- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Sepsis can trigger systemic inflammation and lead to detrimental effects on several organs, with particular emphasis on the lungs. In sepsis-associated lung injury, macrophages
assume a pivotal role, as their overactivation could facilitate the secretion of inflammatory factors and the imbalance of polarization. Hepatocyte nuclear factor 4 alpha (HNF4A) has been
reported its potential involvement in the regulation of inflammatory response and macrophage polarization. This study discusses the role and mechanism of HNF4A in sepsis-induced lung damage.
HNF4A exhibits a decrease in expression by analyzing the differentially expressed genes in the lungs of septic mice from the Gene Expression Omnibus dataset GSE15379. Then, we established a
mouse sepsis model through a cecal ligation and puncture method and observed that the expression of HNF4A was reduced in both lung tissues and alveolar macrophages. To evaluate the function
of HNF4A, we overexpressed HNF4A mediated by adenovirus vectors, which were injected into mice. We found that HNF4A overexpression resulted in a higher survival rate in septic mice and an
amelioration of pulmonary damage. Meanwhile, HNF4A overexpression mitigated the infiltration of inflammatory cells and impeded the M1 polarization but facilitated the M2 polarization of
macrophages in the lung tissues or the alveolar lavage fluid. In vitro, we treated bone marrow-derived macrophages with interleukin-4. Consistent results were obtained that HNF4A
overexpression promoted the M2 polarization of macrophages. Mechanistically, we found that HNF4A transcriptionally regulate the expression of nuclear receptor coactivator 2 (NCOA2) through
binding to its promoter region. NCOA2 interacted with glucocorticoid receptor (GR). Stabilin 1 (STAB1) was selected as a possible target by transcriptome sequencing analysis. Functional
experiments confirmed STAB1 as a downstream target of the HNF4A/NCOA2/GR axis. Overall, this research investigated the potential impact of HNF4A on pulmonary injury in sepsis. It is
suggested that one of the regulatory mechanisms involved in this association may be the NCOA2/GR/STAB1 axis. SIMILAR CONTENT BEING VIEWED BY OTHERS LNCRNA NEAT1 AGGRAVATES SEPSIS-INDUCED
LUNG INJURY BY REGULATING THE MIR-27A/PTEN AXIS Article 08 July 2021 TARGETING HMGB1 FOR THE TREATMENT OF SEPSIS AND SEPSIS-INDUCED ORGAN INJURY Article 26 May 2021 INHIBITION OF TXNDC5
ATTENUATES LIPOPOLYSACCHARIDE-INDUCED SEPTIC SHOCK BY ALTERING INFLAMMATORY RESPONSES Article 04 December 2021 INTRODUCTION Sepsis is a systemic inflammatory response syndrome triggered by
infection that can result in multiple organ dysfunction [1]. It is characterized by symptoms such as fever, tachycardia, dyspnea, and imbalanced white blood cell counts [2]. The lung is the
predominant organ affected by sepsis [3], and the mortality rate associated with acute lung damage (ALI) resulting from sepsis can reach up to 40%, surpassing the fatality rates contributed
by alternative sources of ALI [4]. Macrophages play a key role in the pathogenesis of pulmonary damage in sepsis [5]. Normally, macrophages, as an important part of the immune system, are
responsible for the elimination of infections and apoptotic cells, as well as the preservation of tissue homeostasis [6]. However, these cells become excessively active during the
progression of sepsis, resulting in an exaggerated inflammatory response, an imbalance in polarization, and dysfunction in endophagocytosis, all of which contribute to the acceleration of
the damage response [7]. Macrophages can transition between two primary phenotypes, namely M1 (classical activation, pro-inflammatory) and M2 (replacement activation, anti-inflammatory), in
response to signals received from the microenvironment to maintain tissue homeostasis [8]. However, in the context of sepsis-related danger signals such as pathogen-associated molecular
patterns (PAMPs) and injury-associated molecular patterns (DAMPs), the balance tends to support M1 polarization [9]. The M1-type macrophages further promote the production and release of
pro-inflammatory cytokines and reactive oxygen species (ROS), resulting in heightened lung inflammation and oxidative stress and inducing lung injury [10]. In recent years, intervention of
macrophage polarization with pharmacological or biological agents to promote the transformation of M1 to M2 has emerged as a promising approach for the management of pulmonary injury linked
to sepsis [11,12,13]. However, the molecular targets of sepsis-associated lung injury remain largely unelucidated, which has hindered subsequent development. Hepatocyte nuclear factor 4
alpha (HNF4A) is mainly known for its role in the regulation of hepatic metabolic pathways [14]. It is reported that HNF4A can alleviate acute liver injury generated by acetaminophen [15]
and liver fibrosis caused by toxins and cholestasis [16]. Recently, some studies have suggested that HNF4A may also play a role in the indirect modulation of inflammatory responses [17, 18].
Notably, in Yang et al.’ paper, they proved that HNF4A can exert a protective influence against liver fibrosis through its regulation of liver macrophage polarization [16]. By analyzing the
dataset GSE15379 from Gene Expression Omnibus (GEO) database, we found a notable decrease in the expression of HNF4A in the lung tissue of septic mice with a cecal ligation and puncture
(CLP) method, as compared to the control group (log2FC = −1.26; _p_ < 0.01). Therefore, it is postulated that the down-regulation of HNF4A may potentially play a role in the pathogenesis
of sepsis-associated pulmonary damage by modulating the phenotype transition of macrophages. The glucocorticoid receptor (GR) is a cellular protein that exhibits binding affinity towards
glucocorticoids such as cortisol, hence exerting an influence on the regulation of gene expression [19]. Once the hormone establishes a binding interaction with GR, the resultant complex is
capable of translocating into the nucleus and binding to a specific DNA sequence known as the glucocorticoid response element (GRE), thereby acting as a transcription factor [20]. This
transcription factor possesses its ability to either start or impede the transcription of specific genes, thereby regulating a variety of cellular processes such ad inflammatory response,
immunological function, and metabolism [21]. Nuclear receptor coactivator 2 (NCOA2) is a nuclear receptor coactivator of GR [22]. It can directly bind to the GR monomers or homologous dimers
to participate in the regulation of GR-mediated gene transcription [23]. Moreover, deletion of NCOA2 in obese mice elevated the expression of M1-type factors but reduced the M2-type factors
in macrophages of the adipose tissues [24]. The above literature comprehensively suggests that NCOA2 and GR are significant contributors to the survival and anti-inflammatory M2 phenotype
of macrophages in sepsis. Intriguingly, the JASPAR database indicates binding sites between HNF4A and the NCOA2 promoter. Therefore, we hypothesize that HNF4A may exert a regulatory role in
the sepsis-associated lung injury by influencing NCOA2/GR-mediated gene transcription. In this work, we conducted in vivo and in vitro experiments to investigate the role of HNF4A. In
addition, we employed transcriptome sequencing method to explore the gene regulation mediated by HNF4A/NCOA2/GR axis. RESULTS TRANSCRIPTOMIC ANALYSIS OF LUNG TISSUES FROM CLP-INDUCED SEPTIC
MICE AND THE SHAM MICE IN GSE15379 To investigate the molecular regulatory mechanisms associated with sepsis-induced ALI, we analyzed the profiles of transcriptomics changes in lung tissues
of CLP-induced septic mice compared with the sham mice from GSE15379. The heatmap showed the total differentially expressed genes (DEGs) (∣log2FoldChange∣> 1, and −log10 (_p_-value) >
2.5) (Supplementary Figure 1A). Next, we performed the GO analysis for DEGs. The enrichment results of biological process (BP) showed that the most of DEGs are related to inflammation. The
enrichment of molecular function (MF) identified that the majority of DEGs are related to transcription (Supplementary Figure 1B). We focused on the top 5 GO terms associated with
transcription factors from DEGs. By Venn diagram analysis, we found that 10 overlapping DEGs (CEBP, FOXC1, CEBPD, HNF4A, JUNB, STAT3, FOS, TBX3, CREB1, TRP53) associated with the top 5 GO-MF
terms (Supplementary Figure 1C-D). Notably, HNF4A is a transcription factor in DEGs. Furthermore, the HNF4A expression is down-regulated in lung tissues of CLP-induce septic mice compared
with the sham mice in GSE 15379. Currently, the effect of HNF4A on ALI has not been reported, which is worthy of further investigation. THE EXPRESSION OF HNF4A IS DOWN-REGULATED DURING
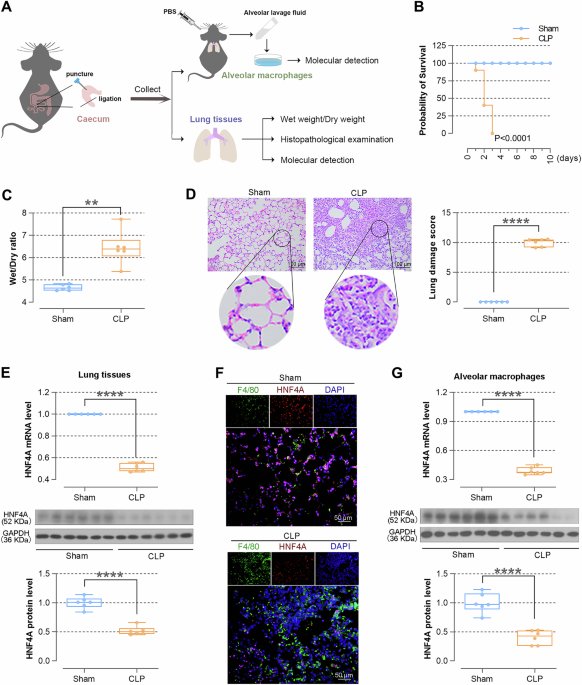
SEPSIS-INDUCED ALI The mouse model of sepsis was constructed by CLP induction to verify the situation of lung injury and changes in HNF4A expression (Fig. 1A). As shown in Fig. 1B, the
mortality rate of CLP-induced mice was higher than the sham mice. The lung tissues of CLP-induced mice showed a higher Wet/Dry ratio, indicating severe edema of lung issues after CLP-induced
sepsis (Fig. 1B). The infiltration of inflammatory cells was obviously observed by HE staining in lung tissues of CLP-induced mice, and the score of lung damage also increased (Fig. 1D).
Next, the alteration of HNF4A expression was determined. We found that the expression levels of HNF4A were down-regulated in lung tissues of CLP-induced mice (Fig. 1E). Alveolar macrophages
were isolated from alveolar lavage fluid. The levels of HNF4A were down-regulated in alveolar macrophages, which were also corroborated by IF staining (Fig. 1F-1G). Collectively, these
results suggested that lung tissues are severely damaged after CLP-induced sepsis, mainly manifested as pulmonary edema and inflammatory cell infiltration, and HNF4A levels are
down-regulated. OVEREXPRESSING HNF4A ALLEVIATES LUNG INJURY AND SUPPRESSES INFLAMMATION IN SEPTIC MICE To clarify the role of HNF4A in ALI, we overexpressed HNF4A mediated by adenovirus
prior to CLP induction, as confirmed by the Real-time PCR and western blot (WB). (Fig. 2A, G and H). On day 3, the probability of survival was 0% for the septic mice instilled with Ad-NC,
while the probability of survival for the septic mice instilled with Ad-HNF4A was 70% (Fig. 2B). Pulmonary edema was improved by HNF4A overexpression in CLP-induced mice, as evidenced by
decreased lung Wet/Dry ratio (Fig. 2C). Compared with the septic mice instilled with Ad-NC, histological results showed the decreased infiltration of inflammatory cells in lung tissues of
the septic mice instilled with Ad-HNF4A (Fig. 2D). Meanwhile, the concentration of total protein in alveolar lavage fluid was decreased by HNF4A overexpression (Fig. 2E). We detected the
levels of M1 and M2 inflammatory cytokines in the alveolar lavage fluid. As shown in Fig. 2F, the levels of IL-4 and IL-10 were increased by HNF4A overexpression, while the levels of TNF-α,
IL-1β, and IL-6 were decreased. Consistently, we found that HNF4A overexpression decreases levels of TNF-α, IL-1β, and IL-6 in alveolar macrophages (Fig. 2I–K). Taken together, these
findings implied that HNF4A overexpression suppresses inflammation and improves ALI in CLP-induced mice. OVEREXPRESSING HNF4A PROMOTES THE POLARIZATION OF MACROPHAGES TOWARDS M2 PHENOTYPE
AND UPREGULATES THE EXPRESSION OF NCOA2 IN SEPTIC MICE To explore the impact of HNF4A on the macrophage polarization, TNF-α and Arg-1 expressions on macrophages (F4/80-positive cells) were
measured by flow cytometry. Compared with septic mice instilled with Ad-NC, HNF4A overexpression resulted in an increase in macrophages towards M2 phenotype and a decrease in macrophages
towards M1 phenotype (Fig. 3A). As shown in Fig. 3B, the number of total leukocytes, neutrophils, and macrophages remarkably reduced in the alveolar lavage fluid of HNF4A-overexpressed mice.
Moreover, the overexpression of HNF4A resulted in an increase in the expressions of NCOA2 in alveolar macrophages of septic mice. (Fig. 3C, D). Immunofluorescence assays (Fig. 3E, F) were
performed to further verify the results of real-time PCR and WB. In sum, these findings indicated HNF4A overexpression promotes the polarization of alveolar macrophages towards M2 phenotype
and up-regulates the levels of NCOA2. OVEREXPRESSING HNF4A PROMOTES THE POLARIZATION OF BMDMS TOWARDS M2 PHENOTYPE AND HNF4A BINDS TO THE NCOA2 PROMOTER We isolated BMDMs from the bone
marrow of C57BL/6J mice and identified the cells by flow cytometry (Fig. 4A). Subsequently, BMDMs were treated with IL-4 for 24 h to induce polarization towards M2 phenotype and treated with
LPS and IFN-γ to induce polarization towards M1 phenotype. The results of WB and Real-time PCR showed that HNF4A levels are up-regulated (Fig. 4B). In order to investigate the impacts of
HNF4A on the polarization of BMDMs, BMDMs were infected with Ad-HNF4A to overexpress HNF4A and then induced polarization towards M2 phenotype (Fig. 4C) and. As shown in Fig. 4D, HNF4A was
overexpressed in IL-4-induced BMDMs, as evidenced by WB and real-time PCR (Fig. 4D). The expression levels of CD206, CD163, and Arg-1 (markers of M2 phenotype) were up-regulated after HNF4A
overexpression, indicating that HNF4A promotes the polarization of BMDMs towards M2 phenotype (Fig. 4E). In contrast, the expression levels of iNOS and MCHII (markers of M1 phenotype) were
down-regulated after HNF4A overexpression in LPS + IFN-γ-induced BMDMs (Supplementary Fig. 2A, B). The levels of TNF-α, IL-1β, and IL-6 were also down-regulated, suggesting that HNF4A
suppressed the polarization of BMDMs towards M1 phenotype (Supplementary Fig. 2C). Furthermore, we found that the expression of NCOA2 is also increased (Fig. 4F). To confirm whether HNF4A
modulates the NCOA2 expression by binding to the NCOA2 promoter, we performed the dual luciferase reporter assay to assess the activity of pGL3-NCOA2 promoter luciferase reporter. The result
showed that HNF4A contributes to the activation of pGL3-NCOA2 promoter luciferase activity (Fig. 4G). To further verify whether HNF4A binds to the NCOA2 promoter directly, we employed an
antibody against HNF4A for immunoprecipitation, followed by PCR analysis in IL-4-induced BMDMs. The results showed that immunoprecipitated products using IgG did not contain the NCOA2
promoter. In contrast, the NCOA2 promoter sequence was detected in the immunoprecipitation product using the HNF4A antibody, suggesting that HNF4A binds to the NCOA2 promoter directly (Fig.
4H). Taken together, these results indicated that HNF4A is able to bind to the NCOA2 promoter, promotes the polarization of BMDMs towards M2 phenotype and suppresses the polarization of
BMDMs towards M1 phenotype. HNF4A AFFECTS THE POLARIZATION OF MACROPHAGES VIA NCOA2/GR PATHWAY To investigate whether HNF4A affects polarization of BMDMs in the activated state of GR
pathway, BMDMs were infected with Ad-HNF4A for 48 h, and Dex was added to activate the GR pathway before inducing polarization (Fig. 5A). After the addition of Dex, HNF4A overexpression
up-regulated the levels of CD206, CD163, and Arg-1, indicating that HNF4A overexpression significantly promotes BMDMs polarization towards M2 phenotype in the activated state of GR pathway
(Fig. 5B, C). On the contrary, HNF4A overexpression down-regulated the levels of iNOS, MCHII, TNF-α, IL-1β, and IL-6, indicating that HNF4A overexpression significantly suppresses BMDMs
polarization towards M1 phenotype in the activated state of GR pathway (Supplementary Fig. 2D-G). Subsequently, we further explored the regulatory mechanism by knocking down NCOA2 or GR.
Under the conditions of adding Dex and overexpressing HNF4A, we found that knocking down GR inhibits BMDMs polarization towards M2 phenotype and promotes BMDMs polarization towards M1
phenotype. Consistently, knocking down NCOA2 also inhibits BMDMs polarization towards M2 phenotype and promotes polarization towards M1 phenotype (Fig. 5D, E, Supplementary Fig. 2H–K). Taken
collectively, HNF4A significantly promoted the polarization of BMDMs towards M2 phenotype and inhibited the polarization of BMDMs towards M1 phenotype via the NCOA2/GR pathway. MRNA-SEQ
WHOLE TRANSCRIPTOME PROFILING IN NCOA2-KNOCKDOWNED BMDMS TREATED WITH AD-HNF4A, IL-4 AND DEX In order to investigate the molecular basis in NCOA2-knock downed BMDMs treated with Ad-HNF4A,
IL-4, and Dex, the mRNA-sequencing was performed. Principal component (PCA) analysis indicated that all samples were clustered in the shNC and shNCOA2 cells treated with Dex and IL-4,
respectively. The difference between the shNC and shNCOA2 cells was statistically significant (Fig. 6B). The volcano plot showed that 1118 DEGs were upregulated and 1080 DEGs were
downregulated under the screening standard of |Log2FC | >1 and Adjust _p_ < 0.01 (Fig. 6C). As shown in the heatmap, it is demonstrated that the clustering relationships among the
samples within each group are reliable. We found that some DEGs are related to the inflammatory response including Dpep1, Hmox1, Ccl25, Tusc2, Csf1, Stab1, and Pld3 (Fig. 6D). To better
understand the role of these DEGs, we performed KEGG and GO enrichment analyses. GO results showed that DEGs are associated with regulation of inflammatory response, regulation of NF-kappaB
transcription factor activity, and cell cycle DNA replication. KEGG results showed that DEGs were mainly related to the TNF, IL-17, and P13K-Akt signaling pathway (Fig. 6E). For the ChIP-seq
database, 625 overlapping peaks were identified between anti-NCOA2 binding peaks and anti-GR binding peaks in GSE99887. The 625 overlapping binding peaks were identified by 533 genes. The
analysis of Venn Diagram was conducted to detect the intersection of 533 genes and DEGs of mRNA-seq. 57 overlapping genes were obtained. Then, the analysis of Venn Diagram was performed to
determine the intersection of 57 genes and top 10% of Genecards-retrieval “Macrophage polarization”. Finally, 10 genes were harvested (Fig. 6F). Of note, STAB1 has been reported to be
associated with anti-inflammatory effects [25]. It has also reported that STAB1 knockdown enhances mortality and lung damage in CLP-induced septic mice [26]. Hence, we focused on exploring
whether the HNF4A/NCOA2/GR axis influences macrophage polarization through STAB1 in a subsequent experiment. HNF4A/NCOA2/GR AFFECTS MACROPHAGE POLARIZATION THROUGH STAB1 To investigate
whether STAB1 is involved in the effect of HNF4A/NCOA2/GR on macrophage polarization, BMDMs were infected with Ad-shSTAB1 to knockdown STAB1 in BMDMs treated with IL-4. This was confirmed by
real-time PCR, WB (Fig. 7E). The results of flow cytometry suggested that knockdown of STAB1 inhibits macrophage polarization towards M2 phenotype (Fig. 7F). Conversely, knockdown of STAB1
increased the level of TNF-α in BMDMs treated with LPS and IFN-γ, indicating that knockdown of STAB1 promotes macrophage polarization towards M1 phenotype (Supplementary Fig. 2L). In the
activated state of GR pathway, STAB1 levels were markedly upregulated after HNF4A overexpression (Fig. 7A). Under the conditions of adding Dex and overexpressing HNF4A, knocking down NCOA2
or GR downregulated the levels of STAB1, indicating that STAB1 may a target of the HNF4A/NCOA2/GR axis (Fig. 7B). Interestingly, ChIP-PCR and dual luciferase assays validated that GR/NCOA2
bind to STAB1 intron 1 (Fig. 7C, D). The above results show that HNF4A/NCOA2/GR affects macrophage polarization through STAB1. Overall, this study revealed that the regulatory axis of
HNF4A/NCOA2/GR/STAB1 affects macrophage polarization, thereby improving sepsis-induced lung damage (Fig. 7G). DISCUSSION Sepsis-induced ALI is a serious clinical complication that can be
life-threatening in severe cases [27]. Currently, the treatment management of ALI in sepsis is progressing towards a more precise and integrated mode, involving advanced methods such as
antioxidant therapy, management of cell death, carbon monoxide delivery strategies, and interventions focused on specific disease processes [28, 29]. In-depth investigations at the molecular
level are crucial for identifying novel therapeutic targets and prediction tools for the lung injury in sepsis. In this study, we identified a new molecule, HNF4A, as a regulator affecting
lung injury in sepsis. By analyzing a CLP-induced microarray of gene expression in lung tissue of sepsis mice, we found that a notable reduction in the expression of HNF4A. HNF4A is a
transcription factor necessary for the development of the liver, playing a significant role in maintaining the benign phenotype [30, 31]. Recent research has also suggested its involvement
in inflammation [18] and macrophage polarization [16]. In this study, we observed diminished HNF4A expression in the lung tissues in a murine model of sepsis. Remarkably, our findings also
revealed that HNF4A was expressed in alveolar macrophages, and its expression was consistently decreased in macrophages obtained from alveolar lavage fluid of the CLP mice as compared to
normal mice. To further verify the role of HNF4A, we treated the septic mice with an adenovirus-mediated HNF4A overexpression intervention. The administration of HNF4A-overexpressed
adenoviral particles in mice raised the expression of HNF4A in the lung and reduced the sepsis-induced mortality. Moreover, HNF4A overexpression mitigates the CLP-induced pulmonary edema and
lung barrier damage and reduces inflammatory cell infiltration, such as neutrophils and macrophages. Macrophages in the lung, involving alveolar macrophages and interstitial macrophages,
are immune cells that reside in lung tissues and play a crucial role in immune defense and maintaining the tissue homeostasis [32]. Through their high plasticity, these macrophages can
display pro-inflammatory or anti-inflammatory characteristics depending on the different microenvironmental stimulation, which is vital for the lung health [33]. Proinflammatory macrophages,
also known as M1-phenotype macrophages, are an activated state of macrophages. In this state, macrophages display strong pro-inflammatory activities and produce pro-inflammatory cytokines
(such as TNF-α, IL-1β, and IL-6) as well as express inducible nitric oxide synthase (iNOS) to eradicate pathogens and promote inflammation. Anti-inflammatory macrophages, commonly referred
to as M2-phenotype macrophages, are another important activation state of macrophages. Macrophages in this state release anti-inflammatory cytokines (such as IL-10) to participate in tissue
repair, immune regulation, anti-inflammatory response, and angiogenesis [34, 35]. They are also involved in the maintenance of tissue homeostasis and immune tolerance by expressing specific
surface markers such as CD206 and CD163 [36]. In this work, we demonstrated that HNF4A overexpression facilitated the transition of macrophages from the M1 phenotype triggered by CLP to the
M2 phenotype. This transition was accompanied by alterations in the expression of pro-inflammatory cytokines TNF-α, IL-1β, IL-6, M1 surface markers MHC II, anti-inflammatory cytokines IL-4
and IL-10, and M2 surface markers CD206, CD163, and Arg-1. In terms of downstream regulators of HNF4A, our attention was directed on NCOA2. As a transcription coactivator, this protein has
been demonstrated to regulate septic injury [37] and macrophage polarization [24]. In this study, we noted the existence of NCOA2 in lung macrophages, and the level of NCOA2 expression was
also elevated in lung tissues and macrophages after HNF4A overexpression. Considering the characteristics of HNF4A as a transcription factor, we employed the dual luciferase assay and
ChIP-PCR to confirm the interaction between the NCOA2 promoter and HNF4A. The experiments demonstrated that HNF4A enhances the expression of NCOA2 by promoting its transcription.
Additionally, as a transcriptional coregulator, the NCOA2 has been defined to bind with GR [23] to regulate gene expression. Notably, the downregulation of NCOA2 in macrophages counteracts
the transcriptional suppression of pro-inflammatory proteins mediated by GR [23]. Previous studies have also shown that conditional knockout of NCOA2 [37] or GR [38] leads to an increased
mortality rate in mice suffering sepsis. These studies suggest that the NCOA2-GR pathway may be a target of the HNF4A impact. To verify this hypothesis, we utilized a glucocorticoid receptor
(GR) ligand, Dex, as an activator of the GR pathway for the investigation. Dex greatly increased the ability of HNF4A to induce macrophages transferring into the M2 phenotype and decreased
the ability of HNF4A to induce macrophages transferring into the M1 phenotype. Nevertheless, when NCOA2 is suppressed, the impact of HNF4A is largely canceled, while Dex could restore the
promotion of HNF4A on the M2 polarization of macrophages and the inhibition of HNF4A on the M1 polarization of macrophages. These phenomena suggest that HNF4A has a regulatory function in
the process of macrophage M2 and M1 polarization through the NCOA2-GR pathway. In addition, we employed transcriptome sequencing to investigate the genes influenced by HNF4A/NCOA2/GR axis.
More than 2000 genes exhibiting differential expression were identified, and following a series of screenings, our attention was directed towards STAB1. STAB1, also known as
macrophage-stimulating 1 receptor, is a transmembrane protein expressed on a variety of cell types, especially macrophages [39]. It is involved in regulating immune response and inflammatory
processes, as well as tissue repair and metabolic balance [40]. By recognizing different ligands such as collagen fragments and heparan sulfate [41], STAB1 can affect the polarization of
macrophages and promote the transformation to M2 phenotype, thus playing a crucial role in inflammation suppression and tissue healing [42]. In the current study, we observed that HNF4A
promoted the expression of STAB1, while NCOA2 or GR silencing inhibited the STAB1 expression raised by HNF4A. Moreover, STAB1 knockdown promoted the M1 phenotypic switch of macrophages and
suppressed the M2 phenotypic switch of macrophages. Unexpectedly, the mechanism by which NCOA2/GR regulates STAB1 is not directly linked to the promoter of STAB1. This is evident from the
fact that overexpression of HNF4A does not significantly affect the luciferase activity of PGL3-pro-luc, which represents the promoter activity of STAB1. However, the NCOA2/GR complex may
bind to the first intron of STAB1, which contains the GR element. The luciferase results confirmed that HNF4A increased the promoter activity containing the first intron of STAB1. In
conclusion, this research validates the potential protective effect of HNF4A against lung injury in sepsis. HNF4A may transcriptionally activate the NCOA2 promoter and increase its
expression, thus, in turn, increasing the formation of NCOA2/GR complex and inducing gene transcription such as STAB1 to promote the polarization of macrophages towards M2 and reduce the
polarization of M1. As a result, the production of inflammatory factors and the infiltration of inflammatory cells are inhibited, leading to the alleviation of lung injury induced by sepsis.
The regulatory axis of HNF4A/NCOA2/GR/STAB1 could potentially serve as a therapeutic target for sepsis-associated lung damage. MATERIALS AND METHODS INDUCTION OF SEPSIS-ASSOCIATED LUNG
INJURY IN MICE Before modeling, female and male C57BL/6 J mice aged 8–12 weeks were adaptively fed for two weeks. The mice had free access to food and water. Mice were maintained under
temperature 20–26 °C, relative humidity 40–60%. The sepsis model was established by CLP. Briefly, C57BL/6 J mice (half male and half female) were randomly allocated to the Sham and CLP
groups (_n_ = 6/group). Mice were anesthetized via inhalation isoflurane. The cecum was ligated and then punctured twice with a 25G-gauge syringe needle in mice of CLP group. After that, the
cecum was placed back in the original position, and the incision was closed. The similar surgery without CLP was performed on mice of the sham group. Besides survival analyses, the mice
were subjected to euthanasia 8 h after operation, and lung tissues along with alveolar lavage fluid were harvested. The concentration of total protein in the alveolar lavage fluid was
quantified utilizing a BCA kit (Beyotime Biotech, Shanghai, China). A portion of the lung tissues was used to assess wet and dry weights, from which the ratio of wet/dry weight was
calculated. Additional lung tissue samples were fixed with neutral formaldehyde solution or frozen with liquid nitrogen for subsequent experiments. For survival assays, the survival status
of mice was observed every 12 h for 10 days. Accidental deaths during the experiment were excluded from the study. Sample sizes were designed with power of 80% at an alpha of 0.05 according
to the literature and our previous studies. In this study, blinding was implemented to minimize bias and ensure the objectivity of the results in histological pathology experiments and
molecular detection experiments. HEMATOXYLIN AND EOSIN (H&E) STAINING AND SCORING The dehydrated lung tissues were embedded in paraffin and cut into 5 μm-thick sections. These sections
were dewaxed in dimethylbenzene and dehydrated in a concentration gradient of alcohol. Subsequently, lung slides were stained with H&E. Lung slides were dehydrated, permeabilized, and
sealed prior to being visualized using a microscope (BX53; Olympus, Tokyo, Japan) under 200× magnifications. The criteria utilized for assessing tissue damage are based on methods
established in prior research [43]. The infiltration of inflammatory cells was observed and the injury of lung tissues was scored. REAL-TIME PCR Total RNA was extracted from lung tissues, or
cells using the TRIpure reagent (BioTeke Corporation, Beijing, China). The concentration of RNA in each sample was determined using ultraviolet spectrophotometer (NANO 2000, Thermo
Scientific, Pittsburgh, PA, USA). RNA samples were reversely transcribed to obtain the corresponding cDNA. After that, the fluorescence quantitative PCR instrument (Bioneer, Daejeon, Korea)
was used to perform the real-time PCR assay. The final relative expression of mRNA was determined utilizing the 2−△△CT method. The primers are provided in Table 1. WESTERN BLOT ANALYSIS
Total proteins in tissues or cells were extracted and quantified using a BCA kit (Solarbio, Beijing, China). The proteins were subjected to separation via SDS-PAGE, followed by transferring
onto PVDF membranes. After blocking, the membranes were incubated with the primary antibodies at 4 °C overnight, and then with the secondary antibodies at 37 °C for 1 h. After visualization
with the ECL reagent (Solarbio, Beijing, China), the optical density values of the protein bands were analyzed using the Gel-Pro-Analyzer software. The primary antibodies were as follows:
HNF4A antibody (1:500, bs-3828R, Bioss, Beijing, China), NCOA2 antibody (1:1000, bs-20558R, Bioss, Beijing, China) and STAB1 antibody (1:500, bs-7510R, Bioss, Beijing, China). The secondary
antibodies were as follows: HRP-labeled goat anti-rabbit (1:3000, SE134, Solarbio, Beijing, China) IgG and HRP-labeled goat anti-mouse IgG (1:3000, SE131, Solarbio, Beijing, China).
IMMUNOFLUORESCENCE (IF) STAINING Lung tissues were made into 5 μm-thick paraffin sections. After dewaxing, antigen repairing, and blocking, the lung sections were incubated overnight with
diluted F4/80 antibody (1:50, Sc-377009, Santa Cruz, Dallas, TX, USA), NCOA2 antibody (1:100, bs-20558R, Bioss, Beijing, China), or HNF4A antibody (1:100, bs-3828R, Bioss, Beijing, China)
overnight at 4 °C. For samples of alveolar macrophages, cell sections were fixed for 15 min and incubated with 0.1% tritonX-100 (Beyotime Biotech, Shanghai, China) for 30 min at room
temperature, followed by incubation with NCOA2 antibody (1:100, bs-20558R, Bioss, Beijing, China). Then sections were incubated with Cy3-labeled goat anti-rabbit IgG (1:200, ab6939, Abcam,
Cambridge, UK) and FITC-labeled goat anti-mouse IgG (1:200, ab6785, Abcam, Cambridge, UK) for 90 min at room temperature. DAPI was used to counterstain the nuclei. After dropping the
anti-fluorescence quenching sealing reagent, the sections were photographed under a fluorescence microscope (BX53; Olympus, Tokyo, Japan) under 400× magnifications. CONSTRUCTION OF
ADENOVIRUS VECTORS Adenoviruses overexpressing HNF4A (Ad-HNF4A) or negative controls (Ad-NC) were generated. For mouse experiments, Ad-NC or Ad-HNF4A of 1 × 109 pfu was instilled into the
trachea of each mouse 48 h prior to establishing the model. For cell experiments, cells were cultured in the medium containing Ad-NC or Ad-HNF4A for 48 h. Bone marrow-derived macrophages
(BMDMs) were treated with IL-4 for 24 h to induce M2 polarization. The 100 nM dexamethasone (Dex) was added to activate the GR pathway. shNCOA2 and shGR were synthesized to knock down the
expression of NCOA2 and GR by adenovirus delivery to infected macrophages. ENZYME-LINKED IMMUNOSORBENT ASSAY (ELISA) To investigate the effect of HNF4A on the inflammation of lung tissue in
septic mice, ELISA was performed on alveolar lavage fluid to detect IL-1β, IL-6, TNF-α, IL-4, and IL-10 via the corresponding kits (Multi Sciences, Hangzhou, China). OD values at 450 nm and
570 nm were determined and the corresponding concentrations were calculated. WRIGHT-GIEMSA STAINING Alveolar macrophages were isolated from alveolar lavage fluid. After washing with PBS, the
alveolar lavage solution was centrifuged at 300 × _g_ for 5 min. Subsequently, cells were seeded in a medium containing 10% fetal bovine serum. Alveolar macrophages were stained with a
Wright-Giemsa stain kit (Jiancheng Bioengineering Institute, Nanjing, China). In brief, alveolar lavage fluid preserved by centrifugation was resuspended. Cell slides were made and fixed in
methanol for 15 min. After drying, the slides were stained with Giemsa A for 1 min and Giemsa B for 7 min. Ultimately, the cells were destained with 80% ethanol until the cells were clear.
Total cells, neutrophils, and macrophages in alveolar lavage fluid were counted respectively. FLOW CYTOMETER ANALYSIS To detect M1-type or M2-type polarization, macrophages in alveolar
lavage fluid of mice were incubated with F4/80 antibody (F21480A03, Multi Sciences, Hangzhou, China), Arg-1 antibody (12-3697-80, Thermo Scientific, Pittsburgh, PA, USA) or TNF-α antibody
(12-27321-41, Thermo Scientific, Pittsburgh, PA, USA) at 4 °C for 30 min in the darkness. BMDMs were incubated with F4/80 antibody and CD11b antibody (F41011b02, Multi Sciences, Hangzhou,
China) to identify the macrophages. BMDMs were incubated with F4/80 antibody and CD206 antibody (12-2061-80, Invitrogen, Carlsbad, CA, USA) to measure M2-type polarization. Subsequently,
flow dye buffer was added and the sample was centrifuged (300 g) for 5 min. The supernatant was removed and resuspended with flow dyeing buffer assaying by flow cytometry. ISOLATION AND
CULTURE OF MOUSE BMDMS BMDMs were obtained using the previous method [44]. In the cell experiment, three biological replicates were performed. The mouse femur was removed in a sterile state
and cleaned with PBS. The two ends of the bone were cut with sterile scissors, and the bone marrow was rinsed from one end with a syringe needle containing serum-free medium so that the
liquid flowed out from the other end and was received into a sterile centrifuge tube for 10 min. After the cell precipitation was suspended with the supernatant of L929 cells, it was
inoculated in a culture dish and cultured in an incubator at 37 °C and 5% CO2. L929 cells were purchased from iCell and cultured in minimum essential medium containing 10% equine serum. The
cells were identified by STR and tested negative for mycoplasma contamination. DUAL-LUCIFERASE REPORTER ASSAY The NCOA2 promoter fragment was inserted into the pGL3-basic luciferase vector
to generate the pGL3-NCOA2 promoter luciferase reporter. 293 T cells were co-transfected with pGL3-NCOA2 promoter luciferase reporter and pRL-TK renilla luciferase reporter plasmid. STAB1
promoter was inserted into the upstream of the transcription unit in the pGL3-basic vector to generate the PGL3-pro-luc promoter luciferase reporter. STAB1 promoter is inserted into the
upstream of the transcription unit in the pGL3-basic vector, and STAB1 intron1 is inserted into the downstream of the transcription unit in the same vector to generate the PGL3-pro-luc-Int1
promoter luciferase reporter. The cells were lysed 48 h after transfection. Firefly luciferase and renilla luciferase activities were determined by the dual luciferase reporter gene assay
kit (KeyGEN, Nanjing, China). The relative luciferase activity was normalized with renilla luciferase activity. 293 T cells were cultured with DMEM containing 10% FBS in a 37 °C, 5% CO2
incubator. The cells were purchased from iCell. CHROMATIN IMMUNOPRECIPITATION (CHIP) ASSAY The binding of HNF4A to NCOA2 promoter was detected using the ChIP assay kit (Beyotime, Shanghai,
China). Briefly, DNA and protein were cross-linked using 1% formaldehyde. The Cells were lysed and sonicated to shear DNA. Cell lysates were immunoprecipitated with control rabbit IgG or
HNF4A antibody (Bioss, Beijing, China). PCR was used to amplify the purified DNA from the cell lysates and the DNA recovered from the immunoprecipitation. The primers for NCOA2 are
5′-GGGAGGCACCTCTGGGACTA-3′ (forward) and 5′-CCTGGCTGGCGAGACTTCA-3′ (reverse); STAB1 intron1: 5′-TCCTTGGCTGACAGTGGG-3′ (forward) and 5′-CTGGTGGGCAGATGAGTG-3′ (reverse). MRNA-SEQ BMDMs were
transfected with Ad-HNF4A and/or Ad-shNCOA2 for 48 h, followed by addition of IL-4 and Dex. Total RNA was isolated from cell samples, after which complementary DNA (cDNA) libraries were
constructed and subjected to mRNA sequencing (mRNA-seq). DEGs were defined as |log2FoldChange | >1 and Adjust _p_ < 0.01. Subsequently, all DEGs were subjected to analysis for Gene
Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment, with the aim of conducting functional annotation. STATISTICAL ANALYSIS Data were presented in the form of mean ±
standard error of mean. GraphPad Prism version 9.5 was utilized for the purposes of data visualization and statistical analysis. The data were tested for normality and homogeneity of
variance. The differences between two groups were evaluated using two-tailed unpaired Student _t_-tests. The differences among three or more groups were evaluated using One-way ANOVA post
hoc Tukey’s tests. The survival curve was compared by the log-rank test and plotted by the Kaplan–Meier method. All analysis with _p_ < 0.05 were considered significant. DATA AVAILABILITY
Data deposition The mRNA-seq data was presented in Dryad database (https://doi.org/10.5061/dryad.gf1vhhn06). Data will be made available on reasonable request from the corresponding author.
CHANGE HISTORY * _ 19 MARCH 2025 There are three instances in this article where “NCOR2” should be corrected to “NCOA2.” The corresponding sentences should be revised as follows: 1. Title:
“HNF4A mitigates sepsis-associated lung injury by upregulating NCOA2/GR/STAB1 axis and promoting macrophage polarization towards M2 phenotype” 2. The last sentence of the abstract: “It is
suggested that one of the regulatory mechanisms involved in this association may be the NCOA2/GR/STAB1 axis.” 3. Discussion in paragraph 5, following reference [42]: “In the current study,
we observed that HNF4A promoted the expression of STAB1, while NCOA2 or GR silencing inhibited the STAB1 expression raised by HNF4A.” _ * _ 14 APRIL 2025 A Correction to this paper has been
published: https://doi.org/10.1038/s41419-025-07519-x _ REFERENCES * Lelubre C, Vincent JL. Mechanisms and treatment of organ failure in sepsis. Nat Rev Nephrol. 2018;14:417–27. Article
PubMed Google Scholar * Gauer R, Forbes D, Boyer N. Sepsis: diagnosis and management. Am Fam Physician. 2020;101:409–18. PubMed Google Scholar * Kumar V. Pulmonary innate immune response
determines the outcome of inflammation during pneumonia and sepsis-associated acute lung injury. Front Immunol. 2020;11:1722. Article PubMed PubMed Central CAS Google Scholar *
Cardinal-Fernández P, Bajwa EK, Dominguez-Calvo A, Menéndez JM, Papazian L, Thompson BT. The presence of diffuse alveolar damage on open lung biopsy is associated with mortality in patients
with acute respiratory distress syndrome: a systematic review and meta-analysis. Chest. 2016;149:1155–64. Article PubMed Google Scholar * Fan EKY, Fan J. Regulation of alveolar macrophage
death in acute lung inflammation. Respir Res. 2018;19:50. Article PubMed PubMed Central Google Scholar * Mosser DM, Hamidzadeh K, Goncalves R. Macrophages and the maintenance of
homeostasis. Cell Mol Immunol. 2021;18:579–87. Article PubMed CAS Google Scholar * Wang Z, Wang Z. The role of macrophages polarization in sepsis-induced acute lung injury. Front
Immunol. 2023;14:1209438. Article PubMed PubMed Central CAS Google Scholar * Aegerter H, Lambrecht BN, Jakubzick CV. Biology of lung macrophages in health and disease. Immunity.
2022;55:1564–80. Article PubMed PubMed Central CAS Google Scholar * Koncz G, Jenei V, Tóth M, Váradi E, Kardos B, Bácsi A, et al. Damage-mediated macrophage polarization in sterile
inflammation. Front Immunol. 2023;14:1169560. Article PubMed PubMed Central CAS Google Scholar * Lee JW, Chun W, Lee HJ, Min JH, Kim SM, Seo JY, et al. The role of macrophages in the
development of acute and chronic inflammatory lung diseases. Cells. 2021;10:897. * Zhang J, Wang C, Wang H, Li X, Xu J, Yu K. Loganin alleviates sepsis-induced acute lung injury by
regulating macrophage polarization and inhibiting NLRP3 inflammasome activation. Int Immunopharmacol. 2021;95:107529. Article PubMed CAS Google Scholar * Zhang Y, Huang T, Jiang L, Gao
J, Yu D, Ge Y, et al. MCP-induced protein 1 attenuates sepsis-induced acute lung injury by modulating macrophage polarization via the JNK/c-Myc pathway. Int Immunopharmacol. 2019;75:105741.
Article PubMed CAS Google Scholar * Luo J, Wang J, Zhang J, Sang A, Ye X, Cheng Z, et al. Nrf2 Deficiency exacerbated clp-induced pulmonary injury and inflammation through autophagy- and
NF-κB/PPARγ-mediated macrophage polarization. Cells. 2022;11:3927. * Walesky C, Gunewardena S, Terwilliger EF, Edwards G, Borude P, Apte U. Hepatocyte-specific deletion of hepatocyte
nuclear factor-4α in adult mice results in increased hepatocyte proliferation. Am J Physiol Gastrointest Liver Physiol. 2013;304:G26–37. Article PubMed CAS Google Scholar * Kotulkar M,
Paine-Cabrera D, Abernathy S, Robarts DR, Parkes WS, Lin-Rahardja K, et al. Role of HNF4alpha-cMyc interaction in liver regeneration and recovery after acetaminophen-induced acute liver
injury. Hepatology. 2023;78:1106–17. Article PubMed Google Scholar * Yang T, Poenisch M, Khanal R, Hu Q, Dai Z, Li R, et al. Therapeutic HNF4A mRNA attenuates liver fibrosis in a
preclinical model. J Hepatol. 2021;75:1420–33. Article PubMed CAS Google Scholar * Kelly C, Jawahar J, Davey L, Everitt JI, Galanko JA, Anderson C, et al. Spontaneous episodic
inflammation in the intestines of mice lacking HNF4A is driven by microbiota and associated with early life microbiota alterations. mBio. 2023;14:e0150423. Article PubMed Google Scholar *
Ehle C, Iyer-Bierhoff A, Wu Y, Xing S, Kiehntopf M, Mosig AS, et al. Downregulation of HNF4A enables transcriptomic reprogramming during the hepatic acute-phase response. Commun Biol.
2024;7:589. Article PubMed PubMed Central CAS Google Scholar * Meijer OC, Koorneef LL, Kroon J. Glucocorticoid receptor modulators. Ann Endocrinol. 2018;79:107–11. Article Google
Scholar * Frank F, Liu X, Ortlund EA. Glucocorticoid receptor condensates link DNA-dependent receptor dimerization and transcriptional transactivation. Proc Natl Acad Sci USA.
2021;118:e2024685118. * Scheschowitsch K, Leite JA, Assreuy J. New insights in glucocorticoid receptor signaling-more than just a ligand-binding receptor. Front Endocrinol. 2017;8:16.
Article Google Scholar * Clarisse D, Van Moortel L, Van Leene C, Gevaert K, De Bosscher K. Glucocorticoid receptor signaling: intricacies and therapeutic opportunities. Trends Biochem Sci.
2024;49:431–44. Article PubMed CAS Google Scholar * Rollins DA, Kharlyngdoh JB, Coppo M, Tharmalingam B, Mimouna S, Guo Z, et al. Glucocorticoid-induced phosphorylation by CDK9
modulates the coactivator functions of transcriptional cofactor GRIP1 in macrophages. Nat Commun. 2017;8:1739. Article PubMed PubMed Central Google Scholar * Coppo M, Chinenov Y, Sacta
MA, Rogatsky I. The transcriptional coregulator GRIP1 controls macrophage polarization and metabolic homeostasis. Nat Commun. 2016;7:12254. Article PubMed PubMed Central CAS Google
Scholar * Cabral F, Al-Rahem M, Skaggs J, Thomas TA, Kumar N, Wu Q, et al. Stabilin receptors clear LPS and control systemic inflammation. iScience. 2021;24:103337. Article PubMed PubMed
Central CAS Google Scholar * Lee W, Park SY, Yoo Y, Kim SY, Kim JE, Kim SW, et al. Macrophagic stabilin-1 restored disruption of vascular integrity caused by sepsis. Thromb Haemost.
2018;118:1776–89. Article PubMed Google Scholar * Bardají-Carrillo M, Martín-Fernández M, López-Herrero R, Priede-Vimbela JM, Heredia-Rodríguez M, Gómez-Sánchez E, et al. Post-operative
sepsis-induced acute respiratory distress syndrome: risk factors for a life-threatening complication. Front Med. 2024;11:1338542. Article Google Scholar * Kim WY, Hong SB. Sepsis and Acute
Respiratory Distress syndrome: recent update. Tuberc Respir Dis. 2016;79:53–7. Article Google Scholar * Sharma A, Yang WL, Ochani M, Wang P. Mitigation of sepsis-induced inflammatory
responses and organ injury through targeting Wnt/β-catenin signaling. Sci Rep. 2017;7:9235. Article PubMed PubMed Central Google Scholar * Hunter AL, Poolman TM, Kim D, Gonzalez FJ,
Bechtold DA, Loudon ASI, et al. HNF4A modulates glucocorticoid action in the liver. Cell Rep. 2022;39:110697. Article PubMed PubMed Central CAS Google Scholar * Thymiakou E, Tzardi M,
Kardassis D. Impaired hepatic glucose metabolism and liver-α-cell axis in mice with liver-specific ablation of the Hepatocyte Nuclear Factor 4α (Hnf4a) gene. Metabolism. 2023;139:155371.
Article PubMed CAS Google Scholar * Melo EM, Oliveira VLS, Boff D, Galvão I. Pulmonary macrophages and their different roles in health and disease. Int J Biochem Cell Biol.
2021;141:106095. Article PubMed CAS Google Scholar * Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and
function in health and disease. J Cell Physiol. 2018;233:6425–40. Article PubMed CAS Google Scholar * Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol.
2020;877:173090. Article PubMed Google Scholar * Wang Y, Smith W, Hao D, He B, Kong L. M1 and M2 macrophage polarization and potentially therapeutic naturally occurring compounds. Int
Immunopharmacol. 2019;70:459–66. Article PubMed CAS Google Scholar * Bhattacharya S, Aggarwal A. M2 macrophages and their role in rheumatic diseases. Rheumatol Int. 2019;39:769–80.
Article PubMed CAS Google Scholar * Chinenov Y, Gupte R, Dobrovolna J, Flammer JR, Liu B, Michelassi FE, et al. Role of transcriptional coregulator GRIP1 in the anti-inflammatory actions
of glucocorticoids. Proc Natl Acad Sci USA. 2012;109:11776–81. Article PubMed PubMed Central CAS Google Scholar * Goodwin JE, Feng Y, Velazquez H, Sessa WC. Endothelial glucocorticoid
receptor is required for protection against sepsis. Proc Natl Acad Sci USA. 2013;110:306–11. Article PubMed CAS Google Scholar * Leonard EJ. Biological aspects of macrophage-stimulating
protein (MSP) and its receptor. Ciba Found Symp. 1997;212:183–91. PubMed CAS Google Scholar * Wang MH, Zhou YQ, Chen YQ. Macrophage-stimulating protein and RON receptor tyrosine kinase:
potential regulators of macrophage inflammatory activities. Scand J Immunol. 2002;56:545–53. Article PubMed CAS Google Scholar * Kzhyshkowska J. Multifunctional receptor stabilin-1 in
homeostasis and disease. ScientificWorldJournal. 2010;10:2039–53. Article PubMed PubMed Central CAS Google Scholar * Patten DA, Wilkinson AL, O’Keeffe A, Shetty S. Scavenger Receptors:
Novel Roles in the Pathogenesis of Liver Inflammation and Cancer. Semin Liver Dis. 2022;42:61–76. Article PubMed CAS Google Scholar * Jiang J, Huang K, Xu S, Garcia JGN, Wang C, Cai H.
Targeting NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox Biology.
2020;36. * Tuckermann JP, Kleiman A, Moriggl R, Spanbroek R, Neumann A, Illing A, et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact
allergy. J Clin Investig. 2007;117:1381–90. Article PubMed PubMed Central CAS Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by the National Natural Science
Foundation of China (82102254 to Tie-Ning Zhang, and 81971810 to Chun-Feng Liu), Liaoning Province Science and Technology Major Special Project (No.2020JH1/10300001 to Chun-Feng Liu), and
345 Talent Program of Shengjing Hospital of China Medical University (M1308 to Tie-Ning Zhang and M1404 to Ni Yang). AUTHOR INFORMATION Author notes * These authors contributed equally:
Yu-Hang Yang, Ri Wen. AUTHORS AND AFFILIATIONS * Department of Pediatrics, PICU, Shengjing Hospital of China Medical University, Shenyang, China Yu-Hang Yang, Ri Wen, Tao Zhang, Ni Yang,
Chun-Feng Liu & Tie-Ning Zhang * Department of Endocrinology, Shanghai Fifth People’s Hospital, Fudan University, Shanghai, China Xin-Mei Huang Authors * Yu-Hang Yang View author
publications You can also search for this author inPubMed Google Scholar * Ri Wen View author publications You can also search for this author inPubMed Google Scholar * Xin-Mei Huang View
author publications You can also search for this author inPubMed Google Scholar * Tao Zhang View author publications You can also search for this author inPubMed Google Scholar * Ni Yang
View author publications You can also search for this author inPubMed Google Scholar * Chun-Feng Liu View author publications You can also search for this author inPubMed Google Scholar *
Tie-Ning Zhang View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS C-FL and T-NZ conceived the study. Y-HY and RW contributed to the design of
the study and wrote the manuscript with help of the other co-authors. Y-HY, RW, X-MH, TZ, and NY contributed to experiments of the study. All authors reviewed the manuscript. CORRESPONDING
AUTHORS Correspondence to Chun-Feng Liu or Tie-Ning Zhang. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interest. ETHICS APPROVAL The animal experiments were
approved by the ethical committees of Shengjing Hospital of China Medical University (2023PS795K), and conducted in accordance with the Guide for the Care and Use of Laboratory Animals. This
work did not involve human subjects. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. Edited by Hans-Uwe Simon SUPPLEMENTARY INFORMATION SUPPLEMENTAL MATERIAL ORIGINAL DATA ORIGINAL REAL-TIME PCR RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under
a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article
are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Yang, YH., Wen, R., Huang, XM. _et al._ HNF4A mitigates
sepsis-associated lung injury by upregulating NCOA2/GR/STAB1 axis and promoting macrophage polarization towards M2 phenotype. _Cell Death Dis_ 16, 120 (2025).
https://doi.org/10.1038/s41419-025-07452-z Download citation * Received: 02 September 2024 * Revised: 15 January 2025 * Accepted: 12 February 2025 * Published: 21 February 2025 * DOI:
https://doi.org/10.1038/s41419-025-07452-z SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative



)



