMed Central
Google Scholar * Amy, S. W., Scott, E. P. & Stephen, T. S. Rapid and selective remodeling of a positioned nucleosome during the induction of IL-12p40 transcription. _Immunity_ 11,
665–675 (1999). Article Google Scholar * Amy, S. W. et al. Nucleosome remodeling at the IL-12p40 promoter is a TLR-dependent, Rel-independent event. _Nat. Immunol._ 2, 51–57 (2001).
Article CAS Google Scholar * Zhu, Y., van Essen, D. & Saccani, S. Cell-type-specific control of enhancer activity by H3K9 trimethylation. _Mol. Cell_ 46, 408–423 (2012). Article CAS
PubMed Google Scholar * Kruidenier, L. et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. _Nature_ 488, 404–408 (2012). Article
CAS PubMed PubMed Central Google Scholar * Stender, J. D. et al. Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. _Mol. Cell_ 48,
28–38 (2012). Article CAS PubMed PubMed Central Google Scholar * Barish, G. D. et al. Bcl-6 and NF-κB cistromes mediate opposing regulation of the innate immune response. _Genes Dev._
24, 2760–2765 (2010). Article CAS PubMed PubMed Central Google Scholar * De Santa, F. et al. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. _EMBO J._
28, 3341–3352 (2009). Article PubMed PubMed Central CAS Google Scholar * Chen, S. et al. The histone H3 Lys 27 demethylase JMJD3 regulates gene expression by impacting transcriptional
elongation. _Genes Dev._ 26, 1364–1375 (2012). Article CAS PubMed PubMed Central Google Scholar * Yan, Q. et al. Jmjd3-mediated epigenetic regulation of inflammatory cytokine gene
expression in serum amyloid A-stimulated macrophages. _Cell Signal._ 26, 1783–1791 (2014). Article CAS PubMed PubMed Central Google Scholar * Kittan, N. A. et al. Cytokine induced
phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. _PLoS ONE_ 8, e78045 (2013). Article CAS PubMed PubMed Central Google Scholar * Chen, X. et
al. Methyltransferase Dot1l preferentially promotes innate IL-6 and IFN-beta production by mediating H3K79me2/3 methylation in macrophages. _Cell Mol. Immunol_. (2018). [Epub ahead of
print]. * Gye, Y. P., Myungsoo, J., Tetyana, P., Timothy, S. B. & John, W. C. Regulation of macrophage cyclooxygenase-2 gene expression by modifications of histone H3. _Am. J. Physiol.
Lung Cell Mol. Physiol._ 286, L956–L962 (2004). Article Google Scholar * Hnin, T. A. et al. LPS regulates proinflammatory gene expression in macrophages by altering histone deacetylase
expression. _FASEB J._ 20, 1315–1327 (2006). Article CAS Google Scholar * Schmeck, B. et al. Histone acetylation and flagellin are essential for Legionella pneumophila-induced cytokine
expression. _J. Immunol._ 181, 940–947 (2008). Article CAS PubMed Google Scholar * Yan, B. et al. HDAC6 deacetylase activity is critical for lipopolysaccharide-induced activation of
macrophages. _PLoS ONE_ 9, e110718 (2014). Article PubMed PubMed Central CAS Google Scholar * Shakespear, M. R. et al. Histone deacetylase 7 promotes toll-like receptor 4-dependent
proinflammatory gene expression in macrophages. _J. Biol. Chem._ 288, 25362–25374 (2013). Article CAS PubMed PubMed Central Google Scholar * Chen, X. et al. Requirement for the histone
deacetylase Hdac3 for the inflammatory gene expression program in macrophages. _Proc. Natl. Acad. Sci. USA_ 109, E2865–E2874 (2012). CAS PubMed PubMed Central Google Scholar * Jia, Y. et
al. IRF8 is the target of SIRT1 for the inflammation response in macrophages. _Innate Immun._ 23, 188–195 (2017). Article CAS PubMed Google Scholar * Schuettengruber, B., Chourrout, D.,
Vervoort, M., Leblanc, B. & Cavalli, G. Genome regulation by polycomb and trithorax proteins. _Cell_ 128, 735–745 (2007). Article CAS PubMed Google Scholar * Qiao, Y., Kang, K.,
Giannopoulou, E., Fang, C. & Ivashkiv, L. B. IFN-gamma induces histone 3 lysine 27 trimethylation in a small subset of promoters to stably silence gene expression in human macrophages.
_Cell Rep._ 16, 3121–3129 (2016). Article CAS PubMed PubMed Central Google Scholar * Kang, K. et al. Interferon-gamma represses M2 gene expression in human macrophages by disassembling
enhancers bound by the transcription factor MAF. _Immunity_ 47, 235–250 e234 (2017). Article CAS PubMed PubMed Central Google Scholar * Wu, C., Li, A., Hu, J. & Kang, J. Histone
deacetylase 2 is essential for LPS-induced inflammatory responses in macrophages. _Immunol. Cell Biol._ 97, 72–84 (2019). Article CAS PubMed Google Scholar * Cao, Q. et al. Histone
deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. _Arterioscler. Thromb. Vasc. Biol._ 34, 1871–1879 (2014). Article CAS
PubMed PubMed Central Google Scholar * Alver, B. H. et al. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. _Nat. Commun._ 8, 14648
(2017). Article PubMed PubMed Central Google Scholar * Cheng, C. et al. SOCS1 hypermethylation mediated by DNMT1 is associated with lipopolysaccharide-induced inflammatory cytokines in
macrophages. _Toxicol. Lett._ 225, 488–497 (2014). Article CAS PubMed Google Scholar * Yan, J. et al. Diabetes impairs wound healing by Dnmt1-dependent dysregulation of hematopoietic
stem cells differentiation towards macrophages. _Nat. Commun._ 9, 33 (2018). * Rajaram, M. V. et al. Mycobacterium tuberculosis lipomannan blocks TNF biosynthesis by regulating macrophage
MAPK-activated protein kinase 2 (MK2) and microRNA miR-125b. _Proc. Natl. Acad. Sci. USA_ 108, 17408–17413 (2011). Article CAS PubMed PubMed Central Google Scholar * Jaiswal, A., Reddy,
S. S., Maurya, M., Maurya, P. & Barthwal, M. K. MicroRNA-99a mimics inhibit M1 macrophage phenotype and adipose tissue inflammation by targeting TNFα. _Cell Mol. Immunol._ 16, 495–507
(2018). Article PubMed CAS PubMed Central Google Scholar * Satoh, T. et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. _Nat.
Immunol._ 11, 936–944 (2010). Article CAS PubMed Google Scholar * Liu, P. S. et al. alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic
reprogramming. _Nat. Immunol._ 18, 985–994 (2017). Article CAS PubMed Google Scholar * Lo Sasso, G. et al. SIRT2 deficiency modulates macrophage polarization and susceptibility to
experimental colitis. _PLoS ONE_ 9, e103573 (2014). Article PubMed PubMed Central CAS Google Scholar * Mullican, S. E. et al. Histone deacetylase 3 is an epigenomic brake in macrophage
alternative activation. _Genes Dev._ 25, 2480–2488 (2011). Article CAS PubMed PubMed Central Google Scholar * Czimmerer, Z. et al. The transcription factor STAT6 mediates direct
repression of inflammatory enhancers and limits activation of alternatively polarized macrophages. _Immunity_ 48, 75–90 (2018). Article CAS PubMed PubMed Central Google Scholar * Yang,
X. et al. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. _Mol. Endocrinol._ 28, 565–574 (2014). Article PubMed PubMed Central CAS Google Scholar * Su, S.
et al. miR-142-5p and miR-130a-3p are regulated by IL-4 and IL-13 and control profibrogenic macrophage program. _Nat. Commun._ 6, 8523 (2015). Article CAS PubMed Google Scholar *
Karo-Atar, D., Itan, M., Pasmanik-Chor, M. & Munitz, A. MicroRNA profiling reveals opposing expression patterns for miR-511 in alternatively and classically activated macrophages. _J.
Asthma_ 52, 545–553 (2015). Article PubMed CAS Google Scholar * Daniel, B. et al. The nuclear receptor PPARgamma controls progressive macrophage polarization as a ligand-insensitive
epigenomic ratchet of transcriptional memory. _Immunity_ 49, 615–626 e616 (2018). Article CAS PubMed PubMed Central Google Scholar * Van Den Bossche, J. et al. Inhibiting epigenetic
enzymes to improve atherogenic macrophage functions. _Biochem. Biophys. Res Commun._ 455, 396–402 (2014). Article PubMed CAS Google Scholar * Bowes, A. J., Khan, M. I., Shi, Y.,
Robertson, L. & Werstuck, G. H. Valproate attenuates accelerated atherosclerosis in hyperglycemic ApoE-deficient mice. _Am. J. Pathol._ 174, 330–342 (2009). Article CAS PubMed PubMed
Central Google Scholar * Markus, H. S. et al. Evidence HDAC9 genetic variant associated with ischemic stroke increases risk via promoting carotid atherosclerosis. _Stroke_ 44, 1220–1225
(2013). Article CAS PubMed PubMed Central Google Scholar * Nazari-Jahantigh, M. et al. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. _J. Clin. Invest._ 122,
4190–4202 (2012). Article CAS PubMed PubMed Central Google Scholar * Li, Z. et al. Oxidized low-density lipoprotein upregulates microRNA-146a via JNK and NF-κB signaling. _Mol. Med.
Rep._ 13, 1709–1716 (2016). Article CAS PubMed Google Scholar * Babu, M. et al. Differential promoter methylation of macrophage genes is associated with impaired vascular growth in
ischemic muscles of hyperlipidemic and Type 2 diabetic mice: genome-wide promoter methylation study. _Circ. Res._ 117, 289–299 (2015). Article CAS PubMed Google Scholar * Wang, X. et al.
Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. _JCI Insight_ 1, e87748 (2016). PubMed PubMed Central Google Scholar * Reddy, M. A. et
al. Regulation of inflammatory phenotype in macrophages by a diabetes-induced long noncoding RNA. _Diabetes_ 63, 4249–4261 (2014). Article CAS PubMed PubMed Central Google Scholar *
Willenborg, S. et al. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. _Blood_ 120, 613–625 (2012). Article CAS PubMed Google Scholar *
Kimball, A., Joshi, A., Henke, P. K., Kunkel, S. & Gallagher, K. PC222. altered histone methylation at the IL-1B promoter in diabetic macrophages enhances inflammation and impairs wound
healing. _J. Vasc. Surg._ 61 176S (2015). * Xu, J. et al. The role of microRNA-146a in the pathogenesis of the diabetic wound-healing impairment: correction with mesenchymal stem cell
treatment. _Diabetes_ 61, 2906–2912 (2012). Article CAS PubMed PubMed Central Google Scholar * Vincent, J.-L., Opal, S. M., Marshall, J. C. & Tracey, K. J. Sepsis definitions: time
for change. _Lancet_ 381, 774–775 (2013). Article PubMed PubMed Central Google Scholar * Li, Y. et al. Inhibition of histone deacetylase 6 improves long-term survival in a lethal septic
model. _J. Trauma Acute Care_ 78, 378–385 (2015). Article CAS Google Scholar * How, C.-K. et al. Expression profile of microRNAs in gram-negative bacterial sepsis. _Shock_ 43, 121–127
(2015). Article CAS PubMed Google Scholar * Chen, Y. et al. Long non-coding RNA NEAT1 plays an important role in sepsis-induced acute kidney injury by targeting miR-204 and modulating
the NF-κB pathway. _Int. Immunopharmacol._ 59, 252–260 (2018). Article CAS PubMed Google Scholar * Zhang, C. C. & Niu, F. LncRNA NEAT1 promotes inflammatory response in
sepsis-induced liver injury via the Let-7a/TLR4 axis. _Int. Immunopharmacol._ 75, 105731 (2019). Article CAS PubMed Google Scholar * Wendeln, A. C. et al. Innate immune memory in the
brain shapes neurological disease hallmarks. _Nature_ 556, 332–338 (2018). Article CAS PubMed PubMed Central Google Scholar * Ivashkiv, L. B. Inflammatory signaling in macrophages:
transitions from acute to tolerant and alternative activation states. _Eur. J. Immunol._ 41, 2477–2481 (2011). Article CAS PubMed PubMed Central Google Scholar * Foster, S. L.,
Hargreaves, D. C. & Medzhitov, R. Gene-specific control of inflammation by TLR-induced chromatin modifications. _Nature_ 447, 972–978 (2007). Article CAS PubMed Google Scholar *
Rodriguez, R. M., Suarez-Alvarez, B. & Lopez-Larrea, C. Therapeutic epigenetic reprogramming of trained immunity in myeloid cells. _Trends Immunol._ 40, 66–80 (2019). Article CAS
PubMed Google Scholar * Vladimir, R. R.-C. et al. Selective and antagonistic functions of SWI/SNF and Mi-2β nucleosome remodeling complexes during an inflammatory response. _Genes Dev._
20, 282–296 (2006). Article CAS Google Scholar * Yan, Q. et al. Nuclear factor-κB binding motifs specify Toll-like receptor-induced gene repression through an inducible repressosome.
_Proc. Natl. Acad. Sci. USA_ 109, 14140–14145 (2012). Article CAS PubMed PubMed Central Google Scholar * Chen, X., El Gazzar, M., Yoza, B. K. & McCall, C. E. The NF-κB factor RelB
and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. _J. Biol. Chem._ 284, 27857–27865 (2009). Article CAS PubMed PubMed
Central Google Scholar * Nahid, M. A. et al. TLR4, TLR7/8 agonist-induced miR-146a promotes macrophage tolerance to MyD88-dependent TLR agonists. _J. Leukoc. Biol._ 100, 339–349 (2016).
Article PubMed CAS Google Scholar * Seeley, J. J. et al. Induction of innate immune memory via microRNA targeting of chromatin remodelling factors. _Nature_ 559, 114–119 (2018). Article
CAS PubMed PubMed Central Google Scholar * Norouzitallab, P., Baruah, K., Biswas, P., Vanrompay, D. & Bossier, P. Probing the phenomenon of trained immunity in invertebrates during
a transgenerational study, using brine shrimp Artemia as a model system. _Sci. Rep._ 6, 21166 (2016). Article CAS PubMed PubMed Central Google Scholar * Quintin, J. et al. Candida
albicans infection affords protection against reinfection via functional reprogramming of monocytes. _Cell Host Microbe_ 12, 223–232 (2012). Article CAS PubMed Google Scholar * Haley, M.
J., Brough, D., Quintin, J. & Allan, S. M. Microglial priming as trained immunity in the brain. _Neuroscience_ 405, 47–54 (2018). * Netea, M. G. et al. Trained immunity: a program of
innate immune memory in health and disease. _Science_ 352, 427 (2016). Article CAS Google Scholar * Bekkering, S. et al. Oxidized low-density lipoprotein induces long-term proinflammatory
cytokine production and foam cell formation via epigenetic reprogramming of monocytes. _Arterioscler Thromb. Vasc. Biol._ 34, 1731–1738 (2014). Article CAS PubMed Google Scholar *
Fanucchi, S. & Mhlanga, M. M. Lnc-ing trained immunity to chromatin architecture. _Front. Cell Dev. Biol._ 7, 2 (2019). * Brown, G. D. et al. Dectin-1 is a major β-glucan receptor on
macrophages. _J. Exp. Med._ 196, 407–412 (2002). Article CAS PubMed PubMed Central Google Scholar * Saeed, S., et al. Epigenetic programming of monocyte-to-macrophage differentiation
and trained innate immunity. _Science_ 345, 1251086 (2014). * Cabezas-Wallscheid, N. et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome,
transcriptome, and DNA methylome analysis. _Cell Stem Cell_ 15, 507–522 (2014). Article CAS PubMed Google Scholar * Kaufmann, E. et al. BCG educates hematopoietic stem cells to generate
protective innate immunity against tuberculosis. _Cell_ 172, 176–190.e119 (2018). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS This work is supported by the
National Natural Science Foundation of China (No. 81602492), the National Key Research and Development Program of China (No. 2016YFA0201402) and the National Major Scientific and
Technological Special Project for “Significant New Drugs Development” (No. 2018ZX09733001). AUTHOR INFORMATION Author notes * These authors contributed equally: Siyuan Chen, Jing Yang
AUTHORS AND AFFILIATIONS * Laboratory of Aging Research and Cancer Drug Target, State Key Laboratory of Biotherapy, National Clinical Research Center for Geriatrics, West China Hospital,
Sichuan University, No. 17, Block 3, Southern Renmin Road, 610041, Chengdu, Sichuan, PR China Siyuan Chen, Jing Yang, Yuquan Wei & Xiawei Wei Authors * Siyuan Chen View author
publications You can also search for this author inPubMed Google Scholar * Jing Yang View author publications You can also search for this author inPubMed Google Scholar * Yuquan Wei View
author publications You can also search for this author inPubMed Google Scholar * Xiawei Wei View author publications You can also search for this author inPubMed Google Scholar
CORRESPONDING AUTHOR Correspondence to Xiawei Wei. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. RIGHTS AND PERMISSIONS OPEN ACCESS This article is
licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in
this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative
Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a
copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Chen, S., Yang, J., Wei, Y. _et al._ Epigenetic
regulation of macrophages: from homeostasis maintenance to host defense. _Cell Mol Immunol_ 17, 36–49 (2020). https://doi.org/10.1038/s41423-019-0315-0 Download citation * Received: 09 June
2019 * Accepted: 28 September 2019 * Published: 29 October 2019 * Issue Date: January 2020 * DOI: https://doi.org/10.1038/s41423-019-0315-0 SHARE THIS ARTICLE Anyone you share the following
link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature
SharedIt content-sharing initiative KEYWORDS * Macrophage * Epigenetics * Polarization * Immune memory

:max_bytes(150000):strip_icc():focal(499x0:501x2)/gettyimages-631246246-0c642aedaed24b7e86890f2a4d198d08.jpg)




