- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
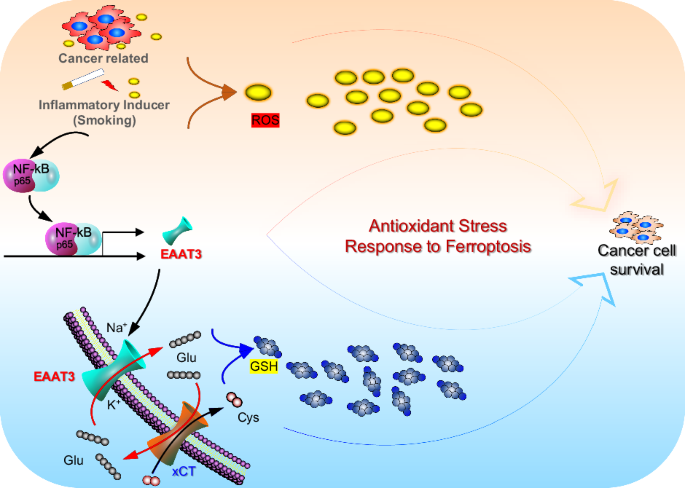
ABSTRACT Cellular glutathione (GSH) in lung cancer cells represents the most abundant antioxidant. GSH production is regulated not only by upregulated cystine/glutamate exchanger (xCT) but
also by the involvement of glutamate transporters, specifically excitatory amino acid transporter 3 (EAAT3). Our prior research established that the uptake of glutamate via EAAT3 plays a
pivotal role in driving cystine uptake through xCT, contributing to GSH biosynthesis during lung tumorigenesis. Nevertheless, the underlying mechanism governing the upregulation of EAAT3
remains enigmatic. In this study, we conducted a comprehensive reanalysis of publicly available data and employed the Gprc5a–/–/SR-IκB mouse model alongside in vitro cell experiments to
elucidate the correlations between NF-κB and EAAT3 in lung cancer. We observed that EAAT3 knockdown, similar to NF-κB inhibition, led to the accumulation of reactive oxygen species (ROS) and
increased sensitivity to ferroptosis induction by RAS-selective lethal 3 (RSL3). Mechanistic insights were obtained through chromatin immunoprecipitation and luciferase reporter assays,
revealing that NF-κB induces EAAT3 expression via two putative cis-elements within its promoter. Furthermore, our investigation unveiled the upregulation of EAAT3 in a subset of clinical
non-small cell lung cancer (NSCLC) tissues, exhibiting a positive correlation with the P65 protein. In addition, the inflammatory factor of smoking was found to augment EAAT3 expression in
both human and murine experimental models. These findings collectively emphasize the pivotal role of the NF-κB/EAAT3 axis in managing antioxidant stress and influencing lung cancer
development. Moreover, this research offers insights into the potential for a combined ferroptosis therapy strategy in lung cancer treatment. HIGHLIGHTS * NF-κB drives EAAT3 expression, key
for GSH biosynthesis in lung cancer cells. * EAAT3 knockdown increases ROS and enhances sensitivity to ferroptosis in lung cancer. * EAAT3 upregulation in NSCLC tissues correlates with P65
protein levels. * Smoking-induced inflammation boosts EAAT3 expression in lung cancer models. * Potential for combined ferroptosis therapy targeting NF-κB/EAAT3 axis in lung cancer. SIMILAR
CONTENT BEING VIEWED BY OTHERS LACTATE DEHYDROGENASE B NONCANONICALLY PROMOTES FERROPTOSIS DEFENSE IN _KRAS_-DRIVEN LUNG CANCER Article Open access 07 December 2024 RETINOIC ACID RECEPTOR
ALPHA INHIBITS FERROPTOSIS BY PROMOTING THIOREDOXIN AND PROTEIN PHOSPHATASE 1F IN LUNG ADENOCARCINOMA Article Open access 20 June 2024 INHIBITION OF LSD1 INDUCES FERROPTOSIS THROUGH THE
ATF4-XCT PATHWAY AND SHOWS ENHANCED ANTI-TUMOR EFFECTS WITH FERROPTOSIS INDUCERS IN NSCLC Article Open access 03 November 2023 INTRODUCTION Lung cancer continues to be one of the most
widespread and lethal malignancies globally, with smoking constituting a significant risk factor for its onset [1]. In recent years, extensive research has unveiled the complex interplay
between chronic inflammation and redox imbalance in the context of lung carcinogenesis [2,3,4]. Chronic exposure to tobacco smoke triggers two significant responses. Firstly, it stimulates
lung epithelial cells. This persistent exposure to various inflammatory stimuli, including environmental toxins, infections, and autoimmunity, creates an inflammatory microenvironment that
enhances oncogenic processes and facilitates tumor initiation [5,6,7]. Within this inflammatory milieu, a complex network of pro-inflammatory cytokines, chemokines, and immune cells
orchestrates activities that promote tumor cell proliferation, angiogenesis, and immune evasion [8,9,10]. Furthermore, chronic inflammation can lead to DNA damage and genomic instability,
further elevating the risk of malignant transformation [4]. A second significant response to chronic tobacco smoke exposure is the generation of reactive oxygen species (ROS), which
overwhelm the cellular antioxidant defense system, primarily centered around glutathione (GSH) [11, 12]. This persistent oxidative stress leads to an ongoing inflammatory response and
subsequent cellular damage, fostering the initiation and progression of lung cancer [13, 14]. One area where the effects of ROS have gained significant attention is in the development of
lung cancer. A comprehensive understanding of the intricate interplay between ROS and antioxidant defense systems, such as GSH, is crucial for elucidating disease progression and developing
potential therapeutic strategies in lung cancer. ROS-induced DNA damage, genomic instability, and altered signaling pathways have all been implicated in the initiation and promotion of lung
carcinogenesis [15,16,17]. In contrast, GSH, a vital intracellular antioxidant, plays a pivotal role in maintaining redox homeostasis by scavenging ROS and safeguarding cells against
oxidative damage [18]. The dysregulation of ROS and GSH balance can tip the scales toward an oxidatively stressed microenvironment, contributing to the transformation and survival of
cancerous cells. This work builds on our previous research, which illustrated that active glutamate uptake through solute carrier family 1 member 1 (EAAT3/SLC1A1) plays a pivotal role in
driving cystine uptake via Xc– for GSH biosynthesis in lung tumorigenesis [19]. Cellular GSH in cancer cells is not only determined by upregulated Xc– but also by dysregulated glutamate
transporters [19]. EAAT3 serves as the predominant amino acid transporter for glutamate and aspartate, making it particularly relevant to cancer development, especially within the realm of
cancer metabolism [20, 21]. While the dysregulation of EAAT3 has been associated with metabolic reprogramming and the progression of various solid tumors, the precise regulatory mechanisms
governing EAAT3 remain insufficiently understood. Drawing on our previous mouse model, Gprc5a-knockout (KO) mice not only developed spontaneous lung tumors [22], but also exhibited an
enhanced inflammatory response within lung tissues [23,24,25]. Recent research has proposed that the NF-κB repressing factor (NKRF) specifically binds to the EAAT3 promoter, resulting in
transcriptional downregulation of EAAT3 expression [26]. When considering our RNA sequencing data, we hypothesize that inflammation may play a role in modulating the expression of EAAT3. In
this study, our objective is to demonstrate the significant role of the NF-κB pathway in the anti-ROS system. We aim to show that the inhibition of NF-κB can induce ROS production by
down-regulating the expression of EAAT3. Utilizing the Gprc5a–/–/SPC-SR-IκBα mouse model, we have discovered that NF-κB induces the expression of EAAT3 through two potential cis-elements in
its promoter. Furthermore, in clinical cases involving smokers, we have verified the correlation between EAAT3 and NF-κB in the context of persistent chronic inflammation caused by smoking.
MATERIALS AND METHODS REAGENTS AND ANTIBODIES REAGENTS QNZ-EVP4593(S4902), BMS-345541 (S8044), BSO (S2433), doxycycline (S5159), puromycin 2HCL (S7417), cocktail (B14002), 2x SYBR Green qPCR
master mix (B21203), NNK (E0651) were from Selleck.cn (Houston, TX, USA). GSH-MEE (G1404), hydrogen peroxide solution (H2O2, 95321), TNFα (SRP3177), and DAPI (D9542) were from Sigma-Aldrich
(St. Louis, MO, USA). Dual-Luciferase® Reporter Assay System (E1910) was from Promega (Madison, WI, USA). CellROX™ Orange Reagent (C10443) was from Thermo Fisher Scientific (Waltham, MA,
USA). RIPA buffer (9806) was from Cell Signaling Technology (Danvers, MA, USA). Immobilon Western Reagents (15015B4) was from Millipore (Burlington, MA, USA). siRNA-P65(sc-29410) and
siRNA-EAAT3(sc-41940) were from Santa Cruz Bio (Dallas, TX, USA). 13C6-15N2-labeled cystine (CNLM-4244-H-PK) was from Cambridge Isotope Laboratories (Tewksbury, MA, USA). ANTIBODIES
Anti-EAAT3 antibody (12686-1-AP) for IHC was from Proteintech (Rosemont, IL, USA). Anti-EAAT3 antibody (14501), anti-BAX (5023), anti-p-P65 (3033), anti-P65 (8242), anti-H3K27ac (8173),
anti-V5-tag (13202) were from Cell Signaling Technology (Danvers, MA, USA). Anti-RNA polymerase II (ab5131) was from Abcam (Cambridge, UK). Anti-RAI3 (Gprc5a; sc-98884) was from Santa Cruz
Bio (Dallas, TX, USA). Anti-β-Actin-HRP (PM053-7), anti-DDDDK-tag (M185-3L) were from MBL (Nagoya, Aichi, Japan). Anti-FLAG® M2 Affinity Gel (A2220) was from Sigma-Aldrich (St. Louis, MO,
USA). The antibodies were diluted according to manufacturers’ instructions. CELL LINES AND CELL CULTURE Mouse tracheal epithelial cells (MTECs) were obtained from normal tracheal tissue of
3-week-old Gprc5a+/+ and Gprc5a–/– mice (C57BL/6 X129sv) as described [27, 28]. The MTECs were cultured in K-SFM supplemented with epithelial growth factor (EGF, 5 ng/ml) and bovine
pituitary extract (50 μg/ml, Invitrogen; Carlsbad, CA, USA). Human embryonic kidney cells HEK293T, normal lung epithelial cells (16HBE, HBEC), and NSCLC cells (A549, PC9) were obtained from
the American Type Culture Collection (ATCC; Manassas, VA, USA) and were tested and authenticated by DNA typing at Shanghai Jiao Tong University Analysis Core (Shanghai, China). The mouse
lung adenocarcinoma cell line 1601 was isolated from lung tumor growth in Gprc5a–/– mice. The HBEC cells were cultured in K-SFM, and the other cells were cultured in DMEM essential medium
with 10% fetal calf serum at 37 °C in a humidified incubator in an atmosphere of 95% air and 5% CO2. IMMUNOPRECIPITATION AND WESTERN BLOTTING Cells were lysed with RIPA buffer, and 500 μg
whole cell lysate was precipitated with 2 μg agarose-conjugated antibody against Flag (Flag beads) and normal IgG (as a negative control). PBS with 0.05% Tween-20 was used to wash the beads
five times. Then, an equal amount of protein loading buffer was added and boiled 5 min. All samples were separated by SDS-PAGE and transferred to NC membranes (GE Healthcare Life Science,
Boston, MA, USA; Lot.G9597136). The 5% (w/v) no-fat dry milk in TBST was used to block non-specific binding sites. The blocked membranes were incubated with the specific primary antibodies
diluted in 5% BSA (Sigma-Aldrich) with 0.05% sodium azide overnight on a shaking bed at 4 °C. HRP-conjugated secondary antibody was incubated at room temperature for 1 h. Finally, the
membranes were visualized by exposure to Immobilon Western reagents. CELLROX (FACS) NSCLC cells were plated in 6-well plates. The cells were transfected with 5 µM siRNA-P65 for 24 h at 37 °C
and treated with GSH-MEE 2 mM for 12 h. The cells were then stained with 5 µM of CellROX® Orange reagent by adding the probe to the complete medium and incubating the cells at 37 °C for 30
min. The cells were then washed with PBS and analyzed on FACS. Finally, the data were analyzed in the FlowJo 7.6 software. CELLROX (IF) NSCLC cells were plated in six-well plates with cover
slip. The cells were transfected with 5 µM si-RNA-P65 for 24 h at 37 °C and treated with GSH-MEE 2 mM for 12 h. The cells were then stained with 5 µM of CellROX® Orange reagent by adding the
probe to the complete medium and incubating the cells at 37 °C for 30 min. The cells were then washed with PBS and put on a glass slide. A Nikon confocal microscope was used for the
analysis and imaging. TRANSFECTION AND CO-IMMUNOPRECIPITATION A549 and PC9 cells were transfected with siRNA-P65 or siRNA-EAAT3 through Lipo 2000 (siRNA: Lipo 1:1). HEK293T cells were
transfected with Flag-EAAT3, V5-SLC7A11, and pcDNA3.1-Vector through PEI (plasmid : PEI 1:4) transfection reagents, which replaced the medium after 4–6 h. After 24 h, we removed the culture
media and washed cells with ice-cold PBS. Cells were centrifuged at 400×_g_ for 10 min at 4 °C. After the second wash, we removed the supernatant completely and resuspended the cell pellets
in 1 mL of ice-cold RIPA lysis buffer containing protease or phosphatase inhibitors. We then placed the tube on ice for 30 min, with occasional mixing. Next, we centrifuged the cell lysate
at 10,000×_g_ for 15–30 min at 4 °C, then added 50 ul Flag-M2-beads and incubated overnight on a rotator at 4 °C. Subsequently, we washed with cold PBS three times and lysed the beads.
Finally, we performed a western blot analysis. QUANTITATIVE REAL-TIME PCR Cells and tissues were first lysed using Trizol, then the total RNA was extracted with RNA Easy Fast Tissue/Cell Kit
(Tiangen, city, ST, Ctry; 4992732) and cDNA was prepared from 1.5 μg of the total. All mRNA was detected by an ABI 7300 real-time PCR machine using the 2x SYBR Green qPCR Master Mix.
Primers used listed in Table 1. ANIMALS Gprc5a–/– mice were generated in a mixed background of 129sv x C57BL/6 as described previously [22, 29]. Crossbreeding _Gprc5a_-ko mice with
SPC-dnIκB-α mice to generated _Gprc5a_–/–/SPC-dnIκB-α mice, in which dominant negative (dn) or super-repressor (SR) IκBα gene is driven by the type II cell marker surfactant protein C (SPC)
promoter [30]. Eight-week-old Gprc5a–/–, Gprc5a+/+, and _Gprc5a_–/–/SPC-dnIκB-α mice were injected with NNK (NNK,100 mg/kg body weight, i.p. dissolved in 0.9% NaCl) for 2 weeks. Ten months
later, all mice were sacrificed for H&E staining analysis, followed by extraction of protein and RNA for metabolic profiling analysis. Eight-week-old C57BL/6 mice were injected with 1601
(5 × 105 cells). BSO (1.5 mg/kg) was orally gavaged every day for 10 days from the day’s injected cells. Two weeks later, tumor volume was measured, and further analysis was conducted.
CONSTRUCTION OF PLASMIDS pcDNA3.1( + )-EAAT3-Flag and pcDNA3.1( + )-SLC7A11-V5 plasmids were constructed as described previously [31]. The mutated luciferase plasmid construction is based on
the traditional plasmid construction protocol. Briefly, we designed primers with desired mutations in the target gene of the luciferase reporter plasmid (pGL3). Then, we conducted PCR
amplification and purification, followed by digestion of the purified PCR product and the original luciferase reporter plasmid with appropriate restriction enzymes. Finally, we performed
ligation and transformation, then selection and sequencing. IHC AND SCORES A tissue microarray was stained to identify EAAT3, GPRC5A, P65, and BAX proteins. The mouse lung tissues were
stained to identify Eaat3. The IHC protocol and score method was performed as previously described [32]. LUCIFERASE ASSAY HEK293T cells were cultured in 24-well plates and transfected with
EAAT3 promoter-driving luciferase plasmid and its mutations (MutA, MutB, and MutAB). After 24 h, cells were lysed in lysis buffer per the instructions of the Dual-Luciferase® Reporter Assay
System. The luminometer reader was used to detect the luciferase activity. CHIP A549 cells were cultured in a 15 cm plate at a density of about 90% and treated with or without Dox. We
performed cross-linking of chromatin and associated proteins using 1% formaldehyde, followed by quenching with glycine. Next, cell samples were lysed in a lysis buffer (1% SDS, 10 mM EDTA,
50 mM Tris, and protease inhibitor mixture, pH 8.0). The chromatin was then sheared into fragments of desired size using enzymatic digestion. To reduce non-specific binding, pre-clearing was
performed by incubating with protein G beads coupled with a non-specific antibody. The chromatin was subsequently immunoprecipitated with specific antibodies (RNA poly II and H3K27ac were
positive control, NIgG was the negative control) against the target protein and incubated overnight at 4 °C with gentle rotation. After capturing the antibody-bound chromatin complexes with
60 ul protein G beads, a series of wash steps were carried out to remove non-specifically bound proteins and DNA. The immunoprecipitated chromatin was eluted from the beads and subjected to
decrosslinking at 65 °C. Proteinase K treatment was then performed to remove proteins, followed by DNA purification. The purified DNA was resuspended in TE buffer for PCR analysis (10 mM
Tris-HCl and 1 mM EDTA, pH 8.0). Enriched DNA was analyzed using quantitative PCR (qPCR) with primers specific to the target genomic regions to identify genome-wide binding sites of the
target protein (Primers used are listed in Table 1). Data analysis involved normalization to appropriate controls and determining the enrichment of the target protein. CSE PREPARATION The
aqueous cigarette smoke extract (CSE) was prepared with slight modifications to a previously established method [33]. In short, 10 mL of serum-free sterile DMEM medium was placed into a 60
mL plastic syringe. Then, 40 mL of cigarette smoke from one puff of a 3R4F filtered cigarette (from the Kentucky Tobacco Research and Development Center, University of Kentucky, Lexington,
KY, USA) was drawn into the syringe. The contents were mixed by vigorous shaking for 30 s. One cigarette was used for every 10 mL of the medium, and each cigarette provided 11 puffs. The
resulting CSE solution, filtered through a sterile 0.22-μm filter, was considered the 100% stock solution. The CSE was then diluted with culture medium before use. GSH COLORIMETRIC DETECTION
Cell lysate: Washed cell pellets are resuspended at 1–10 × 106 cells/mL in cold 5% SSA and are lysed and deproteinized by vigorous vortexing. Incubate cells at 4 °C for 10 min followed by
centrifugation for 10 min at 14,000 rpm and 4 °C. Tumor tissue: Fresh tumor tissue is washed with ice-cold PBS to remove blood then blotted on filter paper before recording wet weight. The
GSH assay was performed following the protocol provided by the GSH Colorimetric Detection Kit (ARBOR ASSAYS, #K006-H1, MI, USA). Briefly, samples were homogenized at a ratio of 10 mg tissue
per 250 µL of ice-cold 5% SSA (sulfosalicylic acid). The homogenate was incubated for 10 min at 4 °C, followed by centrifugation at 14,000 rpm for 10 min at 4 °C to remove precipitated
proteins. The supernatant was then collected and diluted 1:5 with the assay buffer by mixing 1 part supernatant with 4 parts assay buffer, resulting in a final SSA concentration of 1%.
CYSTINE UPTAKE ASSAY For in vivo detection of tumor cell cystine uptake, 13C6-15N2-labeled cystine was administered to mice via intraperitoneal (i.p.) injection. The mice were treated daily
with BSO following the approved animal experiment protocol. The labeled cystine was injected 6 h prior to tumor excision. Fresh tumor tissues were then prepared for UPLC/MS analysis,
following the previously described method in our study [19]. TRANSMISSION ELECTRON MICROSCOPY (TEM) PC9 cells were cultured under standard conditions, harvested at 70–80% confluence, and
fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer at 4 °C for 2 h. After washing with buffer, samples were post-fixed in 1% osmium tetroxide, dehydrated through a graded ethanol
series, and embedded in epoxy resin. Ultrathin sections (70–90 nm) were prepared using an ultramicrotome, stained with 2% uranyl acetate and Reynold’s lead citrate, and imaged using a JEOL
JEM-1400 transmission electron microscope operated at 80 kV. PUBLIC DATASET AND BIOINFORMATICS The GEO datasets were downloaded from NCBI (www.ncbi.nlm.nih.gov). The datasets (GSE17737 and
GSE64614) were used for analysis of the inflammation-inducing factor (smoking), which increased the expression of EAAT3. The dataset (GSE44619) was used for analysis of the correlation
between NF-κB and EAAT3. Fastp software (v0.20.0) was used to trim the adapter and remove low-quality reads to get high-quality clean reads. Clean reads were aligned to the human hg38
reference genome using bowtie2 software (v2.2.4). MACS2 software (v2.2.7.1) was used for peak calling. ChIPSeeker R package (v1.30.3) was used for peak annotation. Homer software (v4.11) was
used to identify motifs. The enriched peaks were visualized in IGV (v2.4.10) software. GO and KEGG pathway analysis were performed based on the promoter-enriched peaks (or promoter-enriched
differentially enriched regions) associated genes using the ClusterProfiler R package (v3.18.1). The EAAT3 and RELA expression heatmaps were from https://xenabrowser.net/. The correlation
of EAAT3 and CXCL1, BCL2, RELA were from http://timer.cistrome.org/ [34]. The transcription factor binding sites were predicted in The Contra v3 (http://bioit2.irc.ugent.be/contra/v3) [35],
and then we re-valued the binding sites using JASPAR (https://jaspar.genereg.net/) [36]. The tHPA website (https://www.proteinatlas.org/) was used for the analysis of the protein expression
in human tumor tissues. STATISTICAL ANALYSES Data were analyzed using SPSS Statistics software (IBM, Version 19) and presented as the mean ± standard deviation. The differences of results
were compared using a two-tailed paired _t_-test assuming unequal distribution. Multiple group comparisons used one-way analysis of variance (ANOVA); a _p_-value < 0.05 was considered
statistically significant. RESULTS NF-ΚB WAS INVOLVED IN THE EXPRESSION OF EAAT3 In our previous study, we reported that GPRC5A deficiency leads to the induction of EAAT3, facilitating
glutamate recycling and GSH biosynthesis. However, the underlying mechanism remained unclear. Notably, the _Gprc5a_-ko mouse lung cancer model exhibits two significant characteristics: the
spontaneous development of lung tumors and a persistent state of inflammation. Extensive data indicate that NF-κB activity is significantly elevated in the lungs of _Gprc5a_-ko mice and
MTEC-KO cells (mouse lung epithelial cells lacking Gprc5a) when compared to wild-type (WT) counterparts [28, 37]. To shed light on the regulatory mechanism of EAAT3 in lung cancer, we
postulated a plausible hypothesis implicating the NF-κB signaling pathway in EAAT3 regulation within lung cancer. In an initial endeavor to substantiate this hypothesis, we conducted a
reanalysis of publicly accessible data from the GSE44619 dataset within the GEO database [38]. This dataset offers insights into the impact of NF-κB inhibition on gene expression in human
lung cancer cell lines. For our analysis, we selected two human lung adenocarcinoma cell lines, HCC827 and PC9. We performed a comparative analysis of differentially expressed genes between
the group treated with IκBɑ super-repressor (SR) mutant and the control group (Fig. 1A). Subsequently, we conducted GO enrichment analysis on the genes that exhibited downregulation,
revealing a statistically significant enrichment of the glutamate receptor pathway in both ADC cell lines (Fig. 1B, C). To gain a deeper understanding of this correlation, we carried out
KEGG analysis on the common differentially expressed genes in both cell lines (Fig. 1D). Among these differential signaling pathways, EAAT3 was enriched within the protein digestion and
absorption pathway. Notably, when the lung cell lines were infected with a retrovirus expressing the IκBɑ-SR mutant, EAAT3 expression exhibited a decrease (Fig. 1E and Supplementary Fig. 1A,
B). We also investigated the connection between NF-κB and EAAT3 in the KP model (KrasG12D/+; Trp53fl/fl) using the publicly available GEO dataset (GSE206644) [39], which provides
comparative RNA-seq gene expression profiling data for KP tumors. Our analysis revealed a positive correlation between the NF-κB pathway and EAAT3 expression in the KP model (Supplementary
Fig. 1C). These results collectively highlight the involvement of the NF-κB signaling pathway in the regulation of EAAT3 expression. NF-ΚB INDUCED THE EXPRESSION OF EAAT3 IN VITRO AND IN
VIVO To investigate the involvement of the NF-κB signaling pathway in the upregulation of EAAT3, we assessed EAAT3 expression in MTEC-KO and MTEC-WT cell lines. Notably, immunoblot analysis
revealed that TNFα induced EAAT3 expression in MTEC-WT cells, while the NF-κB inhibitor BMS 345541 suppressed EAAT3 expression in MTEC-KO cells (Fig. 2A). Additionally, qPCR analysis
demonstrated that TNFα induced EAAT3 expression in human lung epithelial cell lines, HBEC and 16HBE (Fig. 2B, C). Given the presence of several members in the glutamate transporter family,
we also examined the expression of key glutamate transporters in human epithelial cell lines following TNFα treatment. In 16HBE cells, the expression of SLC1A2, SLC1A3, GRM1, GRM5, and GRM8
increased with TNFα treatment (Supplemental Fig. 2A–E). However, in HBEC cells, only GRM1 was induced by TNFα treatment (Supplemental Fig. 2F–J). Furthermore, the expression of EAAT3 in the
NSCLC cell line A549 was dose-dependently repressed by the NF-κB inhibitors BMS 345541 and QNZ-EVP4593 at both the protein and mRNA levels (Fig. 2D–F). Additionally, SLC1A2 and GRM8 were
found to be inhibited by BMS, while several other members, such as SLC1A3, GRM1, and GRM5, exhibited the opposite effect (Supplemental Fig. 2K–O). Moreover, after silencing the expression of
P65 with siRNA, EAAT3 expression decreased (Fig. 2G). In summary, these observations collectively support the notion that EAAT3 expression is induced by NF-κB signaling. To assess whether
the NF-κB pathway plays a crucial role in EAAT3 expression in an in vivo setting, we generated Gprc5a-ko/SPC-dnIκB-α mice by crossbreeding _Gprc5a_-ko mice with SPC-dnIκB-α mice. In the
latter, a dominant negative (dn) or super-repressor (SR) IκBα gene is driven by the type II cell marker surfactant protein C (SPC) promoter (Fig. 2H) [30]. In a prior study, we demonstrated
that TNFα-induced NF-κB activity was significantly suppressed in the lungs of Gprc5a-ko/SPC-dnIκB-α mice when compared to _Gprc5a_-ko mice [37]. Notably, in situ IHC staining revealed that
Eaat3 expression was notably diminished in the tissues from Gprc5a-ko/SPC-dnIκB-α mice compared to those from _Gprc5a_-ko mice (Fig. 2I). Immunoblot analysis further substantiated these
findings by showing a clear reduction in Eaat3 expression in lung tissues of Gprc5a-ko/SPC-dnIκB-α mice in contrast to those of _Gprc5a_-ko mice (Fig. 2J). Similarly, qPCR analysis
demonstrated that the downregulation of Eaat3 occurred at the mRNA level (Fig. 2K). Consequently, these results confirm the essential role of NF-κB signaling in Eaat3 expression within lung
epithelial cells in vivo. INHIBITION OF NF-ΚB INCREASED ROS ACCUMULATION As demonstrated previously, EAAT3 plays a pivotal role in enhancing glutamate uptake for GSH synthesis, contributing
to the aggressive characteristics of lung cancer cells. Consequently, we hypothesize that EAAT3 may function as a sensor and responder to ROS. Exposure to hydrogen peroxide (H2O2) led to a
dose-dependent induction of EAAT3 expression at both the mRNA and protein levels (Fig. 3A, B), while no significant change was observed in the xCT antiporter (Fig. 3B). In the context of
tumor growth, substantial ROS are generated, which can be detrimental to the tumor and necessitate the synthesis of GSH for neutralization. We confirmed this phenomenon through an experiment
involving the subcutaneous transplantation of C57BL/6 mice with 1601 tumor cells. Treatment with the GSH inhibitor BSO significantly suppressed tumor growth (Fig. 3C, D). To confirm the
tumor-suppressive effect of BSO treatment, we utilized a GSH colorimetric assay to demonstrate a significant reduction in GSH levels following BSO exposure (Fig. 3E). Additionally, we
employed 13C6-15N2-labeled cystine to evaluate cystine uptake in tumor cells post-BSO treatment. Given that cystine is rapidly reduced to cysteine intracellularly, we measured the combined
levels of labeled cystine and cysteine to comprehensively estimate cystine uptake. UPLC-MS analysis revealed a decrease in the levels of labeled cystine and cysteine after BSO treatment
(Fig. 3F), indicating reduced cystine uptake. In cancer cells, cellular GSH levels are influenced not only by upregulated xCT (SLC7A11) but also by altered glutamate transporters,
particularly EAAT3. The upregulated glutamate transporter EAAT3 drives cystine uptake via xCT, facilitating GSH synthesis in lung cancer cells. In light of these findings, it is reasonable
to speculate that EAAT3 and xCT might interact at the cell surface. To validate this hypothesis, we conducted a co-immunoprecipitation (co-IP) experiment. However, the results indicated that
these two molecules do not interact with each other (Supplemental Fig. 3). As NF-κB signaling plays a role in EAAT3 expression, we posited that inhibiting NF-κB could result in the
accumulation of ROS. To investigate this hypothesis, we utilized QNZ to inhibit P65 expression in the A549 and PC9 cell lines. Flow cytometry assays revealed a dose-dependent increase in ROS
levels with QNZ treatment (Fig. 3G and Supplementary Fig. 4A). Furthermore, we employed GSH-MEE to neutralize ROS, which demonstrated a reduction in ROS levels after GSH-MEE treatment in
cells previously exposed to QNZ (Fig. 3H and Supplementary Fig. 4B). Immunofluorescence analysis confirmed the elevated ROS levels in cells with an inhibited NF-κB pathway (Fig. 3I and
Supplementary Fig. 4C). Notably, the ROS response markers CHAC1 and NRF2 also increased in cells treated with QNZ. Furthermore, the inhibitory molecules of NRF2, KEAP1, showed reduced levels
(Fig. 3J). Subsequently, we utilized siRNA to silence P65 and EAAT3 expression in the A549 and PC9 cell lines. As demonstrated, silencing either EAAT3 or P65 led to the accumulation of ROS
(Fig. 3K and Supplementary Fig. 4D) and a reduction in GSH levels (Fig. 3L), indicating that NF-κB-mediated induction of EAAT3 plays a key role in regulating GSH synthesis in tumor cells. In
summary, these findings collectively suggest that inhibition of NF-κB activation results in increased ROS production, likely mediated through the involvement of EAAT3. INHIBITION OF
NF-ΚB/EAAT3 INCREASED FERROPTOSIS SENSITIVITY As is widely recognized, the generation of ROS plays a central role in initiating lipid peroxidation, particularly the oxidation of
polyunsaturated fatty acids within cellular membranes. ROS-mediated oxidative stress can lead to ferroptosis. Therefore, we assessed ferroptosis markers in silenced cell lines. Upon
silencing EAAT3, ACSL4 levels increased in PC9 cells, while SLC7A11 levels decreased in A549 cells, suggesting that these cells may become more sensitive to ferroptosis (Fig. 4A). Similarly,
cells with silenced P65 exhibited increased sensitivity to ferroptosis induced by RSL3, with a similar trend observed in EAAT3-silenced cells (Fig. 4B and Supplementary Fig. 5A). It is
worth noting that while cell death also occurred in control cells, siRNA-treated cells experienced a higher rate of cell death compared to the control cells, with a statistically significant
difference. Nevertheless, both groups shared a common outcome, with the majority of cells succumbing to death. To validate this cell death, PI staining revealed more cell death in si-P65
and si-EAAT3 cells following RSL3 treatment (Fig. 4C). Furthermore, when we counted the number of live cells in si-P65 and si-EAAT3 cells exposed to RSL3 treatment, both cell lines exhibited
a dose-dependent increase in cell death in EAAT3-silenced cells (Fig. 4D). It is reasonable to conclude that cells silenced for P65 exhibited even fewer live cells, displaying greater
sensitivity to ferroptosis induced by RSL3 treatment, with a more pronounced trend (Fig. 4E). To further confirm RSL3-induced ferroptosis in EAAT3 and P65 knockdown cell lines, we performed
TEM analysis, a classical imaging technique for detecting ferroptosis-related morphological changes. As shown in Fig. 4F, in si-EAAT3 and si-P65 cells, despite being in a state of oxidative
stress, TEM revealed that mitochondrial morphology remained largely normal, with only a slight increase in electron density and occasional mitochondrial membrane rupture, which appeared to
be within the cells’ adaptive capacity. However, following RSL3 treatment, TEM revealed pronounced mitochondrial shrinkage, significantly increased electron density, extensive membrane
rupture, and loss of continuity, collectively indicating mitochondrial dysfunction and ferroptosis (Fig. 4F). Using the FerrDb database, we analyzed the GEO dataset GSE44619. The
differentially expressed genes related to ferroptosis and suppression displayed varied expression patterns after NF-κB inhibition (Fig. 4G and Supplementary Fig. 5B). Moreover, ROS-related
hypoxia genes also exhibited differing expression patterns in this dataset (Supplementary Fig. 5C). To further strengthen this relationship, we treated cells with the NF-κB inhibitor QNZ. As
shown, the ferroptosis marker ACSL4 was induced in a dose-dependent manner with QNZ treatment, while SLC7A11 was inhibited in the A549 cell line (Fig. 4H). Intriguingly, GPX4, which
protects cells against membrane lipid peroxidation, was induced after QNZ treatment (Fig. 4H). This observation aligns with the rationale that RSL3 is a drug that inhibits GPX4 and thus,
enhances the sensitivity of cells to ferroptosis. An increase in GPX4 expression in tumor cells can make them more susceptible to ferroptosis-inducing agents. As mentioned earlier, NF-κB
inhibition increases ROS levels. Therefore, we employed GSH-MEE to neutralize ROS generation. Following GSH-MEE treatment, the ROS marker CHAC1 decreased in NF-κB inhibition cells, and GPX4,
the ferroptosis marker, was also rescued (Fig. 4I), although ACSL4 showed no significant change. These findings collectively indicate that inhibiting NF-κB/EAAT3 can increase sensitivity to
ferroptosis, particularly when induced by the ferroptosis inducer RSL3. NF-ΚB INDUCES EAAT3 EXPRESSION VIA TWO PUTATIVE CIS-ELEMENTS IN ITS PROMOTER NF-κB is a transcription factor that
plays a central role in mediating inflammatory signaling. The aforementioned observations prompt the question of whether NF-κB directly regulates EAAT3 expression at the transcriptional
level. To investigate whether NF-κB directly activates EAAT3 transcription, we analyzed the DNA sequence of the EAAT3 promoter for potential NF-κB binding cis-elements using The Contra V3
software and the JASPAR website (Fig. 5A, B). The analysis revealed the presence of two candidate NF-κB binding sites in the promoter of EAAT3 (Fig. 5C). To determine if NF-κB could directly
bind to these sites, we conducted ChIP analysis with P65 immunoprecipitation and PCR sequencing in the EAAT3 promoter. The P65 subunit of NF-κB was indeed found to be bound to the two
putative NF-κB binding motifs within the EAAT3 promoter. As a negative control, normal IgG did not bind to the promoter, and as a positive control, P65 was shown to bind to the IL-8 promoter
(Fig. 5D). Furthermore, RNA polymerase II and H3K27ac were also recruited to the EAAT3 promoter in cells with overexpressed exogenous P65 (Dox-induced myc-tagged-p65 expression) (Fig. 5E, F
and Supplementary Fig. 6A). Additionally, we performed a bioinformatic analysis of GSE160856. In this P65 ChIP-Seq data, we observed that P65 was recruited to the EAAT3 promoter,
particularly near the transcription start site (TSS) region (Supplementary Fig. 6B). To determine if these sites could mediate transcription in response to NF-κB stimulation, we constructed
an EAAT3 promoter-derived luciferase reporter plasmid, WT-luc (Fig. 5G). Transfection of HEK293T cells with this WT-luc construct, either alone or in combination with P65, or in treating
cells with TNFα, resulted in the activation of luciferase activity (Fig. 5H). This indicates that the EAAT3 promoter is a direct target of NF-κB signaling. To elucidate the specific role of
each putative binding site, we generated constructs with mutations in the P65 binding motif 1 (MutA), motif 2 (MutB), or both (MutAB) (Fig. 5G–I). We then performed luciferase reporter
analyses by transfecting HEK293T cells with these constructs, either alone or in combination with P65, or by treating the cells with TNFα. Co-transfection of cells with the EAAT3
promoter-driven luciferase reporters, specifically WT-luc, followed by treatment with TNFα or co-transfection with P65, led to the activation of luciferase activity with the wild-type EAAT3
promoter (WT-TNFα or WT-P65). However, the TNFα or P65-mediated activation was significantly reduced when using the EAAT3 promoter-derived luciferase reporter with a single motif mutation,
either Mut A or Mut B. Moreover, the activation was completely abolished when using the double motif-mutated luciferase plasmid, MutAB (Fig. 5J and Supplementary Fig. 6C). Hence, it is
evident that both putative P65 binding motifs in the EAAT3 promoter are essential for NF-κB-induced activation. EAAT3 IS UPREGULATED IN A SUBSET OF NSCLC TISSUES AND POSITIVELY CORRELATED
WITH P65 To investigate the potential correlation between EAAT3 expression and NF-κB signaling, we conducted qPCR analysis on 54 pairs of NSCLC tissues and their respective adjacent normal
tissues. Our findings reveal a significant association, with elevated EAAT3 levels prevalent in 47 out of 54 NSCLC samples, coinciding with increased expressions of RELA and the NF-κB
signaling target gene, BAX (Fig. 6A–C). To comprehensively elucidate this expression correlation, we performed immunohistochemistry (IHC) analyses using data sourced from The Human Protein
Atlas, consisting of 59 paired samples available at https://www.proteinatlas.org/ (Fig. 6D). Remarkably, EAAT3, P65, and BAX exhibited pronounced expression in lung tumor tissues compared to
their adjacent normal counterparts. Conversely, GPRC5A displayed heightened expression predominantly within the adjacent normal tissues (Fig. 6D). The IHC staining scores further
demonstrated a robust correlation within clinical NSCLC samples (Fig. 6E–G and Supplemental Fig. 7A). Moreover, we corroborated our findings using the publicly accessible UCSC Xena database
(https://xenabrowser.net/) to validate the relationship between EAAT3 and RELA. Within the dataset (Raponi 2006) _n_ = 130 instances of lung cancer with elevated RELA expression corresponded
to similarly heightened EAAT3 expression (Fig. 6H). Leveraging TCGA RNA sequencing data, subjected to analysis through TIMER2.0 (http://timer.cistrome.org/), we observed a positive
correlation between EAAT3 and RELA, as well as NF-κB target genes such as BAX and BCL2 (Supplemental Fig. 7B–D). Interestingly, the TCGA pan-cancer relationship analysis showed that the
expression of EAAT3 positively correlated with the expression of NF-κB driver genes and target genes in most cancer types (Fig. 6I, J). Collectively, these extensive datasets substantiate
our hypothesis that increased NF-κB activity potentially contributes to the upregulation of EAAT3, which may not be limited to lung cancer. THE INFLAMMATION-INDUCING FACTOR, SMOKING,
INCREASED THE EXPRESSION OF EAAT3 Given that the NF-κB inflammatory pathway is involved in the regulation of EAAT3 expression, we sought to gather supporting data from the GEO database. As
illustrated in GSE17737, mice received nose-only exposure to 4% mainstream cigarette smoke or air for 2 h/day, 5 days/week over 12 weeks. Mice exposed to smoke, a known inducer of
inflammation, exhibited suppressed Gprc5a expression and increased Eaat3 and RelA (Fig. 7A–C). The target gene of NF-κB signaling, Bax, also displayed elevated expression in the smoking
group, further correlating with EAAT3 (Fig. 7D). In another dataset (GSE64614), the expression of identified distal and proximal signatures in the small airway epithelium (SAE) of smokers
with COPD was compared to that of healthy nonsmokers. The statistics showed that the expression of EAAT3 was enhanced in smokers with COPD compared to healthy nonsmokers, while GPRC5A was
correspondingly suppressed in smokers (Fig. 7E, F) [40]. Furthermore, NF-κB signaling-related genes RELA and BAX exhibited increased expression in smokers (Fig. 7G, H). To further validate
this observation, we treated MTEC and 16HBE separately with cigarette smoke extract (CSE) and garlic oil, as well as 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), a compound also
known as a key carcinogen in tobacco. Both CSE and NNK, major inflammatory inducers in the smoking process, activated the NF-κB pathway, as evidenced by the increased mRNA expression of RELA
and its target gene BAX (Fig. 7I, J). Interestingly, EAAT3 mRNA levels were also upregulated following CSE and NNK stimulation (Fig. 7I, J). A potential mechanism for this induction might
be that the induced EAAT3 enhances GSH production, helping cells combat oxidative stress. Collectively, these data support the notion that increased GSH synthesis via NF-κB-mediated EAAT3
upregulation serves to counteract the high levels of ROS in smokers. DISCUSSION In this study, we have demonstrated the critical role of increased GSH synthesis in countering oxidative
stress during lung tumorigenesis, utilizing the Gprc5a−/− mouse model. Our findings highlight how dysregulated glutamate transporter EAAT3 enhances glutamate uptake, thereby facilitating
cystine uptake and GSH synthesis. This enhancement of GSH production also augments the stemness-like activities of cancer cells. Furthermore, we have identified two putative P65 binding
sites in the EAAT3 promoter that are responsible for NF-κB-mediated induction of EAAT3. Inhibition of NF-κB/EAAT3 leads to increased ROS generation and heightened sensitivity to ferroptosis
induced by RSL3. Lastly, we have shown that glutamate transporters, including EAAT3, are upregulated in the majority of NSCLC samples. This upregulation of glutamate transporters enhances
glutamate uptake, thus facilitating cystine uptake and GSH synthesis, ultimately contributing to lung tumor development. Inflammation has been correlated with a heightened production of ROS.
For instance, TNFα, primarily generated by activated macrophages, has the potential to increase ROS production in lung epithelial cells, which is a plausible mechanism for
inflammation-induced carcinogenesis [41]. As we previously demonstrated, lung tumorigenesis in Gprc5a−/− mice is associated with chronic inflammation. Therefore, it is expected that ROS
production is elevated under conditions of inflammation and tumor development. To counterbalance the elevated ROS levels, malignant cells must acquire the ability to generate a significant
amount of GSH. This increase in GSH levels results from metabolic reprogramming since the standard GSH synthesis system, under normal physiological conditions, may prove inadequate for the
demands of tumorigenesis [42]. In neurons, the primary role of the glutamate transporter EAAT3 is to facilitate the uptake of extracellular glutamate in the central nervous system. This
function is critical for maintaining precise neurotransmission, as it helps regulate extracellular glutamate concentrations to prevent excitotoxic damage. EAAT3 is also integral to
glutathione synthesis in neurons. Studies with Eaat3-knockout mice have demonstrated that the absence of Eaat3 leads to reduced glutathione content in neurons and early onset brain aging due
to oxidative stress. Treatment with the antioxidant N-acetyl-cysteine significantly improves neuronal function in these mice [43]. Glutamate uptake through EAAT3 is closely linked to the
activation of system Xc–. System Xc– imports cystine while exporting glutamate in a 1:1 ratio. Extracellular glutamate acts as a competitive inhibitor for cystine uptake via system Xc–.
Therefore, EAAT3 not only transports extracellular glutamate but also contributes to the intracellular glutamate pool required for cystine import. In fact, transient overexpression of EAAT3
in hippocampal HT22 cells led to increased intracellular GSH levels in the presence of high glutamate concentrations, offering protection against oxidative glutamate toxicity [44, 45]. These
effects were particularly pronounced when EAAT3 was co-overexpressed with xCT [46]. In our current study, we have observed that both xCT and EAAT3 are overexpressed in lung tumors that
develop in _Gprc5a_-ko mice and in a subset of NSCLC tissues. These findings suggest that lung tumor cells have co-opted the system Xc– and EAAT3, a system typically associated with neurons,
to facilitate GSH production. While cystine is generally considered the rate-limiting amino acid for GSH synthesis [47], glutamate also plays a crucial role in GSH production, especially in
cancer cells with a high demand for GSH. For cancer cells lacking upregulated EAAT3, it is plausible that other glutamate transporter systems are elevated, and further investigation is
required to confirm this. Nevertheless, it is clear that the uptake of glutamate and the synthesis of GSH must be maintained at high levels for cancer development. NF-κB is a redox-regulated
sensor that responds to oxidative stress and can be activated by low doses of hydrogen peroxide [48, 49]. In our study, we established a connection between NF-κB and the regulation of
EAAT3. Our analysis has identified two potential NF-κB binding sites in the EAAT3 promoter, indicating that this gene is regulated by signaling pathways associated with inflammation.
Significantly, we have observed that the expression of EAAT3 is suppressed in Gprc5a-/-/SPC-dnIκB-α mice [37], underscoring the requirement for NF-κB in the upregulation of EAAT3. This
regulatory pattern is similar to what has been observed with xCT, as previous studies have reported that xCT expression can be induced by inflammatory stimuli, including substances like LPS
and TNFα. Furthermore, there is a putative NF-κB binding site in the promoter of xCT [50]. While our findings indicate that NF-κB can specifically bind to EAAT3 promoter regions in certain
cell types, multiple pathways and factors are known to regulate EAAT3, including the NRF2-ARE pathway [51], the RFX1 transcription factor [52], NFAT5/TonEBP [53], and ATRA [54]. Therefore,
NF-κB is one of several transcription factors capable of binding to the promoter and activating EAAT3 expression. Besides, several exogenous factors have been shown to activate EAAT3,
primarily by influencing NF-κB signaling, which subsequently upregulates EAAT3 expression. Key factors include: (1) Pro-inflammatory cytokines: Cytokines such as TNFα and interleukin-1 beta
(IL-1β) are known to activate NF-κB, leading to increased EAAT3 expression. (2) Oxidative stress: Due to rapid proliferation and metabolic shifts, lung cancer cells experience high levels of
oxidative stress, which activates NF-κB. This activation upregulates EAAT3 as part of the cell’s defense against ROS. (3)Hypoxia: Under hypoxic conditions, NF-κB activation enhances EAAT3
expression, strengthening antioxidant defenses in lung cancer cells. (4)Environmental toxins and pollutants: Exposure to pollutants and carcinogens, such as cigarette smoke and airborne
particulate matter, can trigger NF-κB signaling in lung tissue, contributing to EAAT3 activation. While our study has shed light on the link between inhibiting NF-κB/EAAT3, ROS accumulation,
and increased ferroptosis sensitivity in lung cancer cells, the precise molecular mechanisms underlying this process remain unclear. Silencing P65 or EAAT3 increases ROS levels and reduces
GSH synthesis, but these changes alone are insufficient to trigger significant cell death due to the robust ferroptosis-resistance mechanisms in cancer cells, including the xCT-GSH-GPX4
system and other protective pathways. This resistance allows cancer cells to tolerate ROS accumulation and maintain survival unless the balance is further disrupted by external ROS inducers
(e.g., H2O2) or inhibitors of ferroptosis-resistance mechanisms (e.g., RSL3 and Erastin). We also have observed an increase in the expression of ACSL4 when cells are treated with an NF-κB
inhibitor, which could explain why ferroptosis becomes more sensitive to RSL3. Additionally, the expression of SLC7A11 decreased, which is another factor contributing to ferroptosis
sensitivity. However, it is worth noting that GPX4, an antioxidant enzyme glutathione peroxidase that protects cells against oxidative stress [55], was also increased when NF-κB was
inhibited. This suggests that the cells rely on GPX4 to resist oxidative stress, and this could potentially increase their sensitivity to RSL3, a ferroptosis inducer that targets GPX4. While
these findings provide valuable insights, we acknowledge that further research is needed to delve into the specific molecular mechanisms at play in this context. DATA AVAILABILITY The data
will be provided by the authors upon reasonable request. CHANGE HISTORY * _ 19 MARCH 2025 The original online version of this article was revised: "The designation of co-first
authorship was clearly stated in our submitted and revised versions, but it is missing in the published article. _ * _ 03 APRIL 2025 A Correction to this paper has been published:
https://doi.org/10.1038/s41419-025-07518-y _ REFERENCES * Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17–48. Article PubMed Google Scholar
* Qin L, Guitart M, Curull V, Sánchez-Font A, Duran X, Tang J, et al. Systemic profiles of microRNAs, redox balance, and inflammation in lung cancer patients: influence of COPD.
Biomedicines. 2021;9:1347. * Zuo L, Wijegunawardana D. Redox role of ROS and inflammation in pulmonary diseases. Adv Exp Med Biol. 2021;1304:187–204. Article CAS PubMed Google Scholar *
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. * Li Y, Hecht SS. Carcinogenic components of tobacco and tobacco smoke: a 2022 update. Food Chem Toxicol.
2022;165:113179. * Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–210. Article CAS PubMed Google Scholar * Akhtar N, Bansal JG. Risk factors of
lung cancer in nonsmoker. Curr Probl Cancer. 2017;41:328–39. Article PubMed Google Scholar * Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Pérez-Gracia JL, et al. Cytokines
in clinical cancer immunotherapy. Br J Cancer. 2019;120:6–15. Article CAS PubMed Google Scholar * Gong Z, Li Q, Shi J, Wei J, Li P, Chang CH, et al. Lung fibroblasts facilitate
pre-metastatic niche formation by remodeling the local immune microenvironment. Immunity. 2022;55:1483–500.e9. Article CAS PubMed PubMed Central Google Scholar * Mantovani A, Allavena
P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44. Article CAS PubMed Google Scholar * Hecht SS. Lung carcinogenesis by tobacco smoke. Int J Cancer.
2012;131:2724–32. Article CAS PubMed PubMed Central Google Scholar * Muri J, Kopf M. Redox regulation of immunometabolism. Nat Rev Immunol. 2021;21:363–81. Article CAS PubMed Google
Scholar * Lin Y, Jiang M, Chen W, Zhao T, Wei Y. Cancer and ER stress: mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed Pharmacother. 2019;118:109249.
Article CAS PubMed Google Scholar * Sfanos KS, Yegnasubramanian S, Nelson WG, De Marzo AM. The inflammatory microenvironment and microbiome in prostate cancer development. Nat Rev Urol.
2018;15:11–24. Article PubMed Google Scholar * Rodríguez-Vargas JM, Ruiz-Magaña MJ, Ruiz-Ruiz C, Majuelos-Melguizo J, Peralta-Leal A, Rodríguez MI, et al. ROS-induced DNA damage and
PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res. 2012;22:1181–98. Article PubMed PubMed Central Google Scholar * Fan J, Ren D, Wang J, Liu X, Zhang H,
Wu M, et al. Bruceine D induces lung cancer cell apoptosis and autophagy via the ROS/MAPK signaling pathway in vitro and in vivo. Cell Death Dis. 2020;11:126. Article CAS PubMed PubMed
Central Google Scholar * Chen B, Song Y, Zhan Y, Zhou S, Ke J, Ao W, et al. Fangchinoline inhibits non-small cell lung cancer metastasis by reversing epithelial-mesenchymal transition and
suppressing the cytosolic ROS-related Akt-mTOR signaling pathway. Cancer Lett. 2022;543:215783. Article CAS PubMed Google Scholar * Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro
AL, Pronzato MA, et al. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev. 2013;2013:972913. Article PubMed PubMed Central Google Scholar * Guo W, Li
K, Sun B, Xu D, Tong L, Yin H, et al. Dysregulated glutamate transporter SLC1A1 propels cystine uptake via Xc(-) for glutathione synthesis in lung cancer. Cancer Res. 2021;81:552–66. Article
CAS PubMed Google Scholar * Wang X, Chen Z, Xu J, Tang S, An N, Jiang L, et al. SLC1A1-mediated cellular and mitochondrial influx of R-2-hydroxyglutarate in vascular endothelial cells
promotes tumor angiogenesis in IDH1-mutant solid tumors. Cell Res. 2022;32:638–58. Article CAS PubMed PubMed Central Google Scholar * Xiong J, Wang N, Zhong HJ, Cui BW, Cheng S, Sun R,
et al. SLC1A1 mediated glutamine addiction and contributed to natural killer T-cell lymphoma progression with immunotherapeutic potential. EBioMedicine. 2021;72:103614. Article CAS PubMed
PubMed Central Google Scholar * Tao Q, Fujimoto J, Men T, Ye X, Deng J, Lacroix L, et al. Identification of the retinoic acid-inducible Gprc5a as a new lung tumor suppressor gene. J Natl
Cancer Inst. 2007;99:1668–82. Article CAS PubMed Google Scholar * Jing B, Wang T, Sun B, Xu J, Xu D, Liao Y, et al. IL6/STAT3 signaling orchestrates premetastatic niche formation and
immunosuppressive traits in lung. Cancer Res. 2020;80:784–97. Article CAS PubMed Google Scholar * Liao Y, Song H, Xu D, Jiao H, Yao F, Liu J, et al.Gprc5a-deficiency confers
susceptibility to endotoxin-induced acute lung injury via NF-κB pathway. Cell Cycle. 2015;14:1403–12. Article CAS PubMed PubMed Central Google Scholar * Song H, Ye X, Liao Y, Zhang S,
Xu D, Zhong S, et al. NF-κB represses retinoic acid receptor-mediated GPRC5A transactivation in lung epithelial cells to promote neoplasia. JCI insight. 2023;8:e153976. * Wang WF, Zhong HJ,
Cheng S, Fu D, Zhao Y, Cai HM, et al. A nuclear NKRF interacting long noncoding RNA controls EBV eradication and suppresses tumor progression in natural killer/T-cell lymphoma. Biochim
Biophys Acta Mol Basis Dis. 2023;1869:166722. Article CAS PubMed Google Scholar * Chen Y, Deng J, Fujimoto J, Kadara H, Men T, Lotan D, et al. Gprc5a deletion enhances the transformed
phenotype in normal and malignant lung epithelial cells by eliciting persistent Stat3 signaling induced by autocrine leukemia inhibitory factor. Cancer Res. 2010;70:8917–26. Article CAS
PubMed PubMed Central Google Scholar * Deng J, Fujimoto J, Ye XF, Men TY, Van Pelt CS, Chen YL, et al. Knockout of the tumor suppressor gene Gprc5a in mice leads to NF-kappaB activation
in airway epithelium and promotes lung inflammation and tumorigenesis. Cancer Prev Res. 2010;3:424–37. Article CAS Google Scholar * Guo W, Hu M, Wu J, Zhou A, Liao Y, Song H, et al.
Gprc5a depletion enhances the risk of smoking-induced lung tumorigenesis and mortality. Biomed Pharmacother. 2019;114:108791. Article CAS PubMed Google Scholar * Skerrett SJ, Liggitt HD,
Hajjar AM, Ernst RK, Miller SI, Wilson CB. Respiratory epithelial cells regulate lung inflammation in response to inhaled endotoxin. Am J Physiol Lung Cell Mol Physiol. 2004;287:L143–52.
Article CAS PubMed Google Scholar * Zhong S, Yin H, Liao Y, Yao F, Li Q, Zhang J, et al. Lung tumor suppressor GPRC5A binds EGFR and restrains its effector signaling. Cancer Res.
2015;75:1801–14. Article CAS PubMed Google Scholar * Guo W, Kuang Y, Wu J, Wen D, Zhou A, Liao Y, et al. Hexokinase 2 depletion confers sensitization to metformin and inhibits glycolysis
in lung squamous cell carcinoma. Front Oncol. 2020;10:52. Article PubMed PubMed Central Google Scholar * Li T, Fanning KV, Nyunoya T, Chen Y, Zou C. Cigarette smoke extract induces
airway epithelial cell death via repressing PRMT6/AKT signaling. Aging. 2020;12:24301–17. Article CAS PubMed PubMed Central Google Scholar * Li T, Fan J, Wang B, Traugh N, Chen Q, Liu
JS, et al. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77:e108–e10. Article CAS PubMed PubMed Central Google Scholar * Kreft L,
Soete A, Hulpiau P, Botzki A, Saeys Y, De Bleser P. ConTra v3: a tool to identify transcription factor binding sites across species, update 2017. Nucleic Acids Res. 2017;45:W490–w4. Article
CAS PubMed PubMed Central Google Scholar * Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Lemma RB, Turchi L, Blanc-Mathieu R, et al. JASPAR 2022: the 9th release of the
open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022;50:D165–d73. Article CAS PubMed Google Scholar * Liao Y, Song H, Xu D, Jiao H, Yao F, Liu J, et al.
Gprc5a-deficiency confers susceptibility to endotoxin-induced acute lung injury via NF-kappaB pathway. Cell Cycle. 2015;14:1403–12. Article CAS PubMed PubMed Central Google Scholar *
Hopewell EL, Zhao W, Fulp WJ, Bronk CC, Lopez AS, Massengill M, et al. Lung tumor NF-κB signaling promotes T cell-mediated immune surveillance. J Clin Investig. 2013;123:2509–22. Article
CAS PubMed PubMed Central Google Scholar * Kong Y, Allison DB, Zhang Q, He D, Li Y, Mao F, et al.The kinase PLK1 promotes the development of Kras/Tp53-mutant lung adenocarcinoma through
transcriptional activation of the receptor RET. Sci Signal. 2022;15:eabj4009. Article CAS PubMed PubMed Central Google Scholar * Yang J, Zuo WL, Fukui T, Chao I, Gomi K, Lee B, et al.
Smoking-dependent distal-to-proximal repatterning of the adult human small airway epithelium. Am J Respir Crit Care Med. 2017;196:340–52. Article CAS PubMed PubMed Central Google Scholar
* Babbar N, Casero RA Jr. Tumor necrosis factor-alpha increases reactive oxygen species by inducing spermine oxidase in human lung epithelial cells: a potential mechanism for
inflammation-induced carcinogenesis. Cancer Res. 2006;66:11125–30. Article CAS PubMed Google Scholar * Moriarty-Craige SE, Jones DP. Extracellular thiols and thiol/disulfide redox in
metabolism. Annu Rev Nutr. 2004;24:481–509. Article CAS PubMed Google Scholar * Cao L, Li L, Zuo Z. N-acetylcysteine reverses existing cognitive impairment and increased oxidative stress
in glutamate transporter type 3 deficient mice. Neuroscience. 2012;220:85–9. Article CAS PubMed Google Scholar * Duerson K, Woltjer RL, Mookherjee P, Leverenz JB, Montine TJ, Bird TD,
et al. Detergent-insoluble EAAC1/EAAT3 aberrantly accumulates in hippocampal neurons of Alzheimer’s disease patients. Brain Pathol. 2009;19:267–78. Article CAS PubMed Google Scholar *
Crino PB, Jin H, Shumate MD, Robinson MB, Coulter DA, Brooks-Kayal AR. Increased expression of the neuronal glutamate transporter (EAAT3/EAAC1) in hippocampal and neocortical epilepsy.
Epilepsia. 2002;43:211–8. Article CAS PubMed PubMed Central Google Scholar * Lewerenz J, Klein M, Methner A. Cooperative action of glutamate transporters and cystine/glutamate
antiporter system Xc- protects from oxidative glutamate toxicity. J Neurochem. 2006;98:916–25. Article CAS PubMed Google Scholar * Hanigan MH. Gamma-glutamyl transpeptidase: redox
regulation and drug resistance. Adv Cancer Res. 2014;122:103–41. Article CAS PubMed PubMed Central Google Scholar * Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress,
inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49:1603–16. Article CAS PubMed PubMed Central Google Scholar * Kriete A, Bosl WJ, Booker G. Rule-based cell
systems model of aging using feedback loop motifs mediated by stress responses. PLoS Comput Biol. 2010;6:e1000820. Article PubMed PubMed Central Google Scholar * Sato H,
Kuriyama-Matsumura K, Hashimoto T, Sasaki H, Wang H, Ishii T, et al. Effect of oxygen on induction of the cystine transporter by bacterial lipopolysaccharide in mouse peritoneal macrophages.
J Biol Chem. 2001;276:10407–12. Article CAS PubMed Google Scholar * Escartin C, Won SJ, Malgorn C, Auregan G, Berman AE, Chen PC, et al. Nuclear factor erythroid 2-related factor 2
facilitates neuronal glutathione synthesis by upregulating neuronal excitatory amino acid transporter 3 expression. J Neurosci. 2011;31:7392–401. Article CAS PubMed PubMed Central Google
Scholar * Ma K, Zheng S, Zuo Z. The transcription factor regulatory factor X1 increases the expression of neuronal glutamate transporter type 3. J Biol Chem. 2006;281:21250–5. Article CAS
PubMed Google Scholar * Miyakawa H, Woo SK, Dahl SC, Handler JS, Kwon HM. Tonicity-responsive enhancer binding protein, a rel-like protein that stimulates transcription in response to
hypertonicity. Proc Natl Acad Sci USA. 1999;96:2538–42. Article CAS PubMed PubMed Central Google Scholar * Bianchi MG, Gazzola GC, Tognazzi L, Bussolati O. C6 glioma cells
differentiated by retinoic acid overexpress the glutamate transporter excitatory amino acid carrier 1 (EAAC1). Neuroscience. 2008;151:1042–52. Article CAS PubMed Google Scholar * Yang
WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31. Article CAS PubMed PubMed Central
Google Scholar Download references FUNDING This study was supported by the National Natural Science Foundation of China (82272408); the Project of Shanghai Science and Technology Committee
(22QA1407700); the Academic Leaders Training Program of Pudong Health Bureau of Shanghai (PWRd2021-14); Clinical Research project of Shanghai Municipal Health Commission (20214Y0247). AUTHOR
INFORMATION Author notes * These authors contributed equally: Donghua Wen, Wenjing Li, Xiang Song, Min Hu. AUTHORS AND AFFILIATIONS * Department of Laboratory Medicine, Shanghai East
Hospital, Tongji University School of Medicine, Shanghai, 200120, China Donghua Wen, Wenjing Li, Min Hu & Wenzheng Guo * Breast Cancer Center, Shandong Cancer Hospital and Institute,
Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, Shandong, 250117, China Xiang Song * College of Life and Environmental Science, Wenzhou University,
Wenzhou, 325035, China Yueling Liao * Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, 200025, China Dongliang Xu * Medical Research Center, Affiliated Hospital of
Binzhou Medical University, Binzhou, 256600, Shandong, China Jiong Deng Authors * Donghua Wen View author publications You can also search for this author inPubMed Google Scholar * Wenjing
Li View author publications You can also search for this author inPubMed Google Scholar * Xiang Song View author publications You can also search for this author inPubMed Google Scholar *
Min Hu View author publications You can also search for this author inPubMed Google Scholar * Yueling Liao View author publications You can also search for this author inPubMed Google
Scholar * Dongliang Xu View author publications You can also search for this author inPubMed Google Scholar * Jiong Deng View author publications You can also search for this author inPubMed
Google Scholar * Wenzheng Guo View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS WG and JD conceptualized the experiment, performed formal
analysis, and wrote the original draft of the manuscript. WG, DW, WL, and MH conducted the cell experiments, including cell culture, protein immunoblotting, FACS, and other cell biology
assays. XS carried out all bioinformatic analyses. YL conducted the molecular cloning experiments, including the construction of luciferase and knockdown plasmids. DX oversaw all mouse
experiments, including the generation of genetically engineered mice and in vivo tumorigenesis assays. WG and DW secured funding for the research. All authors have read and approved the
published version of the manuscript. CORRESPONDING AUTHORS Correspondence to Jiong Deng or Wenzheng Guo. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interest.
ETHICS All mice were maintained according to a protocol approved by Tongji University, School of Medicine Animal Care and Use Committee[experimental animal use permission No: SYXK (Shanghai)
2020-0002] in the specific pathogen-free animal facility in the university. The care of all mice was in accordance with institution guidelines. ADDITIONAL INFORMATION PUBLISHER’S NOTE
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Edited by Guo-Qiang Chen SUPPLEMENTARY INFORMATION SUPPLEMENTAL FIGURE
1 SUPPLEMENTAL FIGURE 2 SUPPLEMENTAL FIGURE 3 SUPPLEMENTAL FIGURE 4 SUPPLEMENTAL FIGURE 5 SUPPLEMENTAL FIGURE 6 SUPPLEMENTAL FIGURE 7 SUPPLEMENTAL FIGURE LEGENDS WESTERN BLOT NO-CUT RAW DATA
RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and
reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes
were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If
material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain
permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Wen, D., Li, W., Song, X. _et al._ NF-κB-mediated EAAT3 upregulation in antioxidant defense and ferroptosis sensitivity in lung cancer. _Cell Death Dis_ 16, 124 (2025).
https://doi.org/10.1038/s41419-025-07453-y Download citation * Received: 29 August 2024 * Revised: 04 February 2025 * Accepted: 12 February 2025 * Published: 22 February 2025 * DOI:
https://doi.org/10.1038/s41419-025-07453-y SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative



