- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Mortality from colorectal cancer (CRC) is significant, and novel CRC therapies are needed. A pseudokinase MLKL typically executes necroptotic cell death, and MLKL inactivation
protects cells from such death. However, we found unexpectedly that MLKL gene knockout enhanced CRC cell death caused by a protein synthesis inhibitor homoharringtonine used for chronic
myeloid leukemia treatment. In an effort to explain this finding, we observed that MLKL gene knockout reduces the basal CRC cell autophagy and renders such autophagy critically dependent on
the presence of VPS37A, a component of the ESCRT-I complex. We further found that the reason why homoharringtonine enhances CRC cell death caused by MLKL gene knockout is that
homoharringtonine activates p38 MAP kinase and thereby prevents VPS37A from supporting autophagy in MLKL-deficient cells. We observed that the resulting inhibition of the basal autophagy in
CRC cells triggers their parthanatos, a cell death type driven by poly(ADP-ribose) polymerase hyperactivation. Finally, we discovered that a pharmacological MLKL inhibitor necrosulfonamide
strongly cooperates with homoharringtonine in suppressing CRC cell tumorigenicity in mice. Thus, while MLKL promotes cell death during necroptosis, MLKL supports the basal autophagy in CRC
cells and thereby protects them from death. MLKL inactivation reduces such autophagy and renders the cells sensitive to autophagy inhibitors, such as homoharringtonine. Hence, MLKL
inhibition creates a therapeutic vulnerability that could be utilized for CRC treatment. SIMILAR CONTENT BEING VIEWED BY OTHERS ABIN-1 IS A KEY REGULATOR IN RIPK1-DEPENDENT APOPTOSIS (RDA)
AND NECROPTOSIS, AND ABIN-1 DEFICIENCY POTENTIATES NECROPTOSIS-BASED CANCER THERAPY IN COLORECTAL CANCER Article Open access 01 February 2021 CULLIN-9/P53 MEDIATES HNRNPC DEGRADATION TO
INHIBIT ERASTIN-INDUCED FERROPTOSIS AND IS BLOCKED BY MDM2 INHIBITION IN COLORECTAL CANCER Article 03 May 2022 TRAF6 REGULATES THE ABUNDANCE OF RIPK1 AND INHIBITS THE RIPK1/RIPK3/MLKL
NECROPTOSIS SIGNALING PATHWAY AND AFFECTS THE PROGRESSION OF COLORECTAL CANCER Article Open access 05 January 2023 INTRODUCTION 20% of patients with colorectal cancer (CRC) have metastatic
disease at the time of diagnosis, and 40% develop metastases after localized disease treatment [1]. Five-year survival of patients with metastatic CRC is below 20% [1]. Hence, novel CRC
therapies are needed. Necroptosis is programmed necrosis driven by a protein kinase MLKL [2]. When activated, MLKL polymerizes, forms pores in the plasma membrane and thus kills the cell
[2]. We noticed unexpectedly that a small molecule MLKL inhibitor necrosulfonamide [3] or MLKL gene knockout (KO) reduce, rather than promote, CRC cell survival. Hence, we tested whether
MLKL inactivation might represent a novel approach for CRC treatment. We found that CRC cell death caused by MLKL inactivation is enhanced by homoharringtonine (HHT), a drug used to treat
chronic myelogenous leukemia [4]. We found that MLKL inactivation and HHT cooperate in blocking the ESCRT machinery-dependent basal autophagy of CRC cells and kill them by a form of cell
death termed parthanatos. HHT is a ribosome inhibitor [5], and such inhibitors trigger ribotoxic stress response, a chain of events that activate protein kinase p38 MAP kinase (p38MAPK)
which often triggers cell death-promoting signals [6]. Autophagy is a process of degradation of the cellular content mediated by the double-membrane vesicles termed autophagosomes [2]. One
of the functions of autophagy is to promote cell survival by eliminating the toxic cellular content, including damaged proteins and organelles [7]. Endosomal sorting complexes required for
transport (ESCRTs) mediate the formation of numerous types of vesicles, including autophagosomes [8] and promote autophagy via several mechanisms [9, 10]. Parthanatos is a form of programmed
necrosis triggered by hyperactivation of poly(ADP-ribose) polymerase 1, an enzyme that promotes the formation of poly(ADP-ribose) [11]. Excessive poly(ADP-ribose) levels cause chromosomal
DNA degradation and cell death [11]. We found that MLKL inactivation renders CRC cell autophagy strongly dependent on the presence of the ESCRT-I complex component VPS37A [8] and that in the
absence of MLKL, HHT activates p38MAPK and thus prevents VPS37A from supporting autophagy. As a result of autophagy inhibition caused by the combined effect of MLKL inactivation and HHT,
CRC cells die by parthanatos. Moreover, we found that a small molecule MLKL inhibitor necrosulfonamide and HHT strongly cooperate in reducing the ability of CRC cells to form tumors in mice.
Hence, pharmacological MLKL inhibition combined with HHT treatment is a potential novel approach for CRC therapy. MATERIALS AND METHODS CELL CULTURE Cell lines DLD1, HCT116, LS180, SW480
and 293 T were cultured in DMEM (GIBCO) and 10% Fetal Bovine Serum (FBS) (Sigma), 100 U/ml penicillin (GIBCO), 100 μg/ml streptomycin (GIBCO), 0.29 mg/ml L-glutamine (GIBCO). Lack of
mycoplasma contamination in all cells was established as published [12]. REAGENTS The following reagents were from Cell Signaling Technology, Danvers, MA. Bafilomycin A1 (catalogue# 54645S),
Z-VAD(OMe)-FMK (catalogue# 60332S), Olaparib (catalogue# 93852S), SB203580 (catalogue# 5633S). The following other reagents were used. Vemurafenib, Santa Cruz Biotechnology, Dallas TX, USA
(catalogue# CAS 918504-65-1), selumetinib, Cedarlane Labs, Burlington ON, Canada (catalogue# ENZ-CHM184-0050), pictilisib, Cedarlane Labs (catalogue# HY-50094-10MG), lapatinib Selleckchem,
Houston, TX, USA (catalogue# S1028), Lipofectamine™ 3000 Transfection Reagent, Thermo Fisher Scientific Waltham, MA, USA (catalogue# L3000008), Tat-BECN1 peptide, Selleckchem Houston, TX,
USA (catalogue# S8595), scrambled peptide, MedChemExpress, Monmouth Junction, NJ, USA (catalogue# HY-P4106). ANTIBODIES The following antibodies used for western blotting were from Cell
Signaling Technology, Danvers, MA, USA. Anti- LC3B (D11) (catalogue# 3868S), anti-DR4 (D9S1R) (catalogue# 42533S) anti-DR5 (D4E9) (catalogue# 8074S), anti-MLKL (D2I6N) (catalogue# 14993S),
anti-alpha-tubulin (DM1A) (catalogue# 3873S), anti-poly/Mono-ADP Ribose (D9P7Z) (catalogue# 89190S), anti-p38 (catalogue# 9212), phospho-p38 (catalogue# 4511). Anti-VPS37A was from
Proteintech, Rosemont, IL, USA (catalogue# 11870-1-AP). Western blotting experiments shown in Figs. 1C, 3C, 5A, B, 6A, C and in Supplementary Fig. 2A, B were performed twice with similar
results. Western blotting experiments shown in Figs. 3A, 4A, B, D, 3B, 5C–F, 6B and 8F were performed three times with similar results. MLKL GENE KNOCKOUT BY CRISPR sgRNA-encoding cDNAs
targeting exon 2 (TTGAAGCATATTATCACCCT) or exon 9 (TTAGCTTTGGAATCGTCCTC) of the MLKL gene were generated by use of CHOPCHOP web tool version 3 [13] and cloned into pX459 expression vector.
DLD-1 cells were transfected with 2ug of pX459 encoding each sgRNA cloned pX459 for 1 day as described [14], the cells were treated with 3 μg/ml of puromycin for 2 days, and clonal cell
lines were isolated by limiting dilution and analyzed for MLKL expression by western blotting. A clone that still expressed MLKL was chosen as a control clone, and clones that did not show
MLKL expression in the case of each sgRNA were selected as MLKL-deficient clones. RNA INTERFERENCE Small interfering (si)RNAs were used as described [15]. In the case of VPS37A knockdown
experiments, the control and VPS37A siRNAs were from Thermo Fisher Scientific (Waltham, MA, USA). Silencer™ Select Negative Control No. 1 siRNA (cat # 4390843) served as a control RNA. The
sequences of VPS37A siRNA1 and 2, respectively, were CGACAAAGAUGACUUAGUATT (cat #s44037) and CGACAUCACUUAAUGGAUATT (cat# s 44038). In the case of ATG12 knockdown experiments, the control and
ATG12 siRNAs were from Horizon Discovery (Cambridge, UK). The sequence of the control ON-TARGETplus non-targeting RNA (cat # D-001810-01) was UGGUUUACAUGUCGACUAA. The sequences of ATG12
siRNA1 and 2, respectively were GGGAAGGACUUACGGAUGU (cat # J-010212-08) and GCAGUAGAGC GAACACGAA (cat # 010212-07). DRUG TREATMENT Fo the clonogenic survival assay, in the case of MLKL KO
clones, cells were plated on day 1, treated with a drug on day 2, and the medium was changed to drug-free medium on day 5. In the case of treatment of cells with NSA, HHT or other drugs
described in the study, cells were plated on day 1, treated with NSA on day 2 to ensure that MLKL is inhibited before the cells are treated further, the second drug was added on day 3, and
the medium was changed to drug-free medium on day 6 in the case of SCH 772984 and on day 7 in the case of other drugs. NSA concentration used in the study is known to effectively inhibit
MLKL-dependent necroptosis [3], while HHT concentration used by us was found to effectively inhibit growth of various cell lines [16]. In the case of other assays, cells were typically
treated with the indicated drugs at several time points, and the results that were reproducibly obtained at a given time point are shown in the paper. Treatment with SCH 772984, vemurafenib,
selumetinib, pictilisib, lapatinib, bafilomycin A1, SBI0206965, SB203580, TAT-Beclin peptide and olaparib was performed as described in the papers cited in the respective sections of the
manuscript since under these conditions the drugs were found to effectively induce the intended effects. Concentrations of all drugs used by us are indicated in figure legends. TUMOR
XENOGRAFTS Animal studies were approved by Dalhousie University Committee on Laboratory Animals. 6-week-old Nu/Nu Nude mice (Charles River Canada, Saint-Constant, QC) were allowed to
acclimatize for 2 weeks. 8 × 106 cells were harvested by trypsin treatment, washed thrice in ice-cold PBS and injected in the muse flank. When tumor volumes reached the average of 80 mm3,
the mice were injected intraperitoneally (IP) with either DMSO or 4.2 mg/kg NSA or 1 mg/kg HHT or both. NSA was injected daily for 7 days and HHT, daily for 5 days, followed by a 2-day break
followed by 5 more days of daily injections. Tumor volumes were measured as published [17]. HYPODIPLOID SUB-G1 DNA CONTENT ANALYSIS 20,000 cells/well were seeded in a 6-well plate,
harvested after drug treatment using 0.25% trypsin at 37 °C for 5 min, followed by trypsin neutralization with equal volume of the respective serum-containing medium. Cells were pelleted at
500 g for 5 min, washed with 2 ml phosphate-buffered saline (PBS), pelleted at 500 × _g_ at 4 °C for 5 min, fixed with 5 ml of 70% ethanol overnight at 4 °C, pelleted at 900 × _g_ for 10
min, washed with 3 ml PBS, pelleted at 900 × _g_ at 4 °C for 5 min, resuspended in 500 μl of PBS containing 100 μg/mL RNase A, incubated for 30 min at room temperature. 200 µl of propidium
iodide was then added to the cells. The cells were analyzed by flow cytometry using BD FACS Celesta instrument, and the data were processed using Flowjo software. ASSESSMENT OF PROBABILITY
OF PATIENT’S OVERALL SURVIVAL The analysis was carried out by use of GEPIA web tool [18]. The cut off for low and high MLKL mRNA expression levels was 78% and 22%, respectively. Western
blotting [12], detection of clonogenic cell survival [19] and detection of the LC3 puncta [20] were performed as published [19]. Representative images of the cells with the LC3 puncta are
shown, and in each case, the scale bar was moved closer to the representative image of the cell from the part of the image not included in the figure. The original western blot images are
shown in Supplementary figures. STATISTICAL ANALYSIS Statistical analysis of the data in Figs. 1a and 9e was performed by the two-sided chi-square test for goodness-of-fit, and statistical
analysis of all other data, by the two-sided Student’s _t_ test. The number of times each experiment was repeated with similar results is indicated in the respective figure legends. RESULTS
HHT ENHANCES CRC CELL DEATH CAUSED BY MLKL INACTIVATION Oncogenic RAS often drives CRC [21, 22]. We found that RAS-induced activation of a protein kinase ERK promotes CRC cell survival by
blocking both apoptotic and non-apoptotic cellular signals [23]. The non-apoptotic events blocked by the RAS/ERK signaling in CRC are understood poorly, and we decided to explore them.
Necroptosis, non-apoptotic programmed cell death driven by plasma membrane permeabilization, is driven by a pseudokinase MLKL which forms an oligomeric structure that disrupts the cell
membrane [24, 25]. To test whether ERK inhibition kills CRC by necroptosis we examined whether death of oncogenic RAS-expressing CRC cells HCT116 [26] caused by the ERK inhibitor SCH 772984
[27] is blocked by necrosulfonamide (NSA), a widely used small molecule MLKL inhibitor that binds MLKL at Cys86 [3] and suppresses necroptosis by preventing MLKL incorporation in the plasma
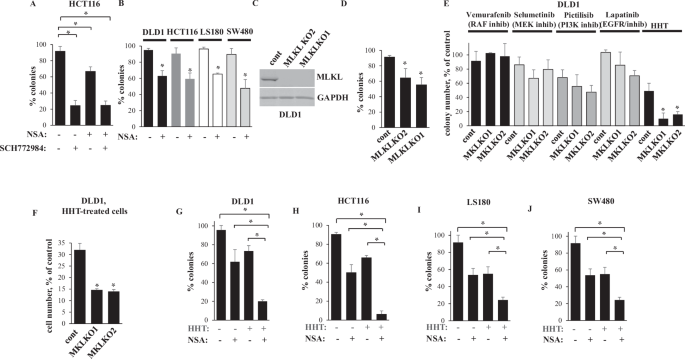
membrane [3]. We assessed cell viability by the clonogenic survival assay often utilized for this purpose [23, 28]. We found that NSA did not block SCH 772984-induced cell death (Fig. 1A).
Thus, necroptosis inhibition is not the mechanism of ERK-dependent CRC cell survival. Unexpectedly, even though MLKL typically promotes cell death [24], we found that NSA reduced clonogenic
survival of HCT116 cells (Fig. 1A). Of note, _MLKL_-deficient mice are viable, healthy, fertile, are born with normal Mendelian frequency and do not show any physical or behavioral
abnormalities [29], i.e. MLKL inactivation is not toxic to the animals. Thus, we decided to test whether MLKL inactivation can potentially serve for CRC treatment. We found that NSA
noticeably reduced clonogenic survival of CRC cells LS180 [23], DLD1 [17] and SW480 [30] (Fig. 1B). We further transfected DLD1, one of these cell lines, with a CRISPR/Cas9 vector encoding
single guide (sg)RNAs targeting MLKL exons 2 or 9 and generated two cell clones in each of which MLKL gene was disrupted by one of these sgRNAs (Fig. 1C). We found that MLKL-deficient cells
are noticeably less clonogenic than the control, MLKL-expressing cells (Fig. 1D). Thus, MLKL inactivation reduces clonogenic survival of CRC cells. Given that MLKL inactivation reduced CRC
cell survival only partially, we searched for approaches enhancing CRC cell death caused by MLKL inactivation. To this end, we tested whether drugs targeting various CRC-promoting
mechanisms, e.g. vemurafenib (a RAF inhibitor) [31], selumetinib (a MEK inhibitor) [32], pictilisib (a PI3 kinase inhibitor) [33] or lapatinib [34] (an EGFR/ERBB2 inhibitor) [35], enhance
death of DLD1 cells caused by MLKL KO. However, none of the drugs enhanced such death (Fig. 1E). In contrast, a ribosomal inhibitor HHT [5], an alkaloid, also known as omacetaxine, used for
treatment of chronic myelogenous leukemia [4, 16], strongly cooperated with MLKL KO in reducing clonogenic survival and the total number of DLD1 cells (Fig. 1E, F). Moreover, HHT and MLKL
inhibitor NSA noticeably cooperated in inhibiting clonogenic survival of CRC cells DLD1, HCT116, LS180 and SW480 (Fig. 1G–J). Thus, MLKL inactivation cooperates with HHT in reducing CRC cell
survival. NSA AND HHT COOPERATE IN REDUCING CRC CELL TUMORIGENICITY IN VIVO We further found that NSA and HHT noticeably cooperated in reducing the ability of HCT116 cells, one of these CRC
cell lines used by us, to grow as subcutaneous tumors in immunodeficient mice (Fig. 2A). During the treatment, we monitored the mice for weight loss, a widely used surrogate marker of
systemic drug toxicity in mouse-based studies [36, 37]. Encouragingly, the mice did not lose weight in response to treatment with each drug alone or both together (Fig. 2B) and did not
display any gross physical abnormalities. Thus, NSA and HHT significantly cooperate in suppressing CRC cell tumorigenicity in vivo. MLKL DOES NOT PROTECT CRC CELLS FROM DEATH BY SECRETING
SOLUBLE FACTORS OR BY PROMOTING TRAIL RECEPTOR DEGRADATION MLKL was suggested to trigger pro-survival signals in certain scenarios, and we tested whether these scenarios apply to our model.
It was proposed that MLKL drives secretion of the pro-survival cytokines by breast tumor cells and that these cytokines induce cellular paracrine survival signals [38]. This notion is based
on the data showing that cell growth blocked by MLKL KO can be restored by the conditioned medium derived from the parental MLKL-expressing cells [38]. However, we found that MLKL-deficient
DLD1 cells cannot be rescued from HHT by the conditioned medium derived from the MLKL-producing cells (Supplementary Fig. 1A). Likewise, the conditioned medium derived from the untreated
HCT116 cells, another CRC cell line, did not rescue them from treatment with the combination of NSA and HHT (Supplementary Fig. 1B). MLKL inactivation was also shown to block lysosomal
degradation of the death receptor DR5 in lung and breast tumor cells and thereby enhance cell death upon treatment with TRAIL, the DR5 ligand [39]. Conceivably, DR5 upregulation in
MLKL-deficient CRC cells could sensitize the cells to the endogenously produced TRAIL [40, 41]. However, we found that MLKL loss upregulates neither DR5 nor DR4, another TRAIL receptor [2],
in CRC cells neither before nor after HHT treatment (Supplementary Fig. 2B, C). Thus, none of the indicated mechanisms mediate the MLKL effect on CRC cells. MLKL INACTIVATION AND HHT
COOPERATE IN KILLING CRC CELLS BY INHIBITING THEIR BASAL AUTOPHAGY Since autophagy is a well-known regulator of cell survival [2], we tested whether MLKL inactivation and HHT treatment
cooperate in blocking the basal autophagy in CRC cells. Lipidation of autophagy-driving protein LC3B is a major autophagy mechanism, and formation of LC3B-II, the lipidated LC3B form, is a
well-established autophagy marker [42]. LC3B-II is eventually degraded after autophagosome-to-lysosome fusion. Hence, while LC3B-II upregulation can signify increased autophagy, it can also
indicate autophagy inhibition caused by reduced fusion of the autophagosome to the lysosome [43]. To ensure that increased LC3B-II levels reflect increased autophagy in our studies, we
tested the effect of HHT and MLKL inactivation on LC3B-II in the absence and in the presence of bafilomycin A1, a lysosomal inhibitor that disrupts autophagosome-to-lysosome fusion and thus
blocks LC3B-II degradation [43]. We found that LC3BII is poorly detectable in bafilomycin A1-untreated cells, and bafilomycin A1 noticeably upregulated LC3BII in DLD1 cells and their MLKL KO
variants (Fig. 3A, B). Hence, the basal rate of autophagy in the cells is sufficiently high to trigger LC3BII lysosomal degradation and thereby render LC3BII poorly detectable, unless
autophagosome-to-lysosome fusion is blocked. We further observed that MLKL KO, NSA or HHT noticeably downregulated LC3BII in bafilomycin A1-treated MLKL-expressing DLD1 cells (Fig. 3A–C).
Remarkably, MLKL KO or NSA significantly cooperated with HHT in downregulating LC3BII (Fig. 3A–C). To confirm our findings by a complementary approach, we tested the ability of
green-fluorescent protein (GFP)-tagged LC3B protein (GFPLC3) to cause puncta formation in the control and MLKL KO CRC cells before and after bafilomycin A1 and HHT treatment. Such puncta are
widely used markers of autophagosome formation [43]. Similar to the LC3BII western blot-based data, GFPLC3 puncta were poorly detectable in bafilomycin A1-untreated cells transiently
transfected with a GFPLC3-encoding expression vector (not shown) but were readily detectable in bafilomycin A1-treated cells (Fig. 3D, E). We found that MLKL KO and HHT significantly
cooperated in reducing the number of the GFPLC3 puncta per cell (Fig. 3D, E). Thus, MLKL inactivation and HHT cooperate in reducing the basal CRC cell autophagy. To test whether autophagy
inhibition triggered by HHT in MLKL-deficient cells is the cause of CRC cell loss, we treated the MLKL KO cells with HHT and the cell permeable peptide composed of the HIV-1 TAT protein
transduction domain attached to either a scrambled amino acid sequence (control peptide) or the peptide derived from a protein Beclin-1, a major autophagy inducer that promotes autophagy by
activating class III PI3 kinase (PI3K) [44]. The TAT domain renders the peptide cell-permeable while the respective Beclin 1 fragment is necessary and sufficient for autophagy induction
[44]. The peptide is being widely used as an autophagy-promoting tool [44,45,46,47]. Similar to Beclin 1, the peptide increases Class III PI3K complexes kinase activity and induces autophagy
in a manner that requires the presence of autophagy drivers ATG 5 and ATG7 [47]. We found that the Beclin-1-derived peptide almost completely prevented MLKL KO from reducing the growth of
HHT-treated DLD1 cells (Fig. 3F). Thus, autophagy inhibition caused by MLKL inactivation combined with HHT is required for CRC cell death induced by this combination treatment. In light of
this, our findings that RAF, MEK, PI3K and EGFR inhibitors did not cooperate with MLKL in killing CRC cells (Fig. 1E) are not surprising since these agents promote, rather than inhibit,
autophagy in various cancer cell types, including CRC cells [48,49,50]. We further knocked down (KD) ATG12, a major autophagy driver [42], in DLD1 cells by two different siRNAs. Of note,
ATG12 forms a covalent complex with a protein ATG5 to promote LC3 lipidation, and ATG12 knockdown downregulated this complex (Fig. 4A). Furthermore, ATG5-ATG12 KD or treatment with
SBI0206965, a specific inhibitor of the critical autophagy driver protein kinase ULK1 [51], noticeably reduced LC3 lipidation in Bafilomycin A1-treated cells (Fig. 4B, D) and suppressed
growth of MLKL-expressing DLD1 cells (Fig. 4C, E). Remarkably, the more efficient ATG5-ATG12 KD was, the more efficiently LC3B lipidation and cell growth were inhibited (Fig. 4A–C). Thus,
inhibition of autophagy is sufficient for suppressing CRC cell growth. These data are consistent with observations that autophagy inhibition substantially reduces growth of colorectal cancer
patient-derived xenografts and multiple human colorectal cancer cell lines [52,53,54,55,56,57,58]. In summary, we conclude that MLKL inactivation and HHT cooperate in killing CRC cells by
inhibiting their basal autophagy. MLKL INACTIVATION RENDERS CRC CELL AUTOPHAGY VPS37A-DEPENDENT The ESCRT machinery includes the ESCRT-I, -II and III multiprotein complexes that promote the
formation of various vesicles, such as endosomes and autophagosomes [8]. During necroptosis, MLKL was found to activate ESCRT-I and III which repaired the MLKL-permeabilized cell membrane
and delayed cell death [59]. To examine the role of ESCRT-I in the effects observed by us, we studied LC3BII levels in bafilomycin A1-treated MLKL-expressing and MLKL KO DLD1 cells before
and after KD of the critical ESCRT-I complex component VPS37A by two different VPS37A-specific siRNAs (Fig. 5A–D). Notably, VPS37A KD did not have any effect on LC3B lipidation of the
control, MLKL-expressing DLD1 cells but strongly reduced LC3B lipidation in MLKL KO DLD1 cells (Fig. 5E, F). Hence, such lipidation becomes VPS37A-dependent in the absence of MLKL. Moreover,
VPS37A was unable to promote/support LC3B lipidation in MLKL-deficient HHT-treated cells (Fig. 5E, F). Thus, MLKL inactivation reduces CRC cell autophagy and renders this autophagy
VPS37A-dependent. However, VPS37A is unable to promote autophagy of HHT-treated cells. HHT CONTRIBUTES TO AUTOPHAGY INHIBITION IN CRC CELLS BY ACTIVATING P38MAPK HHT is a ribosome inhibitor,
and various ribosome inhibitors cause ribotoxic stress response, a chain of signals triggered by the ribosome-bound protein kinase ZAKα. ZAKα further phosphorylates and activates MAP
kinases MKK3 and/or MKK6 which in turn phosphorylate and activate p38MAPK and JNK [6]. Notably, p38MAPK inhibits autophagy via multiple mechanisms, including phosphorylation and inactivation
of the autophagy stimulators ULK1 [60] and ATG5 [61] as well as inactivation of the autophagy stimulator ATG9 [62]. We found that HHT upregulates phospho-p38MAPK, the sign of p38MAPK
activation [63, 64], in both the control and MLKL KO DLD1 cells (Fig. 6A). Four members of the p38MAPK family are known, α, β, γ and δ[64]. We found that treatment of the MLKL KO DLD1 cells
with SB203580, a specific small molecule p38MAPK α and β inhibitor [65], in the presence of Bafilomycin A1 rendered their LC3BII levels comparable to those observed in the untreated cells
(Fig. 6B, C). Hence, HHT-induced p38MAPK activation likely mediates HHT-induced autophagy inhibition in MLKL-deficient CRC cells. MLKL INACTIVATION AND HHT COOPERATE IN KILLING CRC CELLS BY
PARTHANATOS We examined which of the known cell death forms [2] is triggered by the combined inactivation of MLKL and HHT treatment. When using flow cytometry to analyze the distribution of
CRC cells between the phases of the cell cycle based on their DNA content, we noticed that MLKL KO and HHT cooperated in increasing the population of cells with the hypodiploid sub-G1 DNA
content (Fig. 7A–G). The presence of such population indicates chromosomal DNA fragmentation [66], a sign of regulated cell death types, e.g., apoptosis and parthanatos [2]. When we cultured
the MLKL KO DLD1 cells with HHT and the inhibitor of caspases, apoptosis-executing proteases, zVAD-FMK [67], this caused massive chromosomal DNA degradation regardless of MLKL presence (not
shown). Notably, caspase inhibition was found to trigger non-apoptotic cell death in multiple studies [68, 69]. Since the ability of HHT to kill cells strongly depends on the presence of
MLKL (Fig. 1), we concluded that HHT treatment and MLKL inactivation unlikely cooperate in causing CRC cell apoptosis. Parthanatos is programmed necrosis triggered by hyperactivation of
poly(ADP-ribose) polymerase 1 (PARP1), which drives the formation of poly(ADP-ribose) (PAR) [11]. When overproduced, PAR causes the release of the protein AIF from the mitochondria [11]. AIF
then binds the endonuclease MIF, and the AIF–MIF complex triggers chromosomal DNA degradation and cell death [11]. We found that HHT-induced formation of the sub-G1 DNA content in the MLKL
KO DLD1 cells is prevented by the PARP inhibitor olaparib [70] (Fig. 8A–E). Moreover, HHT caused a significant increase in the multiple species of PAR polymers in the cells, a parthanatos
symptom (Fig. 8F) [71]. Furthermore, the autophagy-inducing Beclin1-derived cell-permeable peptide [44] prevented HHT from upregulating PAR in the cells (Fig. 8F). Hence, MLKL inactivation
combined with HHT treatment cooperates in blocking CRC cell autophagy and this autophagy inhibition triggers parthanatos. LOW MLKL MRNA EXPRESSION SIGNIFIES INCREASED CRC PATIENT SURVIVAL To
test whether MLKL expression correlates with CRC patient survival we used GEPIA, an interactive web server for analyzing the RNA sequencing expression data in various tumors derived from
the TCGA and the GTEx projects [18]. We found that reduced levels of MLKL mRNA in CRC correlate with increased patient survival (Fig. 9). These data are consistent with observations
indicating that reduced levels of phosphorylated, i.e., activated, MLKL protein in CRC and esophageal tumors correlate with increased survival of the respective patients [38]. Collectively,
these results support the notion that the lower the levels of active MLKL expression in CRC cells are, the less viable the cells are, the less deadly respective cancers become, the longer
patients survive. In summary, we have shown here that pharmacological or genetic MLKL inactivation, when combined with HHT, blocks the basal CRC cell autophagy and triggers parthanatos of
CRC cells. Moreover, a pharmacological MLKL inhibitor and HHT cooperate in suppressing CRC cell tumorigenicity in vivo. Hence, MLKL inhibition combined with HHT treatment is a potential
novel approach for CRC therapy. DISCUSSION We found that MLKL inactivation when combined with a clinically approved anti-cancer drug HHT represents an effective approach for killing CRC
cells in vitro and suppressing their tumorigenicity in vivo. Hence, combining MLKL inactivation with HHT treatment is a potential novel approach to CRC management. MLKL drives cell death
during necroptosis [2]. We found that MLKL also promotes CRC cell survival by supporting the basal CRC cell autophagy. We discovered that MLKL inactivation and HHT treatment cooperate in
blocking such autophagy and killing CRC cells. Our data indicate that MLKL mediates LC3B lipidation, a critical autophagy step, in CRC cells. The ESCRT-I complex is known to promote
autophagy [10] but apparently, the ESCRT-I component VPS37A is not needed for driving LC3 lipidation in MLKL-expressing CRC cells. However, in MLKL-deficient cells, LC3B lipidation is
strongly reduced and becomes almost entirely VPS37A-dependent. Notably, so far VPS37A was found to drive autophagy by supporting the autophagosomal membrane integrity and/or promoting the
closure of this membrane [9, 10]. To our knowledge, the role of the ESCRT-I machinery in LC3 lipidation has never been demonstrated, which is not surprising since this role becomes apparent
only in the absence of MLKL, a scenario that has not been tested so far. Understanding how VPS37A controls such lipidation and how this process is regulated by MLKL is the subject of our
ongoing studies. We have shown here that the ability of MLKL antagonists to suppress CRC cell autophagy is enhanced by HHT and that this effect is driven by HHT-induced p38MAPK activation.
HHT is a ribosome inhibitor [5], and such inhibitors often activate p38MAPK [6]. Our data are consistent with these findings and with observations that p38MAPK inhibits autophagy by multiple
mechanisms [60,61,62]. Since both NSA and HHT inhibit autophagy, would it be possible to use either drug alone at a higher dose to suppress autophagy and thereby suppress CRC in the clinic?
We found that even complete MLKL loss has only a limited effect on CRC cell autophagy and survival. Moreover, while HHT is effective against CML [4], the use of HHT as a single agent at
non-toxic doses was ineffective against CRC in the clinic [72], and a higher HHT dose was unacceptably toxic [73]. Thus, the use of MLKL antagonists could render the non-toxic HHT doses
sufficient for reducing the basal autophagy in CRC cells below the levels required for cell survival and thereby make this combination treatment efficacious against CRC. We found that
autophagy inhibition caused by MLKL inactivation and HHT treatment kills CRC cells by parthanatos. Oxidative stress and excessive DNA damage are the known consequences of autophagy
inhibition in cancer cells [74, 75] and represent the established stimuli of parthanatos [11]. Testing whether these stimuli trigger parthanatos in CRC cells in response to MLKL inactivation
and HHT treatment represents a promising direction for our future studies. We found that an MLKL inhibitor NSA [3] strongly cooperates with HHT in suppressing growth of tumors formed by CRC
cells in mice. Thus, pharmacological MLKL inhibition combined with HHT treatment is a potential novel approach for CRC management. Importantly, PARP1, a major parthanatos inducer, is
overexpressed in human tumor-derived colon cancer cells compared to normal intestinal epithelium and promotes colon cancer in mice [76]. Conceivably, high PARP1 level in CRC cells could make
them vulnerable to therapies based on MLKL inhibition combined with the use of HHT, and thus create a therapeutic window for CRC treatment. Therefore, PARP1 could be explored as a biomarker
of CRC sensitivity to this combination treatment. DATA AVAILABILITY We will share all data reported in this study upon request. REFERENCES * Biller LH, Schrag D. Diagnosis and Treatment of
Metastatic Colorectal Cancer: A Review. JAMA. 2021;325:669–85. Article CAS PubMed Google Scholar * Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular
mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. Article PubMed PubMed Central Google Scholar * Sun L, Wang
H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27. * Winer ES, DeAngelo DJ. A
Review of Omacetaxine: A Chronic Myeloid Leukemia Treatment Resurrected. Oncol Ther. 2018;6:9–20. Article PubMed PubMed Central Google Scholar * Gurel G, Blaha G, Moore PB, Steitz TA.
U2504 determines the species specificity of the A-site cleft antibiotics: the structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J Mol Biol. 2009;389:146–56.
Article CAS PubMed PubMed Central Google Scholar * Vind AC, Genzor AV, Bekker-Jensen S. Ribosomal stress-surveillance: three pathways is a magic number. Nucleic Acids Res.
2020;48:10648–61. Article CAS PubMed PubMed Central Google Scholar * Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and
restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. Article CAS PubMed PubMed Central Google Scholar * Christ L, Raiborg C, Wenzel EM, Campsteijn C,
Stenmark H. Cellular Functions and Molecular Mechanisms of the ESCRT Membrane-Scission Machinery. Trends Biochem Sci. 2017;42:42–56. Article CAS PubMed Google Scholar * Javed R, Jain A,
Duque T, Hendrix E, Paddar MA, Khan S, et al. Mammalian ATG8 proteins maintain autophagosomal membrane integrity through ESCRTs. EMBO J. 2023;42:e112845. Article CAS PubMed PubMed Central
Google Scholar * Takahashi Y, Liang X, Hattori T, Tang Z, He H, Chen H, et al. VPS37A directs ESCRT recruitment for phagophore closure. J Cell Biol. 2019;218:3336–54. Article CAS PubMed
PubMed Central Google Scholar * David KK, Andrabi SA, Dawson TM, Dawson VL. Parthanatos, a messenger of death. Front Biosci. 2009;14:1116–28. Article CAS Google Scholar * Liu Z, Li H,
Derouet M, Filmus J, LaCasse EC, Korneluk RG, et al. ras Oncogene triggers up-regulation of cIAP2 and XIAP in intestinal epithelial cells: epidermal growth factor receptor-dependent and
-independent mechanisms of ras-induced transformation. J Biol Chem. 2005;280:37383–92. Article CAS PubMed Google Scholar * Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H,
Valen E. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019;47:W171–W174. Article CAS PubMed PubMed Central Google Scholar * Ran FA, Hsu PD,
Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308. Article CAS PubMed PubMed Central Google Scholar * Liu Z, Li H,
Derouet M, Berezkin A, Sasazuki T, Shirasawa S, et al. Oncogenic Ras inhibits anoikis of intestinal epithelial cells by preventing the release of a mitochondrial pro-apoptotic protein
Omi/HtrA2 into the cytoplasm. J Biol Chem. 2006;281:14738–47. Article CAS PubMed Google Scholar * Suo S, Zhao D, Li F, Zhang Y, Rodriguez-Rodriguez S, Nguyen LXT, et al.
Homoharringtonine inhibits the NOTCH/MYC pathway and exhibits antitumor effects in T-cell acute lymphoblastic leukemia. Blood. 2024;144:1343–7. Article CAS PubMed PubMed Central Google
Scholar * Rosen K, Rak J, Leung T, Dean NM, Kerbel RS, Filmus J. Activated Ras prevents downregulation of Bcl-X(L) triggered by detachment from the extracellular matrix. A mechanism of
Ras-induced resistance to anoikis in intestinal epithelial cells. J Cell Biol. 2000;149:447–56. Article CAS PubMed PubMed Central Google Scholar * Tang Z, Li C, Kang B, Gao G, Li C,
Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. Article CAS PubMed PubMed Central Google
Scholar * Khan IA, Yoo BH, McPhee M, Masson O, Surette A, Dakin-Hache K, et al. ErbB2-driven downregulation of the transcription factor Irf6 in breast epithelial cells is required for their
3D growth. Breast Cancer Res. 2018;20:151. Article CAS PubMed PubMed Central Google Scholar * Chipurupalli S, Jiang P, Liu X, Santos JL, Marcato P, Rosen KV. Three-dimensional growth
sensitizes breast cancer cells to treatment with ferroptosis-promoting drugs. Cell Death Dis. 2023;14:580. Article CAS PubMed PubMed Central Google Scholar * Shaw P, Tardy S, Benito E,
Obrador A, Costa J. Occurrence of Ki-ras and p53 mutations in primary colorectal tumors. Oncogene. 1991;6:2121–8. CAS PubMed Google Scholar * Sansom OJ, Meniel V, Wilkins JA, Cole AM,
Oien KA, Marsh V, et al. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc Natl Acad Sci USA. 2006;103:14122–7. Article
CAS PubMed PubMed Central Google Scholar * Yoo BH, Khan IA, Koomson A, Gowda P, Sasazuki T, Shirasawa S, et al. Oncogenic RAS-induced downregulation of ATG12 is required for survival
of malignant intestinal epithelial cells. Autophagy. 2018;14:134–51. Article CAS PubMed Google Scholar * Tait SW, Ichim G, Green DR. Die another way-non-apoptotic mechanisms of cell
death. J Cell Sci. 2014;127:2135–44. Article CAS PubMed PubMed Central Google Scholar * Najafov A, Mookhtiar AK, Luu HS, Ordureau A, Pan H, Amin PP, et al. TAM Kinases Promote
Necroptosis by Regulating Oligomerization of MLKL. Mol Cell. 2019;75:457–68.e454. Article CAS PubMed Google Scholar * Derouet M, Wu X, May L, Yoo BH, Sasazuki T, Shirasawa S, et al.
Acquisition of anoikis resistance promotes the emergence of oncogenic K-ras mutations in colorectal cancer cells and stimulates their tumorigenicity in vivo. Neoplasia. 2007;9:536–45.
Article CAS PubMed PubMed Central Google Scholar * Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, et al. Discovery of a novel ERK inhibitor with activity in models of
acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3:742–50. Article CAS PubMed Google Scholar * Tait SW, Parsons MJ, Llambi F, Bouchier-Hayes L, Connell S, Munoz-Pinedo
C, et al. Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev Cell. 2010;18:802–13. Article CAS PubMed PubMed Central Google Scholar * Wu J,
Huang Z, Ren J, Zhang Z, He P, Li Y, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23:994–1006. Article CAS PubMed PubMed Central
Google Scholar * Dunn EF, Iida M, Myers RA, Campbell DA, Hintz KA, Armstrong EA, et al. Dasatinib sensitizes KRAS mutant colorectal tumors to cetuximab. Oncogene. 2011;30:561–74. Article
CAS PubMed Google Scholar * Joseph EW, Pratilas CA, Poulikakos PI, Tadi M, Wang W, Taylor BS, et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a
V600E BRAF-selective manner. Proc Natl Acad Sci USA. 2010;107:14903–8. Article CAS PubMed PubMed Central Google Scholar * Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et
al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–83. Article CAS
PubMed Google Scholar * Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is
effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009;15:429–40. Article CAS PubMed Google Scholar * Xia W, Mullin RJ, Keith BR, Liu LH, Ma H, Rusnak DW, et al.
Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene. 2002;21:6255–63. Article CAS PubMed
Google Scholar * de Azambuja E, Holmes AP, Piccart-Gebhart M, Holmes E, Di Cosimo S, Swaby RF, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): survival
outcomes of a randomised, open-label, multicentre, phase 3 trial and their association with pathological complete response. Lancet Oncol. 2014;15:1137–46. Article PubMed Google Scholar *
Francia G, Man S, Lee CJ, Lee CR, Xu P, Mossoba ME, et al. Comparative impact of trastuzumab and cyclophosphamide on HER-2-positive human breast cancer xenografts. Clin Cancer Res.
2009;15:6358–66. Article CAS PubMed PubMed Central Google Scholar * Upadhyayula PS, Higgins DM, Mela A, Banu M, Dovas A, Zandkarimi F, et al. Dietary restriction of cysteine and
methionine sensitizes gliomas to ferroptosis and induces alterations in energetic metabolism. Nat Commun. 2023;14:1187. Article CAS PubMed PubMed Central Google Scholar * Liu X, Zhou M,
Mei L, Ruan J, Hu Q, Peng J, et al. Key roles of necroptotic factors in promoting tumor growth. Oncotarget. 2016;7:22219–33. Article PubMed PubMed Central Google Scholar * Park SY, Park
HH, Park SY, Hong SM, Yoon S, Morgan MJ, et al. Reduction in MLKL-mediated endosomal trafficking enhances the TRAIL-DR4/5 signal to increase cancer cell death. Cell Death Dis. 2020;11:744.
Article CAS PubMed PubMed Central Google Scholar * Allen JE, Krigsfeld G, Mayes PA, Patel L, Dicker DT, Patel AS, et al. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear
translocation, TRAIL gene induction, and potent antitumor effects. Sci Transl Med. 2013;5:171ra117. Article Google Scholar * von Karstedt S, Conti A, Nobis M, Montinaro A, Hartwig T,
Lemke J, et al. Cancer cell-autonomous TRAIL-R signaling promotes KRAS-driven cancer progression, invasion, and metastasis. Cancer Cell. 2015;27:561–73. Article Google Scholar * Feng Y, He
D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014;24:24–41. Article CAS PubMed Google Scholar * Yoo BH, Zagryazhskaya A, Li Y, Koomson A, Khan IA, Sasazuki T, et
al. Upregulation of ATG3 contributes to autophagy induced by the detachment of intestinal epithelial cells from the extracellular matrix, but promotes autophagy-independent apoptosis of the
attached cells. Autophagy. 2015;11:1230–46. Article CAS PubMed PubMed Central Google Scholar * Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, et al. Identification of
a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494:201–6. Article CAS PubMed PubMed Central Google Scholar * Vega-Rubin-de-Celis S, Zou Z, Fernandez AF, Ci B, Kim M,
Xiao G, et al. Increased autophagy blocks HER2-mediated breast tumorigenesis. Proc Natl Acad Sci USA. 2018;115:4176–81. Article CAS PubMed PubMed Central Google Scholar * Sharif T,
Martell E, Dai C, Ghassemi-Rad MS, Hanes MR, Murphy PJ, et al. HDAC6 differentially regulates autophagy in stem-like versus differentiated cancer cells. Autophagy. 2019;15:686–706. Article
CAS PubMed Google Scholar * He Y, Lu H, Zhao Y. Development of an autophagy activator from Class III PI3K complexes, Tat-BECN1 peptide: Mechanisms and applications. Front Cell Dev Biol.
2022;10:851166. Article PubMed PubMed Central Google Scholar * Sueda T, Sakai D, Kawamoto K, Konno M, Nishida N, Koseki J, et al. BRAF V600E inhibition stimulates AMP-activated protein
kinase-mediated autophagy in colorectal cancer cells. Sci Rep. 2016;6:18949. Article CAS PubMed PubMed Central Google Scholar * Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth
M, Truong A, et al. Protective autophagy elicited by RAF->MEK->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med. 2019;25:620–7. Article CAS PubMed PubMed
Central Google Scholar * Yang W, Hosford SR, Traphagen NA, Shee K, Demidenko E, Liu S, et al. Autophagy promotes escape from phosphatidylinositol 3-kinase inhibition in estrogen
receptor-positive breast cancer. FASEB J. 2018;32:1222–35. Article CAS PubMed Google Scholar * Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, et al. Small Molecule Inhibition of
the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell. 2015;59:285–97. Article CAS PubMed PubMed Central Google Scholar * Rehman SK, Haynes J, Collignon E, Brown KR,
Wang Y, Nixon AML, et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell. 2021;184:226–42.e221. Article CAS PubMed Google Scholar * Takeda M,
Koseki J, Takahashi H, Miyoshi N, Nishida N, Nishimura J, et al. Disruption of Endolysosomal RAB5/7 Efficiently Eliminates Colorectal Cancer Stem Cells. Cancer Res. 2019;79:1426–37. Article
CAS PubMed Google Scholar * Sakitani K, Hirata Y, Hikiba Y, Hayakawa Y, Ihara S, Suzuki H, et al. Inhibition of autophagy exerts anti-colon cancer effects via apoptosis induced by p53
activation and ER stress. BMC Cancer. 2015;15:795. Article PubMed PubMed Central Google Scholar * Scherr AL, Jassowicz A, Pato A, Elssner C, Ismail L, Schmitt N, et al. Knockdown of Atg7
Induces Nuclear-LC3 Dependent Apoptosis and Augments Chemotherapy in Colorectal Cancer Cells. Int J Mol Sci. 2020;21:1099. * Devenport SN, Singhal R, Radyk MD, Taranto JG, Kerk SA, Chen B,
et al. Colorectal cancer cells utilize autophagy to maintain mitochondrial metabolism for cell proliferation under nutrient stress. JCI Insight. 2021;6:e138835. * Mazouffre C, Geyl S,
Perraud A, Blondy S, Jauberteau MO, Mathonnet M, et al. Dual inhibition of BDNF/TrkB and autophagy: a promising therapeutic approach for colorectal cancer. J Cell Mol Med. 2017;21:2610–22.
Article CAS PubMed PubMed Central Google Scholar * Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative
metabolism and tumorigenesis. Genes Dev. 2011;25:460–70. Article CAS PubMed PubMed Central Google Scholar * Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, et al. ESCRT-III
Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell. 2017;169:286–300.e216. Article CAS PubMed PubMed Central Google Scholar * He Y, She H, Zhang T, Xu
H, Cheng L, Yepes M, et al. p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J Cell Biol. 2018;217:315–28. Article CAS PubMed PubMed
Central Google Scholar * Keil E, Hocker R, Schuster M, Essmann F, Ueffing N, Hoffman B, et al. Phosphorylation of Atg5 by the Gadd45beta-MEKK4-p38 pathway inhibits autophagy. Cell Death
Differ. 2013;20:321–32. Article CAS PubMed Google Scholar * Webber JL, Tooze SA. Coordinated regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP. EMBO J. 2010;29:27–40.
Article CAS PubMed Google Scholar * Rosen K, Shi W, Calabretta B, Filmus J. Cell detachment triggers p38 mitogen-activated protein kinase-dependent overexpression of Fas ligand. A novel
mechanism of Anoikis of intestinal epithelial cells. J Biol Chem. 2002;277:46123–30. Article CAS PubMed Google Scholar * Cuenda A, Rousseau S. p38 MAP-kinases pathway regulation,
function and role in human diseases. Biochim Biophys Acta. 2007;1773:1358–75. Article CAS PubMed Google Scholar * Sicard P, Clark JE, Jacquet S, Mohammadi S, Arthur JS, O’Keefe SJ, et
al. The activation of p38 alpha, and not p38 beta, mitogen-activated protein kinase is required for ischemic preconditioning. J Mol Cell Cardiol. 2010;48:1324–8. Article CAS PubMed PubMed
Central Google Scholar * Darzynkiewicz Z, Juan G, Li X, Gorczyca W, Murakami T, Traganos F. Cytometry in cell necrobiology: analysis of apoptosis and accidental cell death (necrosis).
Cytometry. 1997;27:1–20. Article CAS PubMed Google Scholar * Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA. Inhibition of human caspases by peptide-based
and macromolecular inhibitors. J Biol Chem. 1998;273:32608–13. Article CAS PubMed Google Scholar * Christofferson DE, Li Y, Hitomi J, Zhou W, Upperman C, Zhu H, et al. A novel role for
RIP1 kinase in mediating TNFalpha production. Cell Death Dis. 2012;3:e320. Article CAS PubMed PubMed Central Google Scholar * Koike A, Hanatani M, Fujimori K. Pan-caspase inhibitors
induce necroptosis via ROS-mediated activation of mixed lineage kinase domain-like protein and p38 in classically activated macrophages. Exp Cell Res. 2019;380:171–9. Article CAS PubMed
Google Scholar * Penning TD. Small-molecule PARP modulators-current status and future therapeutic potential. Curr Opin Drug Discov Devel. 2010;13:577–86. CAS PubMed Google Scholar *
Andrabi SA, Umanah GK, Chang C, Stevens DA, Karuppagounder SS, Gagne JP, et al. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad
Sci USA. 2014;111:10209–14. Article CAS PubMed PubMed Central Google Scholar * Neidhart JA, Young DC, Kraut E, Howinstein B, Metz EN. Phase I trial of homoharringtonine administered by
prolonged continuous infusion. Cancer Res. 1986;46:967–9. CAS PubMed Google Scholar * Witte RS, Lipsitz S, Goodman TL, Asbury RF, Wilding G, Strnad CM, et al. A phase II trial of
homoharringtonine and caracemide in the treatment of patients with advanced large bowel cancer. Invest New Drugs. 1999;17:173–7. Article CAS PubMed Google Scholar * Mathew R, Karp CM,
Beaudoin B, Vuong N, Chen GH, Chen HY, et al. Autophagy Suppresses Tumorigenesis through Elimination of p62. Cell. 2009;137:1062–75. Article CAS PubMed PubMed Central Google Scholar *
Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–81. Article CAS
PubMed PubMed Central Google Scholar * Dorsam B, Seiwert N, Foersch S, Stroh S, Nagel G, Begaliew D, et al. PARP-1 protects against colorectal tumor induction, but promotes
inflammation-driven colorectal tumor progression. Proc Natl Acad Sci USA. 2018;115:E4061–E4070. Article PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS 90% of
this study was supported by a grant from The Cancer Research Society and 10%, by a grant from Canadian Institutes of Health Research (KVR is a principal investigator on both grants). XL and
SC were recipients of Cancer Research Training Program postdoctoral fellowships via Beatrice Hunter Cancer Research Institute. SC and PJ were recipients of the IWK Health Sciences Centre
postdoctoral fellowships. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Departments of Pediatrics & Biochemistry and Molecular Biology, Dalhousie University, Halifax, NS, Canada Peijia
Jiang, Sandhya Chipurupalli, Byong Hoon Yoo, Xiaoyang Liu & Kirill V. Rosen Authors * Peijia Jiang View author publications You can also search for this author inPubMed Google Scholar *
Sandhya Chipurupalli View author publications You can also search for this author inPubMed Google Scholar * Byong Hoon Yoo View author publications You can also search for this author
inPubMed Google Scholar * Xiaoyang Liu View author publications You can also search for this author inPubMed Google Scholar * Kirill V. Rosen View author publications You can also search for
this author inPubMed Google Scholar CONTRIBUTIONS PJ, BHY, SC and XL performed the experiments. KVR conceived and supervised the study as well as wrote the initial manuscript draft.
CORRESPONDING AUTHOR Correspondence to Kirill V. Rosen. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ETHICS APPROVAL AND CONSENT TO PARTICIPATE Animal
studies were approved by Dalhousie University Committee on Laboratory Animals, protocol 20-081. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to
jurisdictional claims in published maps and institutional affiliations. Edited by Boris Zhivotovsky SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURES ORIGINAL WESTERN BLOTS RIGHTS AND
PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any
medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Jiang, P.,
Chipurupalli, S., Yoo, B.H. _et al._ Inactivation of necroptosis-promoting protein MLKL creates a therapeutic vulnerability in colorectal cancer cells. _Cell Death Dis_ 16, 118 (2025).
https://doi.org/10.1038/s41419-025-07436-z Download citation * Received: 26 September 2024 * Revised: 27 January 2025 * Accepted: 06 February 2025 * Published: 20 February 2025 * DOI:
https://doi.org/10.1038/s41419-025-07436-z SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative