- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Clear cell renal cell carcinoma (ccRCC) is the most lethal subtype of renal cancer, and its treatment options remain limited. Therefore, there is an urgent need to discover
therapeutic agents for ccRCC treatment. Here, we demonstrate that dimethyl fumarate (DMF), an approved medication for multiple sclerosis [1] and psoriasis, can inhibit the proliferation of
ccRCC cells. Mechanistically, hepatocyte nuclear factor 1β (HNF1B), a transcription factor highly expressed in ccRCC, is succinated by DMF at cysteine residues, leading to its proteasomal
degradation. Furthermore, HNF1B interacts with and stabilizes Yes-associated protein (YAP), thus DMF-mediated HNF1B degradation decreases YAP protein level and the expression of its target
genes, resulting in the suppression of ccRCC cell proliferation. Importantly, oral administration of DMF sensitizes ccRCC to sunitinib treatment and enhances its efficacy in mice. In
summary, we provide evidences supporting DMF as a potential drug for clinical treatment of ccRCC by targeting HNF1B and reveal a previously unrecognized role of HNF1B in regulating YAP in
ccRCC. SIMILAR CONTENT BEING VIEWED BY OTHERS DIMETHYL FUMARATE ABROGATES HEPATOCELLULAR CARCINOMA GROWTH BY INHIBITING NRF2/BCL-XL AXIS AND ENHANCES SORAFENIB’S EFFICACY Article Open access
14 May 2025 ISCA2 INHIBITION DECREASES HIF AND INDUCES FERROPTOSIS IN CLEAR CELL RENAL CARCINOMA Article Open access 12 September 2022 HMGCS2 SERVES AS A POTENTIAL BIOMARKER FOR INHIBITION
OF RENAL CLEAR CELL CARCINOMA GROWTH Article Open access 05 September 2023 INTRODUCTION Renal cell carcinoma (RCC) which arises from renal tubular epithelial cells, is one of the malignant
tumors of the urinary system. According to its pathological features, RCC primarily consists of three subtypes: chromophobe renal cell carcinoma (chRCC), clear cell renal cell carcinoma
(ccRCC) and papillary renal cell carcinoma (pRCC). Among these, ccRCC is the most prevalent subtype and is characterized by the presence of abundant cytoplasmic glycogen and lipids [2, 3].
ccRCC accounts for ~75–80% of all RCC cases, and has a poorer prognosis compared to other subtypes [4]. Over the past few decades, various therapeutic strategies have been employed for ccRCC
treatment, including surgical management, chemotherapy and radiotherapy [5]. Among these treatments, surgery remains the primary treatment option for ccRCC due to its limited response to
chemotherapy and radiotherapy. Since early-stage ccRCC often presents with few noticeable clinical symptoms, and in several cases, metastasis is already present at the time of diagnosis,
which contributes to a poor prognosis with the 5-year survival rate is only 10% [6,7,8]. Therefore, it is crucial to develop new strategies for ccRCC treatment. HNF1B, a 65-kDa protein, is a
member of the superfamily of transcription factors that includes homeodomain proteins such as Pit-1, Oct-1/2, and POU. HNF1B consists of three functional regions: an amino-terminal domain,
a DNA-binding domain and a carboxy-terminal domain. The gene encoding HNF1B is located on the long arm of chromosome 17 [9,10,11]. During the early embryonic development, the expression of
HNF1B is expressed in several organs, including liver, kidney, pancreas, lung and urinary tract. Heterozygous mutations in _HNF1B_ are associated with several congenital diseases including
renal cysts, pancreatic hypoplasia, abnormal liver function tests, and urogenital tract abnormalities, suggesting that HNF1B is crucial for the normal development of these organs [1, 12].
Although research on HNF1B has increased in recent years, its precise role in carcinogenesis remains insufficiently understood. Interestingly, HNF1B functions as a protooncogene in some
tumors, while it acts as a tumor suppressor in others, and whether it behaves as an oncogene or tumor suppressor appears to depend on the type and histogenesis of the tumor [13, 14].
Dimethyl fumarate (DMF) is a small molecule compound, a dimethyl ester of fumaric acid, which was first approved by the U.S. Food and Drug Administration in 2013 under the brand name
Tecfidera [15]. It is registered as an anti-inflammatory drug for the treatment of autoimmune diseases, such as multiple sclerosis and psoriasis [16, 17]. Several studies have demonstrated
that the molecular mechanisms underlying these effects can be mainly attributed to two aspects. First, DMF disrupts the interaction between Kelch-like ECH-associated protein 1 (KEAP1) and
erythroid 2–related factor 2 (Nrf2), leading to activation of Nrf2 and subsequent expression of antioxidant genes, which protects cells from reactive oxygen species that is generated during
inflammation [16, 18, 19]. Second, DMF inhibits NF-κB signaling pathway by suppressing the translocation of NF-κB family members into the nucleus, resulting in a significant decrease in the
production of proinflammatory cytokines [16, 17, 20, 21]. However, the function of DMF in ccRCC and its underlying mechanisms remain unknown. The present study uncovers a previously unknown
role of DMF in ccRCC, embodying the concept of “old drugs, new uses”, which offers a more time- and cost-efficient alternative compared with the new drug discovery [22, 23]. We demonstrate
that DMF targets HNF1B for ubiquitin-dependent proteasomal degradation and effectively suppresses the proliferation of ccRCC cells by impairing HNF1B mediated stabilization of Yes-associated
protein (YAP). Notably, oral administration of DMF decreases HNF1B levels in mice and enhances the sensitivity of ccRCC to sunitinib treatment. Overall, our study unveils a new regulatory
axis involving DMF, HNF1B and YAP, and demonstrates that DMF is a potential candidate for clinical treatment of ccRCC. RESULTS HNF1B FACILITATES CELL PROLIFERATION IN CCRCC To investigate
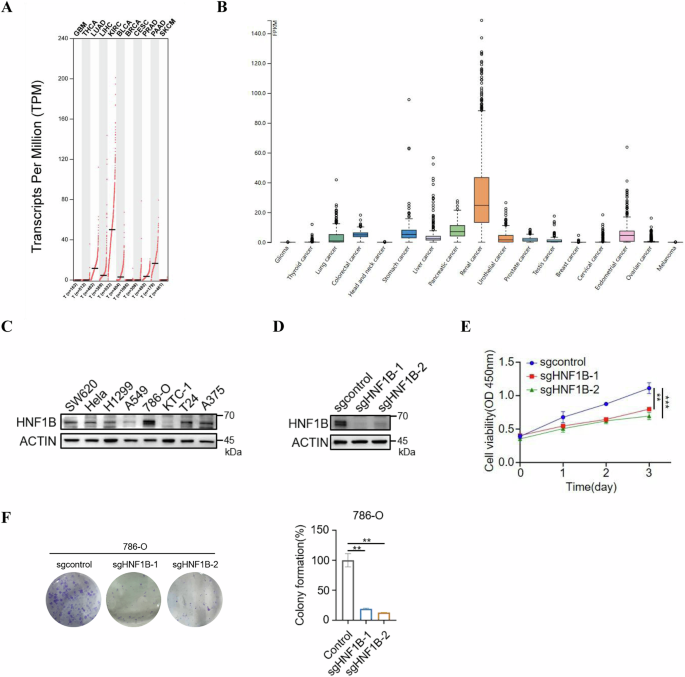
the role of HNF1B in tumorigenesis, we utilized the GEPIA and HPA databases to determine the expression level of HNF1B in various types of tumors. Notably, HNF1B shows the highest expression
in ccRCC compared to other cancers across both databases (Fig. 1A, B). Consistently, the protein level of HNF1B is also highest in 786-O ccRCC cell line compared to other cancer cell lines,
such as SW620 (colorectal cancer), HeLa (cervical carcinoma), KTC-1 (thyroid cancer), T24 (bladder cancer), A375 (melanoma), H1299 and A549 (non-small cell lung cancer) (Fig. 1C). A
previous study reported that HNF1B functions as a tumor suppressor and inhibits cell proliferation in prostate cancer [24]. However, another study suggested that HNF1B may act as an
oncogene, even though the underlying mechanism remains unclear [25]. Given these findings, we wanted to explore what role did HNF1B play in the cell proliferation of ccRCC. HNF1B knockout
cells were generated using the CRISPR/Cas9 system (Fig. 1D), and we found that HNF1B knockout significantly suppressed both proliferation and colony formation of ccRCC cells (Fig. 1E, F).
Together, these results indicate that HNF1B positively regulates cell proliferation in ccRCC. DMF SUPPRESSES CELL PROLIFERATION AND DECREASES HNF1B PROTEIN LEVEL IN CCRCC Previous studies
reported that DMF inhibits breast cancer and melanoma, which prompted us to investigate if DMF has the same impact on ccRCC. Indeed, we observed that DMF significantly inhibited the
proliferation and colony formation of 786-O and RCC4 cells in a dose-dependent manner (Fig. 2A, B). Given the high expression of HNF1B in ccRCC and HNF1B knock out negatively regulates cell
proliferation of ccRCC, we next explored whether the impact of DMF on cell proliferation was related to HNF1B. We generated ccRCC cells stably expressing Flag-HNF1B (Fig. 2C), treated these
cells with DMF, and examined the effect of DMF on exogenous Flag-HNF1B. The results showed that exogenous Flag-HNF1B protein level was significantly reduced upon DMF treatment (Fig. 2D).
Furthermore, we assessed the expression of endogenous HNF1B after DMF treatment and it showed that DMF also decreased endogenous HNF1B protein level in both 786-O and RCC4 cells (Fig. 2E).
To further confirm these results, we treated these two cell lines with DMF for varying durations and found that DMF reduced HNF1B levels in a time-dependent manner (Fig. 2F). Similar effects
were observed when cells were treated with increasing concentrations of DMF, and the expression of HNF1B decreased in a dose-dependent manner (Fig. 2G). To further validate the effects of
DMF on HNF1B, we examined the expression levels of HNF1B target genes under DMF treatment. The results revealed that DMF significantly downregulated the mRNA levels of _CRB3_, _KIF12_ and
_PKHD1_ in 786-O and RCC4 cells (Fig. 2H, I), further confirming the role of DMF on the downregulation of HNF1B. Together, these data indicate that DMF treatment decreases HNF1B protein
levels in ccRCC. DMF DOWNREGULATES HNF1B VIA UBIQUITIN-PROTEASOME PATHWAY To determine how DMF decreases the expression level of HNF1B, we examined the effects of DMF treatment on the mRNA
level of _HNF1B_ in ccRCC cells, and there was no alteration in _HNF1B_ mRNA level upon DMF treatment (Fig. 3A). Furthermore, DMF has no effect on the mRNA stability of HNF1B (Fig. 3B),
indicating that DMF did not regulate HNF1B expression through transcription. Interestingly, in the presence of cycloheximide, a protein synthesis inhibitor, the half-life of HNF1B under DMF
treatment was shorter than that of the control group in both 786-O and RCC4 cells (Fig. 3C), suggesting that DMF may reduce the protein stability of HNF1B. To investigate the mechanism how
HNF1B was degraded, we treated cells with NH4Cl, a lysosome pathway inhibitor and MG132, a proteasome pathway inhibitor, respectively, to determine which inhibitor could block HNF1B
degradation in the presence of DMF. The results showed that only MG132 could rescue DMF-induced degradation of HNF1B, suggesting that DMF reduces HNF1B protein level via the
ubiquitin-proteasome pathway (Fig. 3D, E). To confirm this hypothesis, we examined the ubiquitination level of HNF1B in HEK-293T, 786-O and RCC4 cells, and found that both exogenous and
endogenous HNF1B ubiquitination levels significantly increased upon DMF treatment (Fig. 3F, G). We next determined whether MLN4924, an inhibitor of cullin-RING E3 ligases, plays a role in
DMF-induced HNF1B degradation and found that it did not rescue the expression levels of HNF1B in the presence of DMF (Fig. 3H). We then explored the potential deubiquitinases (DUBs) that may
contribute to the DMF induced reduction of HNF1B. Analysis of the BioGRID database revealed three DUBs—BAP1, USP54 and OTUD3—that show potential interactions with HNF1B (Fig. 3I). We then
determined the interactions of Flag-HNF1B with these three DUBs via coimmunoprecipitation (co-IP) in HEK-293T cells. The results confirmed the interactions between Flag-HNF1B and these three
DUBs with different degrees (Fig. 3J). Moreover, overexpression of HA-OTUD3 increased the protein levels of Flag-HNF1B, while HA-BAP1 and HA-USP54 had no obvious effects (Fig. 3K),
indicating that HA-OTUD3 may play a crucial role in HNF1B stability regulation upon DMF treatment. To confirm this hypothesis, we performed further co-IP assays and found that DMF treatment
downregulated the interaction between Flag-HNF1B and HA-OTUD3 (Fig. 3L). Collectively, these data suggest that OTUD3 binds to and stabilizes HNF1B, and that DMF treatment impairs this
interaction, thereby promoting the degradation of HNF1B via ubiquitin-proteasome pathway. DMF-INDUCED HNF1B SUCCINATION ENHANCES ITS UBIQUITINATION As a thiol-reactive electrophile, DMF can
induce covalent modifications of thiols and directly reacts with cysteine residues of different kinds of proteins, including KEAP1, NF-κB, LYN and GSDMD, leading to the inactivation of these
proteins [21, 26,27,28]. We hypothesized that DMF may also affect HNF1B by modification of its cysteine residues. Indeed, HNF1B was identified as a target of succination following DMF
treatment (Fig. 4A). To investigate this further, we pretreated cells with N-acetyl-L-cysteine (NAC), a cell-permeable thiol that mimics the role of cysteines, prior to DMF treatment [29,
30]. Interestingly, NAC suppressed DMF induced decrease of HNF1B protein levels in both 786-O and RCC4 cells, suggesting that NAC may protect the cysteines of HNF1B from modification by DMF,
thereby preventing subsequent ubiquitination and degradation of HNF1B (Fig. 4B). To determine which cysteine residues of HNF1B are succinated by DMF and contribute to its degradation, we
generated cysteine mutants of HNF1B and assessed the effects of DMF on these mutants. Surprisingly, the protein levels of wild-type (WT) and C223S, C268S, C273S and C552S HNF1B mutants all
decreased under DMF treatment, while the 4 CS mutant exhibited no response to DMF (Fig. 4C). More importantly, unlike the WT form, the 4CS mutant HNF1B could not be succinated by DMF (Fig.
4D), and DMF impaired the interaction between OTUD3 and WT HNF1B, while there was no detectable change in the interaction between OTUD3 and 4CS mutant HNF1B upon DMF treatment (Fig. 4E).
Consistently, DMF significantly increased the ubiquitination level of WT HNF1B, while the ubiquitination level of 4CS mutant HNF1B did not change under DMF treatment (Fig. 4F). Collectively,
these results indicate that DMF promotes succination of HNF1B, impairing its interaction with OTUD3, which in turn enhances its ubiquitination and degradation. DMF-HNF1B AXIS SUPPRESSES YAP
ACTIVITY It is well established that YAP plays a critical role in promoting cell proliferation, metastasis and chemoresistance in multiple solid tumors [31]. Notably, HNF1B was identified
as a potential interacting protein of YAP by mass spectrum [32], which prompted us to investigate whether HNF1B inhibits cell proliferation through YAP. To examine this hypothesis, we
conducted a co-IP assay, which revealed that HNF1B indeed interacted with YAP (Fig. 5A). Interestingly, this interaction could be inhibited by DMF (Fig. 5B). Given that the cooperation of
YAP-TEAD4 complex is enhanced by HHEX, which promoted cell proliferation [33], we examined whether HNF1B plays a similar role, and found that HNF1B did not affect the interaction between YAP
and TEAD4 (Fig. 5C). Moreover, we examined the effects of DMF on YAP phosphorylation and succination levels and results showed that there were no significant changes upon DMF treatment
(Fig. 5D, E). Surprisingly, we observed that DMF downregulated YAP protein levels, but not its mRNA levels, in both 786-O and RCC4 cells (Fig. 5F, G). Consistently, immunofluorescence assays
also revealed that DMF decreased the protein levels of YAP both in cytoplasm and nucleus (Fig. 5H). Moreover, the mRNA levels of _CYR61_ and _AXL_, the downstream target genes of YAP, were
also decreased following DMF treatment (Fig. 5I, J). We further determined the expression level of YAP in HNF1B knockout cells. Western blot analysis demonstrated that YAP expression level
was reduced in the absence of HNF1B (Fig. 5K). Notably, qPCR analysis revealed that HNF1B knockout also significantly decreased YAP target gene expression (Fig. 5L, M). These data suggest
that DMF-induced HNF1B degradation may trigger YAP instability, leading to the inactivation of YAP downstream targets. A previous study reported that YAP can act as a transcriptional
regulator of MYC, which in turn controls cell cycle [34]. We therefore evaluated the impact of DMF on MYC mRNA levels via qPCR. Our findings revealed a marked reduction in MYC mRNA levels
after DMF treatment (Fig. 5N), with a similar outcome observed in HNF1B knockout cells (Fig. 5O). Overall, these findings demonstrate that DMF promotes HNF1B degradation, leading to the
suppression of YAP and its downstream target gene expression. Next, we determine how DMF regulates YAP stability and results showed that DMF mediated YAP degradation can be rescued by the
overexpression of HNF1B in ccRCC cells (Fig. 5P), suggesting DMF mediated YAP degradation is dependent on HNF1B. To confirm which degradation pathway was involved in this process, we treated
cells with MG132 or NH4Cl in the presence of DMF. The results showed that only MG132 could rescue DMF-induced degradation of YAP, suggesting that DMF reduces YAP protein level via the
ubiquitin-proteasome pathway (Fig. 5Q). Furthermore, overexpression of HNF1B significantly decreased YAP ubiquitination levels (Fig. 5R), while knocking down HNF1B led to an increase of its
ubiquitination in both 786-O and RCC4 cells (Fig. 5S). Taken together, these results demonstrate that HNF1B decreases the ubiquitination level of YAP and enhances its stability, whereas DMF
mediated HNF1B succination promotes YAP ubiquitination and degradation. DMF ENHANCES THE SENSITIVITY OF CCRCC CELLS TO SUNITINIB Sunitinib, a tyrosine kinase inhibitor (TKI), has been
approved as a first-line treatment for ccRCC. Recently, several studies have associated YAP activation with resistance to sunitinib therapy, presenting a major obstacle to improve survival
of patients with ccRCC [35]. Based on our finding that DMF reduced HNF1B mediated YAP stabilization, we wonder whether DMF can enhance the sensitivity of ccRCC to sunitinib treatment. The
results showed that treatment with DMF or sunitinib alone moderately inhibited cell proliferation and colony formation. Notably, the combination of DMF and sunitinib exhibited a strong
synergistic effect on both cell proliferation and colony formation (Fig. 6A, B). To further validate these findings in vivo, we employed a cell-derived xenograft tumor model. 786-O cells
were injected subcutaneously into mice, which were then treated with DMF and/or sunitinib once the tumors became palpable (Fig. 6C). Consistent with the in vitro observations, the
combination of DMF and sunitinib significantly suppressed tumor growth and reduced tumor weight compared to single-agent treatments (Fig. 6D–F). Immunohistochemistry (IHC) staining revealed
that oral administration of DMF significantly decreased the protein levels of tumoral HNF1B (Fig. 6G, H). Moreover, the combined treatment with DMF and sunitinib dramatically reduced Ki67
levels in tumor tissues (Fig. 6G, H). Collectively, these data reveal an important role of DMF in enhancing the sensitivity of ccRCC to sunitinib treatment (Fig. 6I). DISCUSSION Despite the
availability of several treatment options for ccRCC, patients remain susceptible to develop drug resistance, which is a key factor in disease progression and contributes to high global
mortality rates. As a result, there is a critical need to explore novel therapeutic drugs and strategies. However, drug discovery is an expensive and time-consuming process, typically
costing around $2 billion and taking over a decade, with most drugs failing in clinical trials due to inadequate efficacy or adverse side effects [36]. Recently, drug repurposing, granting
approved drugs new therapeutic uses, has garnered significant attention. Researchers have identified several existing drugs with new applications in other diseases. For example, metformin, a
first-line drug for the treatment of type 2 diabetes mellitus, possesses immunomodulatory properties in cancer and inflammatory diseases, while chloroquine, known as an anti-malaria
medication, shows anti-tumor activity in several types of cancer [37, 38]. DMF has been approved for the treatment of MS and psoriasis for several years, and in the current study, we
identified its novel role in ccRCC therapy. As shown in Fig. 6I, DMF succinates HNF1B, leading to its proteasomal degradation and subsequent inactivation of YAP, which inhibits cell
proliferation of ccRCC. This suggests that DMF is a promising candidate for ccRCC treatment, given that its pharmacokinetics and safety profile have been well-established. ccRCC is
characterized by mutations of Von Hippel–Lindau (_VHL_), which lead to abnormal activation of hypoxia inducible factors, a group of transcription factors that regulate various genes,
including vascular endothelial growth factor (VEGF), facilitating neovasculogenesis in tumors [2, 39]. Sunitinib, one of the TKIs, was used to target VEGF signaling pathway for the treatment
of ccRCC [39]. However, most of ccRCC patients can develop resistance to sunitinib treatment. In this study, we demonstrated that DMF can enhance the sensitivity of ccRCC to sunitinib
treatment, and the combination of DMF and sunitinib dramatically inhibited the cell proliferation and tumor growth of ccRCC, providing a potential therapeutic strategy to overcome sunitinib
resistance in ccRCC. Several studies have highlighted the critical role of post-translational modifications (PTMs) in regulating protein stability. For instance, D-mannose induces
phosphorylation of PD-L1, which disrupts its glycosylation and triggers ubiquitin-dependent degradation [40, 41]. Similarly, acetylation of FGL1 enhances its ubiquitination and degradation
[40, 41]. In this study, we elucidated the molecular mechanism by which DMF, a metabolite, can promote succination and subsequent ubiquitin-proteasome-mediated degradation of HNF1B via
OTUD3, leading to destabilization and inactivation of YAP. These findings provide new evidence for metabolites regulating protein stability and cell signaling via PTMs. In summary, we report
a previously unrecognized PTM and function of HNF1B in ccRCC, and provide a potential drug and strategy for clinical treatment of ccRCC by targeting HNF1B-YAP axis. MATERIALS AND METHODS
CELL CULTURE AND CELL TRANSFECTION The cell lines 786-O, RCC4, SW620, Hela, T24, A375 and HEK-293T cells were maintained in DMEM (Meilun Biotechnology, China). A549 cells were cultured in
F-12 medium, while H1299 and KTC-1 cells were maintained in RPMI 1640. All media were supplemented with 10% fetal bovine serum (FBS) (Biological Industries, Israel) and 1%
penicillin/streptomycin (P/S). For cell transfection, EZ Trans (Life-iLab, China) was used according to manufacturer’s protocols. REAGENTS AND ANTIBODIES DMF (624-49-7), CCK-8 (C0005),
sunitinib (557795-19-4), MLN4924 (905579-51-3) were purchased from TargetMol (Shanghai, China). MG132 (HY-13259) was obtained from MCE (New Jersey, USA). Antibodies were listed as follows:
HNF1B (12533-1-AP, Proteintech), Actin (81115-1-RR, Proteintech,), GAPDH (60004-1-Ig, Proteintech), Flag-tag (HOA012FL01, AbHO), HA-tag (HOA012HA01, AbHO), YAP (ET1608-30, HUABIO), 2SC
(crb2005017, Discovery Antibodies), pYAP (ET1611-69, HUABIO), mouse secondary antibody (L3032, SAB), rabbit secondary antibody (L3012, SAB). MUTAGENESIS AND GENE KNOCKOUT Human cDNAs of
protein HNF1B were cloned into pLVX-2Flag lentiviral expression vector. Various site-directed mutants of HNF1B were generated by PCR using KOD Fx (TOYOBO, Japan). HNF1B plasmids were
amplified, and the products were digested with DpnI enzyme (Takara, Japan) before being transformed into NcmDH5-α (NCM Biotech, China) for amplification. HNF1B knockout cells were generated
through the CRISPR/Cas9 system. HEK-293T cells were co-transfected with LentiCRISPRv2, psPAX2, and pMD2.G in a ratio of 4:3:1. The supernatant containing virus particles was collected twice,
at 48 and 72 h post transfection, and then filtered using 0.45 μm filter. For lentiviral infection, cells were incubated in lentivirus-containing medium with polybrene (10 μg/mL) for 48 h
and then selected by puromycin for 7 days. The guide RNA (gRNA) sequence targeting HNF1B were as follows: 5′- AGGGCTGCTAAAATGATCAA -3; 5′- GACGTACCAGGTGTACAGAG -3. QPCR ANALYSIS Total RNA
was extracted using EZ-press RNA purification Kit (EZ Bioscience, USA). 1 μg of purified RNA was reverse transcribed into cDNA using 4× Reverse Transcription Master Mix (EZ Bioscience). qPCR
was performed with 2× SYBR qPCR Mix (KTSM, AlpaLife) using an Applied Biosystems 7300 Plus Sequence Detection System. The human ACTIN gene was utilized for normalization. Primers used in
qPCR analysis are listed below: ACTIN (5′-GGCATAGAGGTCTTTACGGATGTC-3′; 5′-TATTGGCAACGAGCGGTTCC-3′); HNF1B (5′-GGCAATTGCACAAATGTCCTCT-3′; 5′-ATTGTCTGAGGTGCCAGCAG-3′);
CYR61(5′-AAGAAACCCGGATTTGTGAG-3′; 5′- GCTGCATTTCTTGCCCTTT-3′); AXL (5′-GTGGGCAACCCAGGGAATATC-3′; 5′-GTACTGTCCCGTGTCGGAAAG-3′); MYC (5′-GGCTCCTGGCAAAAGGTCA-3′; 5′-CTGCGTAGTTGTGCTGATGT-3′).
WESTERN BLOT AND COIMMUNOPRECIPITATION Cells were washed with ice-cold PBS and lysed with 0.5% NP-40 lysis buffer containing 1% protease inhibitor and 1% phosphatase inhibitor at 4 °C for 30
min. Cell lysates were heated with Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) sample loading buffer at 100 °C for 10 min and then subjected to Western blot
according to standard protocol. Briefly, protein samples were separated by SDS-PAGE, transferred onto nitrocellulose filter membranes and incubated with primary antibodies overnight at 4 °C,
followed by HRP-conjugated anti-mouse secondary antibodies or anti–rabbit secondary antibodies at room temperature for one hour. Images were captured using Tanon 5200 imaging system (Tanon,
China). For co-IP, cell lysates were collected and centrifuged at 12,000 × _g_ for 15 min at 4 °C. The supernatants were incubated with anti-Flag (AbHO) beads overnight at 4 °C. The
following day, beads were washed five times with NP-40 buffer, heated with SDS-PAGE sample loading buffer, and then subjected to western blot analysis as description above. CELL
PROLIFERATION AND COLONY FORMATION ASSAYS Cell proliferation was assessed via CCK-8 assay. Cells were seeded in 96-well plates at a density of 1000 cells/well, and the cell viability was
measured four times with 24-h intervals by CCK-8 using a microplate reader at optical density (OD) of 450 nm. For colony formation assay, a total of 1000 cells were seeded into per well of
6-well plate and cultured for 14 days, with media changed every other day. Subsequently, colonies were washed using PBS, fixed in 10% methanol at room temperature for 15 min, and stained
with 0.1% crystal violet for 30 min. After washing with PBS, images of the colonies were captured. Decolorization was performed using 33% acetic acid, and the resulting solution was analyzed
to measure OD value at 570 nm for statistical analysis. ANIMAL EXPERIMENTS All animal experiments were approved by the Animal Care and Use Committee at Fudan University. Five-week-old male
NSG mice were purchased from Model Organisms Center, Shanghai, China. The mice were housed in specific pathogen-free conditions at temperature of 22–23 °C, with free food and water access in
a 12 h light/dark cycle. A total of 1 × 107 786-O cells in 50% matrigel were injected subcutaneously into mice. Once the tumors became palpable, the mice were randomly assigned into four
groups and treated with different drugs by oral gavage: (1) control group (0.1% DMSO); (2) DMF group (30 mg/kg of DMF everyday); (3) sunitinib group (20 mg/kg of sunitinib every other day);
(4) combined treatment group (30 mg/kg of DMF every day and 20 mg/kg of sunitinib every other day). Tumor growth was measured every four days by calipers, and tumor volume was estimated
using the formula: 0.5 × L (longer diameter) × W2 (shorter diameter). At the experimental endpoint of experiments, the mice were euthanized, and the tumors were excised and weighed. DATA
AVAILABILITY Further information and requests for reagents may be directed to, and will be fulfilled by, the author Yanping Xu ([email protected]). CHANGE HISTORY * _ 28 APRIL 2025 The
original online version of this article was revised: In the original publication, Figure 1F (sgcontrol) was incorrectly inserted. The accurate image is provided instead. This correction
does not affect the conclusions of the article. _ * _ 19 MAY 2025 A Correction to this paper has been published: https://doi.org/10.1038/s41419-025-07675-0 _ REFERENCES * Dubois-Laforgue D,
Cornu E, Saint-Martin C, Coste J, Bellanne-Chantelot C, Timsit J, et al. Diabetes, associated clinical spectrum, long-term prognosis, and genotype/phenotype correlations in 201 adult
patients with Hepatocyte Nuclear Factor 1B (HNF1B) molecular defects. Diabetes Care. 2017;40:1436–43. Article CAS PubMed Google Scholar * Jonasch E, Walker CL, Rathmell WK. Clear cell
renal cell carcinoma ontogeny and mechanisms of lethality. Nat Rev Nephrol. 2021;17:245–61. Article CAS PubMed Google Scholar * Zhao L, Wang Z, Xu Y, Zhang P, Qiu J, Nie D, et al.
Sphingosine kinase 1 regulates lipid metabolism to promote progression of kidney renal clear cell carcinoma. Pathol Res Pract. 2023;248:154641. Article CAS PubMed Google Scholar *
Ljungberg B. Prognostic markers in renal cell carcinoma. Curr Opin Urol. 2007;17:303–8. Article PubMed Google Scholar * Singh D. Current updates and future perspectives on the management
of renal cell carcinoma. Life Sci. 2021;264:118632. Article CAS PubMed Google Scholar * Long Z, Sun C, Tang M, Wang Y, Ma J, Yu J, et al. Single-cell multiomics analysis reveals
regulatory programs in clear cell renal cell carcinoma. Cell Discov. 2022;8:68. Article CAS PubMed PubMed Central Google Scholar * Zhu M, Li Y, Wang Y, Lin P, Mi J, Zhong W. Multi-omics
analysis uncovers clinical, immunological, and pharmacogenomic implications of cuproptosis in clear cell renal cell carcinoma. Eur J Med Res. 2023;28:248. Article CAS PubMed PubMed
Central Google Scholar * Yang RN, Zhou FR, Wang HY, Wang QH, Ji JL, Huang T, et al. Antitumor activity of RUNX3: upregulation of E-cadherin and downregulation of the epithelial-mesenchymal
transition in clear-cell renal cell carcinoma. Open Life Sci. 2022;17:1579–90. Article CAS PubMed PubMed Central Google Scholar * Bockenhauer D, Jaureguiberry G. HNF1B-associated
clinical phenotypes: the kidney and beyond. Pediatr Nephrol. 2016;31:707–14. Article PubMed Google Scholar * Barbacci E, Chalkiadaki A, Masdeu C, Haumaitre C, Lokmane L, Loirat C, et al.
HNF1beta/TCF2 mutations impair transactivation potential through altered co-regulator recruitment. Hum Mol Genet. 2004;13:3139–49. Article CAS PubMed Google Scholar * Hiesberger T, Shao
X, Gourley E, Reimann A, Pontoglio M, Igarashi P. Role of the hepatocyte nuclear factor-1beta (HNF-1beta) C-terminal domain in Pkhd1 (ARPKD) gene transcription and renal cystogenesis. J Biol
Chem. 2005;280:10578–86. Article CAS PubMed Google Scholar * Okorn C, Goertz A, Vester U, Beck BB, Bergmann C, Habbig S, et al. HNF1B nephropathy has a slow-progressive phenotype in
childhood-with the exception of very early onset cases: results of the German Multicenter HNF1B Childhood Registry. Pediatr Nephrol. 2019;34:1065–75. Article PubMed Google Scholar * Dundr
P, Bartu M, Hojny J, Michalkova R, Hajkova N, Struzinska I, et al. HNF1B, EZH2 and ECI2 in prostate carcinoma. Molecular, immunohistochemical and clinico-pathological study. Sci Rep.
2020;10:14365. Article CAS PubMed PubMed Central Google Scholar * Bartu M, Dundr P, Nemejcova K, Ticha I, Hojny H, Hajkova N. The Role of HNF1B in Tumorigenesis of Solid Tumours: a
Review of Current Knowledge. Folia Biol. 2018;64:71–83. Article CAS Google Scholar * Owjfard M, Karimi F, Mallahzadeh A, Nabavizadeh SA, Namavar MR, Saadi MI, et al. Mechanism of action
and therapeutic potential of dimethyl fumarate in ischemic stroke. J Neurosci Res. 2023;101:1433–46. Article CAS PubMed Google Scholar * Burness CB, Deeks ED. Dimethyl fumarate: a review
of its use in patients with relapsing-remitting multiple sclerosis. CNS Drugs. 2014;28:373–87. Article CAS PubMed Google Scholar * Bruck J, Dringen R, Amasuno A, Pau-Charles I,
Ghoreschi K. A review of the mechanisms of action of dimethylfumarate in the treatment of psoriasis. Exp Dermatol. 2018;27:611–24. Article PubMed Google Scholar * Itoh K, Tong KI,
Yamamoto M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med. 2004;36:1208–13. Article CAS PubMed Google Scholar
* Gillard GO, Collette B, Anderson J, Chao J, Scannevin RH, Huss DJ, et al. DMF, but not other fumarates, inhibits NF-kappaB activity in vitro in an Nrf2-independent manner. J
Neuroimmunol. 2015;283:74–85. Article CAS PubMed Google Scholar * Vandermeeren M, Janssens S, Wouters H, Borghmans I, Borgers M, Beyaert R, et al. Dimethylfumarate is an inhibitor of
cytokine-induced nuclear translocation of NF-kappa B1, but not RelA in normal human dermal fibroblast cells. J Invest Dermatol. 2001;116:124–30. Article CAS PubMed Google Scholar *
Schmitt A, Xu W, Bucher P, Grimm M, Konantz M, Horn H, et al. Dimethyl fumarate induces ferroptosis and impairs NF-kappaB/STAT3 signaling in DLBCL. Blood. 2021;138:871–84. Article CAS
PubMed Google Scholar * Takeda T, Tsubaki M, Asano R, Itoh T, Imano M, Satou T, et al. Dimethyl fumarate suppresses metastasis and growth of melanoma cells by inhibiting the nuclear
translocation of NF-kappaB. J Dermatol Sci. 2020;99:168–76. Article CAS PubMed Google Scholar * Yang B, Shi J. Developing new cancer nanomedicines by repurposing old drugs. Angew Chem
Int Ed Engl. 2020;59:21829–38. Article CAS PubMed Google Scholar * Lu W, Sun J, Zhou H, Wang F, Zhao C, Li K, et al. HNF1B inhibits cell proliferation via repression of SMAD6 expression
in prostate cancer. J Cell Mol Med. 2020;24:14539–48. Article CAS PubMed PubMed Central Google Scholar * Patel SA, Hirosue S, Rodrigues P, Vojtasova E, Richardson EK, Ge J, et al. The
renal lineage factor PAX8 controls oncogenic signalling in kidney cancer. Nature. 2022;606:999–1006. Article CAS PubMed PubMed Central Google Scholar * Humphries F, Shmuel-Galia L,
Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin D and blocks pyroptosis. Science. 2020;369:1633–7. Article CAS PubMed PubMed Central Google Scholar
* Saidu NEB, Kavian N, Leroy K, Jacob C, Nicco C, Batteux F, et al. Dimethyl fumarate, a two-edged drug: current status and future directions. Med Res Rev. 2019;39:1923–52. Article PubMed
Google Scholar * Cheng J, Liu Y, Yan J, Zhao L, Zhou Y, Shen X, et al. Fumarate suppresses B-cell activation and function through direct inactivation of LYN. Nat Chem Biol.
2022;18:954–62. Article CAS PubMed Google Scholar * Wright DJ, Gray LJ, Finkelstein DI, Crouch PJ, Pow D, Pang TY, et al. N-acetylcysteine modulates glutamatergic dysfunction and
depressive behavior in Huntington’s disease. Hum Mol Genet. 2016;25:2923–33. CAS PubMed Google Scholar * Kastrati I, Siklos MI, Calderon-Gierszal EL, El-Shennawy L, Georgieva G, Thayer
EN, et al. Dimethyl fumarate inhibits the nuclear factor kappaB pathway in breast cancer cells by covalent modification of p65 protein. J Biol Chem. 2016;291:3639–47. Article CAS PubMed
Google Scholar * Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. 2016;29:783–803. Article CAS PubMed PubMed Central Google Scholar * Rosenbluh J,
Mercer J, Shrestha Y, Oliver R, Tamayo P, Doench JG, et al. Genetic and proteomic interrogation of lower confidence candidate genes reveals signaling networks in beta-catenin-active cancers.
Cell Syst. 2016;3:302–16.e4. Article CAS PubMed PubMed Central Google Scholar * Guo Y, Zhu Z, Huang Z, Cui L, Yu W, Hong W, et al. CK2-induced cooperation of HHEX with the YAP-TEAD4
complex promotes colorectal tumorigenesis. Nat Commun. 2022;13:4995. Article CAS PubMed PubMed Central Google Scholar * Choi W, Kim J, Park J, Lee DH, Hwang D, Kim JH, et al. YAP/TAZ
Initiates gastric tumorigenesis via upregulation of MYC. Cancer Res. 2018;78:3306–20. Article CAS PubMed Google Scholar * Nguyen CDK, Yi C. YAP/TAZ signaling and resistance to cancer
therapy. Trends Cancer. 2019;5:283–96. Article CAS PubMed PubMed Central Google Scholar * Prasad S, Gupta SC, Aggarwal BB. Serendipity in cancer drug discovery: rational or coincidence?
Trends Pharmacol Sci. 2016;37:435–50. Article CAS PubMed Google Scholar * Foretz M, Guigas B, Viollet B. Metformin: update on mechanisms of action and repurposing potential. Nat Rev
Endocrinol. 2023;19:460–76. Article CAS PubMed Google Scholar * Baradaran Eftekhari R, Maghsoudnia N, Dorkoosh FA. Chloroquine: a brand-new scenario for an old drug. Expert Opin Drug
Deliv. 2020;17:275–7. Article PubMed Google Scholar * Kase AM, George DJ, Ramalingam S. Clear cell renal cell carcinoma: from biology to treatment. Cancers. 2023;15:665. Article PubMed
PubMed Central Google Scholar * Lin M, He J, Zhang X, Sun X, Dong W, Zhang R, et al. Targeting fibrinogen-like protein 1 enhances immunotherapy in hepatocellular carcinoma. J Clin Invest.
2023;133:e164528. Article PubMed PubMed Central Google Scholar * Zhang R, Yang Y, Dong W, Lin M, He J, Zhang X, et al. D-mannose facilitates immunotherapy and radiotherapy of
triple-negative breast cancer via degradation of PD-L1. Proc Natl Acad Sci USA. 2022;119:e2114851119. Article PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We
are grateful to the members of L.L. laboratory for discussion throughout this study. This work was supported by the National Key R&D Program of China (2022YFA0807100,
2020YFA0803400/2020YFA0803402), the National Natural Science Foundation of China (82172936, 82472850, 82121004, 82372754, 82073128 and 32000918), the Shanghai Rising-Star Program
(24QA2709900), the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning and the Fundamental Research Funds for the Central Universities.
AUTHOR INFORMATION Author notes * These authors contributed equally: Yue Dai, Hongchen Li. AUTHORS AND AFFILIATIONS * Fifth People’s Hospital of Shanghai, MOE Key Laboratory of Metabolism
and Molecular Medicine, Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Fudan University, Shanghai, China Yue Dai, Shiyin Fan, Xue Sun, Mingen Lin, Jiaxi
Li, Yi Gao, Ziyin Tian & Lei Lv * Tongji Hospital, Frontier Science Center for Stem Cell Research, School of Life Sciences and Technology, Tongji University, Shanghai, China Hongchen Li,
Ziyi Cui, Xinyu Zhao & Yanping Xu * Department of Endocrinology, Fifth People’s Hospital of Shanghai, Fudan University, Shanghai, China Kai Wang & Bingbing Zha * Department of
Neurosurgery, Huashan Hospital, Institute for Translational Brain Research, MOE Frontiers Center for Brain Science, Shanghai Medical College, Fudan University, Shanghai, China Hui Yang
Authors * Yue Dai View author publications You can also search for this author inPubMed Google Scholar * Hongchen Li View author publications You can also search for this author inPubMed
Google Scholar * Shiyin Fan View author publications You can also search for this author inPubMed Google Scholar * Kai Wang View author publications You can also search for this author
inPubMed Google Scholar * Ziyi Cui View author publications You can also search for this author inPubMed Google Scholar * Xinyu Zhao View author publications You can also search for this
author inPubMed Google Scholar * Xue Sun View author publications You can also search for this author inPubMed Google Scholar * Mingen Lin View author publications You can also search for
this author inPubMed Google Scholar * Jiaxi Li View author publications You can also search for this author inPubMed Google Scholar * Yi Gao View author publications You can also search for
this author inPubMed Google Scholar * Ziyin Tian View author publications You can also search for this author inPubMed Google Scholar * Hui Yang View author publications You can also search
for this author inPubMed Google Scholar * Bingbing Zha View author publications You can also search for this author inPubMed Google Scholar * Lei Lv View author publications You can also
search for this author inPubMed Google Scholar * Yanping Xu View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS YD played major roles in
designing and doing the experiments, analyzing the results and organizing the figures; HL did the revision experiments; SF, KW, ZC, XZ, XS, ML, JL, YG, and ZT helped to conduct the
experiments; HY designed and supervised the animal study; BZ, LL, and YX conceived and designed the study; LL and YX supervised the study; YD, HL, and YX wrote the manuscript. CORRESPONDING
AUTHORS Correspondence to Hui Yang, Bingbing Zha, Lei Lv or Yanping Xu. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ETHICS APPROVAL AND CONSENT TO
PARTICIPATE All animal experiments were approved by the Animal Care and Use Committee at Fudan University. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard
to jurisdictional claims in published maps and institutional affiliations. Edited by Angelo Peschiaroli SUPPLEMENTARY INFORMATION UNCROPPED WESTERN BLOTS RIGHTS AND PERMISSIONS OPEN ACCESS
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as
long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third
party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the
article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright
holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Dai, Y., Li, H., Fan, S. _et al._
Dimethyl fumarate promotes the degradation of HNF1B and suppresses the progression of clear cell renal cell carcinoma. _Cell Death Dis_ 16, 71 (2025).
https://doi.org/10.1038/s41419-025-07412-7 Download citation * Received: 07 July 2024 * Revised: 19 December 2024 * Accepted: 30 January 2025 * Published: 06 February 2025 * DOI:
https://doi.org/10.1038/s41419-025-07412-7 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative







