- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT MitoKATP is a channel of the inner mitochondrial membrane that controls mitochondrial K+ influx according to ATP availability. Recently, the genes encoding the pore-forming (MITOK)
and the regulatory ATP-sensitive (MITOSUR) subunits of mitoKATP were identified, allowing the genetic manipulation of the channel. Here, we analyzed the role of mitoKATP in determining
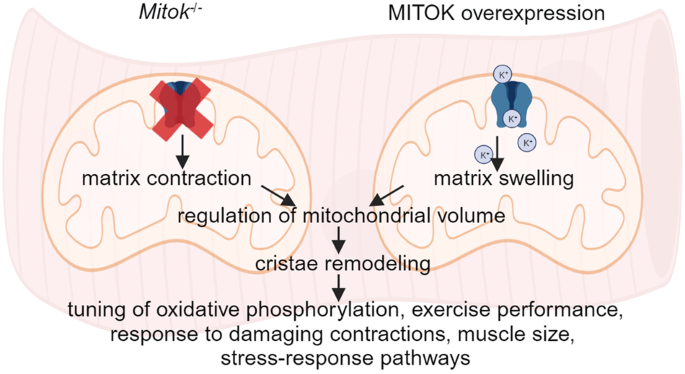
skeletal muscle structure and activity. _Mitok_−/− muscles were characterized by mitochondrial cristae remodeling and defective oxidative metabolism, with consequent impairment of exercise
performance and altered response to damaging muscle contractions. On the other hand, constitutive mitochondrial K+ influx by MITOK overexpression in the skeletal muscle triggered overt
mitochondrial dysfunction and energy default, increased protein polyubiquitination, aberrant autophagy flux, and induction of a stress response program. MITOK overexpressing muscles were
therefore severely atrophic. Thus, the proper modulation of mitoKATP activity is required for the maintenance of skeletal muscle homeostasis and function. SIMILAR CONTENT BEING VIEWED BY
OTHERS MCU-INDEPENDENT CA2+ UPTAKE MEDIATES MITOCHONDRIAL CA2+ OVERLOAD AND NECROTIC CELL DEATH IN A MOUSE MODEL OF DUCHENNE MUSCULAR DYSTROPHY Article Open access 21 March 2024 A KINASE
INTERACTING PROTEIN 1 REGULATES MITOCHONDRIAL PROTEIN LEVELS IN ENERGY METABOLISM AND PROMOTES MITOCHONDRIAL TURNOVER AFTER EXERCISE Article Open access 01 November 2023 PERM1 INTERACTS WITH
THE MICOS-MIB COMPLEX TO CONNECT THE MITOCHONDRIA AND SARCOLEMMA VIA ANKYRIN B Article Open access 12 August 2021 INTRODUCTION Within mitochondria, dynamic regulation of K+ fluxes warrants
structural and functional organelle integrity. Despite the huge driving force of the negative membrane potential across the inner mitochondrial membrane (IMM), in physiological conditions
the [K+] inside and outside mitochondria is similar, due to the low activity of K+ channels coupled with the activity of the electroneutral K+/H+ antiporter. Increased mitochondrial K+
influx is accompanied by osmotically obligated water entering through mitochondrial aquaporins, resulting in matrix swelling. By controlling matrix volume, K+ cycle contributes to the
coupling between respiration and ATP synthesis and to the maintenance of both the structure and the function of the organelle [1]. Thus, K+ homeostasis is thought to play a major role in the
regulation of mitochondrial activity, including reactive oxygen species (ROS) production, with implications in cell fate and pathophysiological conditions [2]. This is particularly relevant
in the response to ischemic/reperfusion injury, which is due to the sudden increase in reactive oxygen species (ROS) production once O2 supply is restored. In these settings, the
pharmacological opening of mitochondrial K+ channels exerts a protective effect [3, 4]. Despite the uncertainty on the precise mechanism, mitochondrial uncoupling preventing ROS generation
during reperfusion may be involved. Mitochondrial K+ fluxes must be tightly controlled. High intracellular [K+] indeed potentially represents a serious threat for mitochondrial integrity,
since excessive K+ entry in mitochondria can quickly dissipate the large electrochemical gradient with severe consequences at both organelle and cellular level. Despite this, multiple K+
channels are located at the IMM, with different structure, molecular composition, and biochemical features. The need for this redundancy and the specific roles of individual channels are
still unclear, but emerging evidence indicates that each of them is controlled by different physiological stimuli, allowing a fine-tuned modulation of the mitochondrial function [1]. Our
current understanding of the functions and roles of mitochondrial K+ channels was historically limited by several constraints. The most evident one is the lack of biochemical information
since the molecular identity of many mitochondrial K+ channels is unknown. In other cases where molecular information is available, the same K+ channel has been shown to be located at both
plasma and inner mitochondrial membranes, but the sorting mechanism remains a mystery. This promiscuous localization prevents the direct investigation of the specific contribution of
mitochondrial versus plasma membrane K+ channels to organelle and cell physiology. As a consequence, until recently the comprehension of mitochondrial K+ fluxes has been largely based on
pharmacological approaches that lack specificity and proper genetic validation. This gridlock has been recently solved, at least in part, by the recognition of a K+ channel specifically
located at the IMM, i.e. the mitochondrial ATP-sensitive K+ channel (mitoKATP). The molecular identification of the mitoKATP channel showed that the assembly of the pore-forming subunit
(MITOK) with a regulatory (MITOSUR) subunit is able to mediate ATP-dependent mitochondrial K+ entry [5]. _MITOK_ ablation causes instability of the mitochondrial membrane potential, widening
of the intracristae space and impaired oxidative phosphorylation. Ex vivo, in an ischemia/reperfusion model, _MITOK_ deletion suppresses cardioprotection elicited by preconditioning induced
by diazoxide, an opener of the mitoKATP channel. On the opposite side, MITOK overexpression in cell lines triggers mitochondrial fragmentation, swelling, and a drop in mitochondrial
membrane potential. Concerning the skeletal muscle, until recently it was not even clear whether mitoKATP channels were expressed or not. The first indications were provided by
pharmacological modulation of the channel in isolated rat skeletal muscle mitochondria. Those organelles responded to potassium channel openers (KCOs) in terms of increased mitochondrial
swelling, oxygen consumption and depolarization, and these effects were counteracted by the addition of KATP channel inhibitors [6], indicating the existence of mitoKATP in skeletal muscle,
although with different pharmacological properties compared to other tissues. As for respiration, experiments in chicken skeletal muscle mitochondria recorded an inhibition rather than
activation by KCOs [7], indicating that different fibre types and metabolic properties could impinge on these responses. Additionally, in adult mouse skeletal muscle bundles, diazoxide
reduced muscle fatigue, i.e. the decrease in muscle tension upon repetitive electrical stimulation. The effects of mitoKATP opening were prevented by 5-HD, a blocker of the channel [8]. In
chick muscles, again some differences were observed. In line with the effects detected in the mouse muscles, diazoxide increased post-fatigue tension, but no effect was detected by the
addition of 5-HD [9]. Also in this case, the different muscle models (slow versus fast) and the different fatigue protocols may have played a role. Considering the pivotal role of
mitochondrial ion fluxes in the control of skeletal muscle structure and function, here we analysed the consequence of the genetic manipulation of mitoKATP activity in the mouse skeletal
muscle, by deleting or overexpressing MITOK, respectively. Structural and functional assays, corroborated by metabolomics data, indicate a primary role of mitochondrial K+ influx to maintain
organelle homeostasis, essential for optimal skeletal muscle activity. On the other hand, MITOK overexpression offers a genetic tool, an alternative to protonophore agents, to dissect the
outcomes of mitochondrial depolarization and uncoupling. In the skeletal muscle, uncontrolled potassium entry triggers mitochondria dysfunction and energy crisis. Polyubiquitinated proteins
accumulate, the autophagic flux is blocked, and a cellular stress response program is induced determining muscle atrophy, despite the persistent activation of the IGF1/AKT1 pathway.
Altogether, the genetic manipulation of the mitoKATP channel uncovers the role of mitochondrial K+ fluxes in skeletal muscle structure and function. RESULTS _MITOK_ DELETION IN SKELETAL
MUSCLE ALTERS MITOCHONDRIA CRISTAE SHAPE, METABOLITE PROFILE AND RESPIRATION Previous work demonstrated that _MITOK_ deletion in HeLa cells causes the accumulation of aberrant mitochondria
characterized by widening of the intra-cristae space and decreased respiration. Mitochondrial membrane potential (ΔΨm) is overall intact, but unstable, as shown by the fact that mitochondria
undergo asynchronous, rapid and transient depolarizations (i.e. mitochondrial ‘flickering’ or ‘flashes) [5]. Thus, we wondered whether the deletion of _Mitok_ triggers similar effects in
the skeletal muscle and if muscle function is consequently affected. To this aim, we took advantage of the _Mitok__−/−_ mouse, whose heart was characterized by a slight increase in
sensitivity to I/R injury and loss of pharmacological preconditioning by the mitoKATP opener diazoxide [5]. Western blot analyses confirmed the absence of MITOK protein in the skeletal
muscle of _Mitok__−/−_ mouse (Fig. 1A, S4). Skeletal muscle mitochondria of _Mitok__−/−_ animals were characterized by increased cristae width (Fig. 1B), indicating that the hallmarks of
MITOK deficiency are conserved in post-mitotic tissues. To get further insights into the metabolic consequences of defective mitochondrial K+ homeostasis, we performed an unbiased
metabolomics analysis of 662 compounds in _Mitok__−/−_ muscles compared to WT counterparts (Fig. 1C). _Mitok__−/−_ muscles were characterized by decreased levels of phospholipids, including
cytidine 5’-monophosphate, phosphatidylcholines, phosphatidylethanolamines, phosphatidylglycerols, sphingolipids, and phosphatidylinositols, suggesting decreased phospholipid synthesis
and/or increased phospholipid breakdown. Notably, phosphatidylglycerols are the main component of the mitochondria-specific lipid cardiolipin and have important roles in membrane remodeling
and stress responses [10], suggesting these data may reflect _Mitok__−/−_-induced decreased cardiolipin/mitochondrial biogenesis and/or membrane remodeling. Also, the sphingolipids synthesis
intermediates sphinganine and sphingadienine were significantly decreased along with several ceramides, sphingomyelins, and sphingosine in MITOK-deficient muscles relative to controls,
suggesting decreased sphingolipid and ceramides synthesis. In addition, cholesterol showed significant decreases in MITOK-deficient muscle. Collectively, the reduced levels of phospholipids,
sphingolipids, and cholesterol suggest large-scale remodeling of muscle cell membranes in response to MITOK ablation, in accordance with EM data. Next, we wished to further characterize the
consequence of defective mitochondrial K+ homeostasis, by measuring mitochondrial function parameters, including ΔΨm, mitochondrial Ca2+ uptake, and oxygen consumption rate (OCR). Deletion
of MITOK did not affect the ΔΨm (Fig. 1D) or mitochondrial Ca2+ uptake (Fig. S1A-B), while basal, ATP-linked, and maximal respiration were all decreased in myofibres with impaired
mitochondrial K+ flux (Fig. 1E). In addition, according to the metabolite profile, _Mitok__−/−_ muscles were characterized by an increase in glucose-6-phosphate that may reflect an increase
in hexokinase activity. Moreover, glycolytic intermediates were elevated, including fructose 1,6-diphosphate/glucose 1,6-diphosphate and dihyroxyacetone phosphate (DHAP), suggesting
increased glycolysis (Fig. 1F). Thus, defective mitoKATP activity alters membrane structure, triggers mitochondria cristae widening, and reduces oxidative metabolism in skeletal muscle.
_MITOK_ DELETION TRIGGERS EXERCISE INTOLERANCE AND SUSCEPTIBILITY TO DAMAGING MUSCLE CONTRACTIONS The above observations, pointing to defective mitochondrial membrane remodelling and
oxidative metabolism, lead us to verify the consequences of _Mitok_ deletion on muscle structure and activity. In _Mitok_−/− muscles neither signs of damage were detected (Fig. 2A), nor
differences in fibre size were observed, both in fast-twitch (i.e. extensor digitorum longus, EDL) and in slow-twitch (i.e. soleus) muscles (Fig. 2B), suggesting that MITOK ablation does not
alter the overall histological features of the skeletal muscle. Next, we wished to know whether mitoKATP was required for muscle activity. We detected an impaired running capacity on a
treadmill until exhaustion, both uphill (Fig. 2C) and downhill (Fig. 2D), indicating that MITOK was required to sustain endurance exercise. We then performed in vivo measurements to assess
the force of hindlimb muscles when subjected to contractions at increasing frequency until tetanus is reached. No difference was observed between the two genotypes, indicating that
mitoKATP-dependent mitochondrial K+ entry was unnecessary to sustain a single bout of tetanic force production (Fig. 2E). During exercise, muscles are also subjected to the lengthening
effects of eccentric contractions, that cause sarcolemma damage progressively decreasing force production. The role of mitochondrial K+ homeostasis was assessed by measuring in vivo the
residual tetanic force of the hindlimb upon repetitive lengthening contractions (Fig. 2F). This assay highlighted the increased force reduction of _Mitok__−/−_ mice compared to WT controls
when subjected to the tearing effects of lengthening contractions, indicating that physiological mitochondria K+ entry is required to counteract muscle damage. MITOK OVEREXPRESSION ALTERS
SKELETAL MUSCLE MITOCHONDRIAL STRUCTURE AND FUNCTION In HeLa cells, MITOK overexpression causes mitochondrial fragmentation, swelling and collapse of cristae [5]. To investigate the effects
of MITOK overexpression on mitochondrial morphology and function in skeletal muscle during postnatal development, we injected hindlimbs of newborn mice with adeno-associated viral particles
overexpressing Flag-tagged MITOK (AAV9-MITOK). Muscles were collected and analyzed 4 weeks later (Fig. 3A), and western blot analysis confirmed the expression of AAV9-MITOK (Fig. 3B, S4).
Electron microscopy images of longitudinal sections of EDL muscles revealed that MITOK overexpression strongly affects mitochondrial morphology. Many mitochondria were swollen, and internal
cristae were collapsed (Fig. 3C), as previously observed in HeLa cells, in which the constitutive opening of the mitoKATP channel triggers a net influx of potassium cations leading to water
entry, subsequent mitochondrial swelling and accumulation of aberrant mitochondria [5]. In line with the alteration of mitochondrial morphology, immunoblot analysis of mitochondrial proteins
upon MITOK overexpression suggested a decreased content of proteins localized at the inner mitochondrial membrane, including IMMT and COXIV. We also observed an enrichment in GRP75, HSP60,
and TFAM, which mainly localize into the matrix, possibly due to expression induction or decreased degradation for reasons that may be related to cell stress activation pathways (Fig. 3D,
S4). Analysis of ETC components revealed that MITOK overexpression significantly decreases the expression of the subunit NDUFB8 of NADH Dehydrogenase complex I, of the SDHB subunit of
complex II, and of the subunit I of complex IV (Fig. 3E, S4). We further evaluated the consequence of the mitochondria structural defects exerted by MITOK overexpression by assessing ΔΨm,
that was decreased in AAV-MITOK infected myofibres compared to controls (Fig. 3F). Moreover, we determined SDH activity on tibialis anterior (TA), EDL, and soleus muscle cryosections. The
number of SDH activity-positive myofibres in MITOK-overexpressing muscles strongly decreased compared to control muscles (Fig. 3G). The negative impact of MITOK overexpression on
mitochondrial function was also revealed by the increased phosphorylation of acetyl CoA carboxylase (ACC), a target of the energy stress sensor AMPK that is activated by a decrease in the
energy charge triggering catabolic responses (Fig. 3H, S4). Thus, a bioenergetic defect occurs in consequence of dysregulated mitochondrial K+ entry and organelle structure alterations. The
mitochondrial dysfunction of MITOK-overexpressing muscles prompted us to determine the involvement of the PINK1-Parkin pathway, which participates in the response to mitochondria stress.
PINK1, which is continuously degraded in physiological conditions, accumulates on the mitochondrial outer membrane (OMM) upon IMM depolarization and mitochondrial damage. On the
mitochondrial surface, PINK1 recruits, phosphorylates, and thus activates, the E3 ubiquitin ligase Parkin, which triggers polyubiquitination of several targets at the OMM, priming the
selective removal of aberrant organelles by mitophagy. Thus, we thought of determining whether PINK1 accumulates upon MITOK overexpression, and whether Parkin is activated. We observed
increased PINK1 protein levels in MITOK-overexpressing muscles compared to controls (Fig. 3I, S4). However, assessment of Parkin recruitment to mitochondria was hampered by the fact that
MITOK overexpression alters Parkin protein levels (Figure S2A, S5). Nonetheless, the mitochondrial fraction of MITOK-overexpressing muscles was enriched in polyubiquitinated proteins,
indicating the accumulation of ubiquitin-targeted mitochondria (Fig. 3J, S4). Moreover, general activation of the ubiquitin-proteasome system occurs, as demonstrated by the accumulation of
polyubiquitinated proteins in the total cell lysates. Overall, these data indicate that MITOK overexpression induces a drastic alteration of mitochondrial ultrastructure, a decrease of
mitochondrial function, and activation of mitochondrial damage-sensing pathways. MITOK OVEREXPRESSION IMPINGES ON SKELETAL MUSCLE CATABOLISM AND STRESS RESPONSE PROGRAMS Damaged or
dysfunctional mitochondria lead to defective energy production and altered signaling pathways impinging on nuclear gene programs that regulate muscle mass [11]. Accordingly, mitochondria
quality control pathways play a central role in regulating the cell programs that underlie muscle homeostasis. In this context, the accumulation of polyubiquitinated proteins at the OMM
represents a signaling event for the selective removal of aberrant mitochondria and, in this process, both proteasome and autophagy pathways are involved. Thus, we first thought of verifying
the activity levels of the ubiquitin-proteasome system, which plays a key role in muscle atrophy [12]. As anticipated in Fig. 3J, total protein ubiquitination was increased in
MITOK-overexpressing muscles (Fig. 4A, S5). However, the expression of muscle-specific E3 ubiquitin ligases, including Atrogin1, Murf1, Mul1, Traf6, and MUSA1, was unaffected (Fig. 4B),
suggesting that a steady-state activation level was reached. We next checked the activity of the autophagy-lysosome system, which on one side is required for continuous turnover of aberrant
proteins and organelles, maintaining muscle homeostasis, on the other hand is activated upon catabolic signals [12]. The protein levels of the autophagy marker LC3-II were increased in
MITOK-overexpressing TA muscles compared to controls (Fig. 4C, S5). The protein levels of p62/SQSTM1, which is an autophagy receptor bridging polyubiquitinated proteins of the outer membrane
of depolarized mitochondria with LC3 on the autophagosome surface, were also increased in MITOK overexpressing muscles (Fig. 4C-D, S5), and this was accompanied by an increase in p62/SQSTM1
transcription (Fig. 4E). However, the mRNA levels of other autophagy genes were unaltered (Fig. 4F), suggesting that the increase in LC3-II levels could be due to hampered
autophagosome-lysosome fusion and degradation rather than to autophagy induction. To discern between these two possibilities, we measured the autophagy flux in MITOK-overexpressing muscles
compared to controls. As illustrated in Fig. 4G, hindlimbs of newborn mice were injected with AAV9-MITOK or control viral particles. Four weeks later mice were i.p. injected with colchicine
twice at 12 h interval, to block autophagosome-lysosome fusion, and sacrificed 12 h after the second injection. As expected, in control muscles colchicine triggered the accumulation of
LC3-II indicating an increase in the number of autophagosomes due to a block in their degradation (Fig. 4H). The levels of the p62 protein demonstrated a tendency to rise as well (Fig. 4H).
In MITOK overexpressing muscles either treated with colchicine or not, LC3-II and p62 levels were comparable, indicating that MITOK overexpression causes a block in the autophagic flux (Fig.
4H). The accumulation of dysfunctional mitochondria and the impairment in their removal is causative of cell stress activation. In MITOK overexpressing muscles, different genes implicated
in cellular stress response and DNA repair were induced, including growth arrest and DNA damage-inducible 45α (GADD45a), which is upregulated by fasting and immobilization-induced atrophy
[13, 14], p53 and its downstream target p21, both involved in muscle atrophy [15, 16], and FANCL, one of the genes of the Fanconi Anemia (FA) family, required for the activation of the FA
pathway, that is involved in DNA repair [17] (Fig. 4I). In addition, heat shock proteins HSP27 and HSP90, that play a fundamental role in the maintenance of muscle homeostasis [18], were
also induced (Fig. 4J). Thus, mitochondrial dysfunction, altered quality control systems and catabolic pathways, and a cell-stress response characterize MITOK-overexpressing muscles. We
thought of further investigating muscle protein regulation in conditions of constitutive mitochondrial K+ entry by monitoring the IGF1/AKT1 pathway, which controls muscle mass both by
inducing protein synthesis and by inhibiting protein degradation. In AAV9-MITOK injected mice, AKT1 pathway was activated, as demonstrated by the increase in AKT1 phosphorylation and of its
downstream effectors (Fig. 4K, S5). In particular, MITOK-overexpressing muscles showed an increase in the phosphorylation of 4EBP1, an inhibitor of protein translation, inactivated by
AKT-dependent phosphorylation. The phosphorylated form of S6, a positive regulator of protein translation, also increased in MITOK-overexpressing muscles. Finally, GSK3β, a positive
regulator of protein synthesis and AKT target was also hyperphosphorylated in MITOK-overexpressing muscles, suggesting that protein synthesis is active. MITOK OVEREXPRESSION CAUSES MUSCLE
MASS LOSS The complex scenario comprising altered mitochondrial function, activation of mitophagy-related pathways in concert with block of the autophagic flux, and induction of AKT1
pathway, prompt the investigation of the effects of constitutive mitoKATP opening on muscle mass and architecture. We analyzed muscles both four (Fig. 5) and eight (Figure S3A) weeks after
AAV9-MITOK infection to evaluate the progression of the phenotype. MITOK overexpression negatively affected muscle size during post-natal growth. In particular, the reduction in muscle
weight of AAV9-MITOK injected muscles was more than 50% both 4 and 8 weeks after the infection (Fig. 5A-B, S3B), which was due to great decrease of the mean fibre area compared to controls
(Fig. 5C, S3C). Additionally, as demonstrated by H&E (Fig. 5D) and Sirius red (Fig. 5E, S3D) staining, fibrotic area were not detected, suggesting that MITOK overexpression triggers
muscle atrophy as a consequence of a specific reduction in fibre size. MITOK OVEREXPRESSION IN ADULTHOOD TRIGGERS MUSCLE ATROPHY Postnatal growth of skeletal muscle relies on both protein
synthesis and myonuclear accretion. During adulthood, myofibre size depends on protein synthesis/degradation balance, while myonuclear number remains mainly constant [19, 20]. We wished to
understand whether the effects of MITOK overexpression are limited to the growing muscle or rather are detectable also when mitoKATP is constitutively open exclusively during adulthood. To
this aim, EDL muscles of adult mice were infected with AAV9-MITOK and analyzed 2 weeks later (Fig. 6A). MITOK expression was confirmed by western blot analyses (Fig. 6B, S5). In adult
muscles upon MITOK overexpression neither signs of degeneration were present (Fig. 6C), nor SDH staining indicated major mitochondrial dysfunction (Fig. 6D). However, fibre size was
decreased in MITOK-overexpressing muscles compared to controls, although less than in muscles injected with AAV9-MITOK soon after birth (Fig. 6E). In addition, similarly to post-natal
overexpression, MITOK in adulthood altered the expression of mitochondrial markers and caused an increase in p62 and LC3-II protein levels (Fig. 6F, S5). Finally, MITOK overexpression in the
adult muscle triggered the activation of the AKT1 pathway, including phosphorylation of S6 and 4EBP1 (Fig. 6G, S5). In conclusion, MITOK overexpression in adulthood exerted similar effects
compared to post-natal growth, including atrophy, alterations of mitochondria and autophagy protein expression, and activation of AKT1 pathway, indicating that the myofibre is the primary
target of the increased mitochondrial K+ entry. DISCUSSION This represents the first account of the effects of mitoKATP deletion, and thus of impaired mitochondrial K+ fluxes in the living
animal. As previously reported, mitoKATP opening elicits cardioprotection in ischemia/reperfusion injury, and this effect is lost in the hearts of _Mitok_−/− mice. _Mitok__-/-_ animals are
otherwise viable and fertile, with no overt phenotype. Here, we aimed at clarifying whether skeletal muscle is susceptible to the effects of _Mitok_ deletion previously observed in HeLa
cells, including altered mitochondrial cristae width and metabolism, and whether loss of mitochondrial K+ homeostasis exerts any consequence on muscle structure and activity. Skeletal muscle
mitochondria were as susceptible as those of cell lines to the widening effects of _Mitok_ deletion on mitochondrial cristae, and these data were corroborated by a metabolomic analysis
highlighting the role of mitochondria K+ homeostasis on phospholipid, sphingolipid, and cholesterol content, suggestive of membrane remodeling, that may negatively affect muscle contraction,
as previously suggested [21]. Similarly to what observed in cell lines, the defects in mitochondrial shape were causative of altered oxidative metabolism which, together with membrane
remodeling, translated into a reduced running capacity both uphill, testing endurance capacity, and downhill, where also damaging lengthening contractions take place. It is important to
highlight that the diminished exercise performance observed in _Mitok__−/−_ animals on the treadmill may be linked, at least partially, to the disturbance of mitochondrial K+ homeostasis in
tissues beyond skeletal muscle, notably in the heart. To gain a clearer understanding of the specific contribution of skeletal muscle to this reduced performance, strength determinations of
the hindlimb are instrumental. In vivo force measurements in the anesthetized mouse revealed a major role of mitoKATP in preserving muscle force upon repetitive lengthening contractions,
while it was dispensable for single bouts of tetanic force production. Thus, in agreement with the cytoprotective role of mitoKATP, both downhill running and direct force measurements of the
hindlimb highlighted the importance of mitochondrial K+ entry during damaging lengthening contractions. On the other hand, the ATP demand occurring during endurance training requires prompt
mitoKATP-dependent response, while short-term tetanic force production does not require acute mitochondrial K+ entry. These data are in line with the low activity of mitoKATP channels in
the presence of resting levels of cytosolic ATP and unveil the need of mitochondrial volume adjustments to meet the energy requirements of the working muscle. However, it should be noted
that adaptations occurring in constitutive knockout animals may hinder phenotypic manifestations that may instead result from acute gene deletion. Moreover, ischemic preconditioning has been
observed in skeletal muscle [22]. Thus, whether mitoKATP plays a role in the protection of skeletal muscle during hypoxia and reoxygenation that may occur during muscle work remains to be
established. At the opposite side, MITOK overexpression triggers mitochondrial swelling and loss of membrane potential, thus representing an unvaluable genetic tool to study the effects of
mitochondrial poisoning in a specific and tightly controlled manner. MITOK overexpression alters the organelle architecture in muscle and causes energy default with detrimental effects on
organ physiology, highlighted by the dramatic atrophy. As for the signaling pathways involved, mitochondrial stress triggered the accumulation of PINK1 and of polyubiquitinated proteins at
mitochondria, and the activation of the energy sensor AMPK. Mitochondria dysfunction and polyubiquitination of OMM proteins are signals for the recruitment of autophagy receptors and,
together with AMPK induction, are supposed to additionally induce macroautophagy, the degradative system responsible for the removal of bulk aberrant proteins and organelles. However, no
induction of autophagy-related genes is detected in MITOK overexpressing muscles. At the opposite, the autophagy flux is blocked, determining the accumulation of undigested material and
aberrant mitochondria, and further contributing to atrophy in a vicious cycle. Given that autophagy operates continuously within the cell to generate metabolites crucial for sustaining
cellular metabolism, eliminate damaged components, and enhance repair and stress resistance, any disruption in autophagy can lead to muscle degeneration and weakness [12]. Autophagy
deficiency is indeed per se sufficient to trigger loss of muscle mass, as demonstrated by the atrophic phenotype of muscles depleted of the essential autophagy gene Atg7 [23]. Additionally,
the Akt pathway that sustains protein synthesis and hampers protein degradation, is induced. Akt inhibits autophagosome formation, suggesting that, in MITOK-overexpressing muscles, Akt
activation most likely represents a compensatory response, still frustrated, to the detrimental effects of excessive mitochondrial K+ entry. Moreover, polyubiquitinated proteins are
increased, suggesting both an accumulation due to the defective autophagic process, and an augmented delivery of aberrant proteins to the proteasome system. However, mRNA levels of
muscle-specific ubiquitin ligases are unchanged, indicating that a steady-state response to the long-term mitochondrial damage is reached. Thus, excessive accumulation of K+ in mitochondria
results in organelle dysfunction and disruption of the main degradative systems necessary for maintaining mitochondrial health. Similarly, an imbalance in mitochondrial quality control
pathways leads to impaired mitophagy and the buildup of malfunctioning organelles [11]. Therefore, the overexpression of MITOK initiates a conserved catabolic program. This scenario also
involves activation of a cell-stress response transcriptional program. GADD45a is a nuclear protein that has been described as a stress sensor implicated in the regulation of many cellular
functions including DNA repair, cell cycle arrest and apoptosis. In skeletal muscle, GADD45a expression is increased by fasting [13] and immobilization [14] and is required for atrophy
induced by different stimuli [14]. In MITOK-overexpressing muscles, GADD45a mRNA levels were increased, indicating that it may play a primary role. Various mechanisms may be responsible for
GADD45a induction. For example, heterodimers of ATF4 with C/EBPβ mediate muscle atrophy by binding and activating an evolutionary-conserved regulatory element in the Gadd45a gene [24].
Moreover, HDAC4 is upstream of GADD45a in denervation-induced atrophy [25], and additional undetermined factors are also involved. In addition, p53, p21, FANC-L, and the HSP genes HSP27 and
HSP90, involved in growth suppression, DNA repair or stress-response pathways in skeletal muscle, were upregulated, suggesting a coordinated transcriptional response to the primary atrophy
trigger. In skeletal muscle, p53 activates muscle atrophy, while loss of p53 expression partially protects from immobilization-induced atrophy [16]. p21 promotes atrophy by reducing the
expression of spermine oxidase, which is involved in the maintenance of muscle fibre size in physiological conditions [15]. Finally, HSPs represent an important skeletal muscle defense
system induced by a variety of environmental burdens such as oxidative stress, inflammation and damage [18]. Thus, the gene expression profile induced by MITOK overexpression points to a
multi-faceted stress-response pattern. In summary, mitoKATP activity controls skeletal muscle mitochondria volume and cristae shape, impinging on metabolism and energy production.
Accordingly, loss of ATP-regulated mitochondrial K+ entry translates into defective muscle activity, loss of cytoprotection and increased sensitivity to damaging triggers. On the other side,
uncontrolled K+ entry by overexpression of MITOK leads to the dissipation of the electrochemical gradient for protons, resulting in mitochondrial damage and triggering a cellular response
that involves the activation of specific signaling pathways responsible for muscle mass loss. Overall, the fine-tuned modulation of mitoKATP activity is required for the maintenance of
skeletal muscle homeostasis and function. Thus, our study not only validates prior evidence regarding the role of mitoKATP in regulating mitochondrial volume but also underscores the
function of mitoKATP-dependent potassium flux in skeletal muscle pathophysiology. Further studies are required to determine the regulation of MitokATP activity in diverse atrophy conditions,
such as disuse, denervation, cachexia, or myopathies—whether of mitochondrial origin or seemingly unrelated. Additionally, a comprehensive exploration is warranted to determine if
modulating MitoKATP could serve as a therapeutic target in these conditions. Furthermore, future investigations are necessary to unravel the intricate interplay between mitoKATP and other
mitochondrial potassium channels in various scenarios of skeletal muscle activity and disease. This includes elucidating whether their activities synergize in controlling potassium fluxes
and understanding the mechanisms of their modulation. MATERIALS AND METHODS ANIMALS _Mitok__−/−_ mice were generated by genOway on a C57BL/6 N background. Two LoxP sites flanking exon 4 of
the mouse _Mitok_ gene were introduced by homologous recombination. The genotype was verified by PCR with the following primers: Mitok knockout fw 1 GCACCTTGTCAGCACCATGACAACTC Mitok knockout
fw 2 GAGGGATCGCTGTGGAAGGCTGTAT Mitok knockout rv GCGGACAAAGATTGTGTCACTGTTTGC The knockout allele yields an amplification product of 769 bp, whereas the wild-type allele generates a 278-bp
fragment. CD1 mice was provided by Charles River. All mouse experiments were performed in accordance with the Italian law D. L.vo n°26/2014. METABOLOMICS ANALYSIS Gastrocnemius muscles of
adult mice were harvested and underwent untargeted metabolomics analysis performed by Metabolon, Inc. Briefly, samples were prepared using the automated MicroLab STAR® system from Hamilton
Company. The resulting extract was divided into five fractions: two for analysis by two separate reverse phase (RP)/ultra-performance liquid chromatography tandem mass spectrometry
(UPLC-MS/MS) methods with positive ion mode electrospray ionization (ESI), one for analysis by RP/UPLC-MS/MS with negative ion mode ESI, one for analysis by hydrophilic interaction liquid
chromatography/UPLC-MS/MS with negative ion mode ESI, and one sample was reserved for backup. MITOCHONDRIAL MEMBRANE POTENTIAL (ΔΨM) MEASUREMENTS FDB Muscles were digested in collagenase A
(4 mg/ml) (Roche) dissolved in Tyrode’s salt solution (pH 7.4) (Sigma-Aldrich) containing 10% fetal bovine serum (Thermo Fisher Scientific). Single fibres were isolated, plated on
laminin-coated glass coverslips and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 25 mM HEPES (Thermo Fisher Scientific), 10% fetal bovine serum, penicillin (100
U/ml) and streptomycin (100 μg/ml). Fibres were maintained at 37 °C in a humidified incubator with 5% CO2. After 24 h, fibres were incubated with 20 nM tetramethylrhodamine methyl ester
(TMRM, Molecular Probes) in Krebs-Ringer modified buffer (KRB, 135 mM NaCl, 5 mM KCl, 1 mM MgCl2, 20 mM HEPES, 1 mM MgSO4, 0.4 mM KH2PO4, 1 mM CaCl2, 5.5 mM glucose, pH 7.4) for 30 min at 37
°C. During the experiments, myofibres were maintained in KRB at RT in the presence of 75 μM N-benzyl-P-toluenesulfonamide (BTS, Sigma-Aldrich) to avoid fibre contraction. Images were
acquired by confocal microscopy. At the end of each experiment, 10 μM CCCP was added to collapse the ΔΨm. After background correction, the fluorescence value after addition of CCCP was
subtracted for each fibre. Analysis was performed by using the Fiji distribution of Image J [26]. OCR (OXYGEN CONSUMPTION RATE) MEASUREMENTS FDB muscles were digested in collagenase A (4
mg/ml) (Roche) dissolved in Tyrode’s salt solution (pH 7.4) (Sigma-Aldrich) containing 10% fetal bovine serum (Thermo Fisher Scientific). Single fibres were isolated, plated on
laminin-coated XF24 microplate wells and cultured in DMEM (D5030 Sigma-Aldrich), supplemented with 1 mM NaPyr, 5 mM glucose, 33 mM NaCl, 15 mg phenol red, 25 mM HEPES, and 1 mM of L-Glu.
Fibres were maintained for 2 h at 37 °C in 5% CO2. The rate of oxygen consumption was assessed in real-time with the XF24 Extracellular Flux Analyzer (Agilent), which allows to measure OCR
changes after up to four sequential additions of compounds. A titration with the uncoupler CCCP was performed to utilize the CCCP concentration (0.8 μM) that maximally increases OCR. To
calculate basal and maximal respiration, non-mitochondrial O2 consumption was subtracted from absolute values. ATP-linked respiration was calculated as the difference between basal and
oligomycin-insensitive O2 consumption. The results were normalized for the fluorescence of Calcein (Sigma-Aldrich). Fibres were loaded with 2 μM Calcein for 30 min. Fluorescence was measured
using a Perkin Elmer EnVision plate reader in well scan mode using 480/20 nm filter for excitation and 535/20 nm filter for emission. REAL-TIME IMAGING OF MITOCHONDRIAL CA2+ IN FDB FIBRES
FDB muscles were digested in collagenase A (4 mg/ml) (Roche, #COLLA-RO) dissolved in Tyrode’s salt solution (pH 7.4) (Sigma Aldrich, #T2145) containing 10% fetal bovine serum (Thermo
Fisher). Single fibres were isolated, plated on laminin-coated glass coverslips, and cultured in DMEM supplemented with 25 mM HEPES (Thermo Fisher, #42430), 10% fetal bovine serum, 100 U/ml
penicillin and 100 mg/ml streptomycin (Thermo Fisher, #15070063). Fibres were maintained at 37 °C with 5% CO2. During the experiments, myofibres were maintained in Krebs-Ringer modified
buffer (135 mM NaCl, 5 mM KCl, 1 mM MgCl2, 20 mM HEPES, 1 mM MgSO4, 0.4 mM KH2PO4, 1 mM CaCl2, 5.5 mM glucose, pH 7.4) containing 0.02% pluronic acid for 20 min at 37 °C and then washed with
Krebs-Ringer modified buffer in presence of 75 μM N benzyl-P-toluenesulfonamide (Sigma Aldrich, #BL3H160B8B26) to avoid fibre contraction. 30 mM caffeine (Sigma Aldrich, #C0750) was added
to elicit Ca2+ release from intracellular stores. Experiments were performed on a Zeiss Axiovert 200 microscope equipped with a 40×/1.3 N.A. PlanFluor objective. Excitation was performed
with a DeltaRAM V high-speed monochromator (Photon Technology International) equipped with a 75 W xenonarc lamp. Images were captured with a high-sensitivity Evolve 512 Delta EMCCD
(Photometrics). The system is controlled by MetaMorph 892 7.5 (Molecular Devices) and was assembled by Crisel Instruments. To measure mitochondrial Ca2+ uptake, fibres were dissected and
loaded with 2 μM mt-fura-2.3, an optimized version of the recently developed mitochondria targeted fura-2 Ca2+ probe [27]. Images were collected by alternatively exciting the fluorophore at
340 and 380 nm and by recording fluorescence emission through a 515/30 nm band-pass filter (Semrock). Exposure time was set to 100 ms. The acquisition was performed at binning 1 with 200 of
EM gain. Image analysis was performed with Fiji distribution of the ImageJ software [26]. Images were background subtracted. Changes in fluorescence (340/380 nm ratio) was expressed as R.
MOUSE TREADMILL EXPERIMENTS For acute concentric exercise experiments, 3- or 6-month-old mice were acclimated to and trained on a 10° uphill LE8700 treadmill (Harvard apparatus) for 2 days.
On day 1, mice ran for 5 min at 8 m/min and on day 2 mice ran for 5 min at 8 m/min followed by 5 min at 10 m/min. On day 3, mice were subjected to a single bout of running starting at the
speed of 10 m/min. Forty minutes later, the treadmill speed was increased at a rate of 1 m/min every 10 min for a total of 30 min and then increased at the rate of 1 m/min every 5 min until
mice were exhausted. Exhaustion was defined as the point at which mice spent >5 s on the electric shocker without attempting to resume running. Total running time and total running
distance were recorded for each mouse. For eccentric exercise experiments, 3-month-old mice were acclimated to and trained on a 10° downhill treadmill for 2 days. On day 1, mice ran for 5
min at 8 m/min and on day 2 mice ran for 5 min at 8 m/min followed by 5 min at 10 m/min. On days 3–5, mice were subjected to a single bout of running starting at the speed of 10 m/min. Forty
minutes later, the treadmill speed was increased at a rate of 1 m/min every 10 min for a total of 30 min and then increased at the rate of 1 m/min every 5 min until mice were exhausted.
Exhaustion was defined as above. Total running time and total running distance were recorded for each mouse. FORCE MEASUREMENTS Muscle function in vivo was assessed for the gastrocnemius
muscle, as described previously [28]. Briefly, mice were anesthetized with a mixture of tiletamine/zolazepam (40 mg/kg) and xilazine (7 mg/kg) and the foot was mounted on a 305B muscle lever
system (Aurora Scientific, ON Canada). A lever arm of 2,1 mm was used for all groups, as no major differences in body weight between various groups was observed. Electrodes were then placed
on either side of the sciatic nerve, while the common peroneal nerve was cut. The knee was blocked, and an electrical stimulation was applied to the sciatic nerve, inducing the isometric
plantar flexion of the foot. The force-frequency curve was obtained by stimulating at increasing frequencies (starting with a single depolarization up to 150 Hz). Force was then normalized
to the weight of gastrocnemius and plantaris muscles, as an estimate of specific force. Eccentric contractions were performed by moving the foot backward at a velocity of 40 mm/s while the
gastrocnemius was stimulated with a frequency sufficient to induce full tetanic fusion (100 Hz). Contractions were repeated once every 20 seconds to void inducing fatigue. Experimental data
were analyzed using a self-compiled program in LabView. AAV9-MITOK ADMINISTRATION AAV9-MITOK was purchased from Vector Biolabs. For experiments in the newborn, 1010 vg were injected into the
hindlimb of 4- to 6-day-old CD1 male mice. Muscles were subsequently analyzed 4- or 8 weeks postinjection as reported in the Result section. An average of 70% of fibres were positive for
the AAV infections. For experiments in the adult, CD1 male mice were used. EDL muscles were isolated through a small hindlimb incision, and 1010 vg were injected along the muscle length. FDB
muscles were injected with 5 ×109 vg. Muscles were analyzed 15 days post-injection. AUTOPHAGY FLUX ANALYSIS Autophagic flux analysis was performed as previously described [29] with some
modifications. Hindlimbs muscle of newborn mice were injected with AAV9-MITOK. Colchicine was dissolved in water and stored at -20 °C, as a stock solution, at a concentration 4 mg/ml. On the
day of treatment, colchicine was diluted to 0.1 mg/ml in water and 0.1 mg/Kg colchicine was i.p injected. Control mice received an equal volume of NaCl 0.9%. The treatment was repeated 12
hours after the first injection. Mice were sacrificed 24 hours after the first injection, TA muscles were harvested and frozen in liquid nitrogen-cooled isopentane. RNA EXTRACTION, REVERSE
TRANSCRIPTION, AND QUANTITATIVE REAL-TIME PCR Total RNA was extracted from TA muscles using SV Total RNA isolation kit (Promega) following the manufacturer’s instructions. The RNA was
quantified with Nanodrop (Thermo Fisher Scientific). From 400 ng of total RNA of each sample, complementary DNA was generated with a cDNA synthesis kit (SuperScript II, Thermo Fisher
Scientific) and analysed by real-time PCR using IQ5 thermocycler and SYBR green chemistry (Bio-Rad). The primers were designed and analysed with Primer3 [30]. Real-time PCR standard curves
were constructed by using serial dilution of cDNAs of the analysed samples, using at least four dilution points and the efficiency of all primer sets was between 85 and 102%. Actin was used
as an internal control for cDNA quantification and normalization of the amplified products. Real-time PCR primer sequences were as follow: Atrogin-1: FW: GCAAACACTGCCACATTCTCTC RV:
CTTGAGGGGAAAGTGAGACG MuRF-1: FW: CCTTCCTCTCAAGTGCCAAG RV: CCTCAAGGCCTCTGCTATGT Mul-1: FW: AGGGCATTCTTTCAGAAGCA RV: GGGGTGGAACTTCTCGTACA Traf6: FW: GCAGTGAAAGATGACAGCGTGA REV:
TCCCGTAAAGCCATCAAGCA Fanc-L: FW: GCACGCAGGATTGCATTAGG RV: GCTACCACTCAGCTTCATTCC p21: FW: CGGTGTCAGAGTCTAGGGGAA RV: GAACAGGTCGGACATCACCA P53: FW: GGCGTAAACGCTTCGAGATG
RV:TCAGGTAGCTGGAGTGAGCCGADD45α: FW: GAAAGTCGCTACATGGATCAGT RV: AAACTTCAGTGCAATTTGGTTC HSP27: FW: TGACCCAGGCTGGAGTAGAA RV: TGGCTCGGGACAACAACATT HSP90: FW: GGAGATTTTCCTCCGCGAGT RV:
GTCATGCCAATGCCTGTGTC MITOCHONDRIA FRACTIONATION TA and EDL muscles (approx. 250 mg of tissue) were minced and homogenized with a motor-driven pestle (20 strokes) in an isotonic buffer (mIB-
mitochondrial Isolation Buffer - 250 mM sucrose, 10 mM KCl, 20 mM HEPES, 1 mM EDTA, pH 7.4). The total homogenate was centrifugated at 1000× _g_ for 5 min to remove nuclei and entire cells.
The supernatant was then centrifuged at 10000×g for 10 min to pellet the crude mitochondrial fraction. The mitochondrial fraction was washed twice with mIB and centrifuged for 10 min at
10000xg after each washing. The second supernatant was further centrifuged at 40,000× _g_ for 20 min to remove light membranes and clear the cytosolic fraction. Proteins of the total
homogenate, of the mitochondria fraction, and of the cytosolic fraction were extracted in RIPA buffer (150 mM NaCl, 50 mM TRIS, 1% Triton-X100, 0.1% Na-deoxycholate and 0.1% SDS, pH 7.4) and
quantified using the BCA Protein Assay Kit (Thermo Fisher Scientific) following the manufacturer instructions. 20 μg of protein were separated by SDS-PAGE, transferred onto nitrocellulose
membranes and probed using the indicated antibodies. WESTERN BLOTTING AND ANTIBODIES To monitor protein levels, frozen muscles were dissolved by means of Qiagen Tissue Lyser and protein
extracts were prepared in a buffer containing: 50 mM Tris pH 7.50, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 10% glycerol, 2% SDS, 1% Triton X-100, Roche Complete Protease Inhibitor Cocktail, 1 mM
PMSF, 1 mM NaVO3, 5 mM NaF and 3 mM β-glycerophosphate. 40-60 mg of total proteins were loaded, according to BCA quantification. Proteins were separated by SDS-Page electrophoresis, in
commercial 4-12% acrylamide gels (Thermo Fisher Scientific) and transferred onto nitrocellulose membranes (Thermo Fisher Scientific) by semidry electrophoretic transfer. Blots were blocked 1
hour at RT with 5% non-fat dry milk (Bio-Rad) in TBS-tween (0.5 M Tris, 1.5 M NaCl, 0.01% Tween) solution and incubated overnight at 4 °C with primary antibodies. Secondary antibodies were
incubated 1 h at RT. After each antibody incubation, three washes of 10’ each were performed with TBS-0.01% tween. The following antibodies were used: anti-pAKT1 (1:100 Cell Signalling
#4060), anti-Actin (1:10000 Santa Cruz sc-47778, 1:10000 Proteintech 20536-1-AP), anti-LC3B (1:500 Cell Signalling #2775), anti-p62 (1:3000, Sigma P0067), anti-TOM20 (1:20000 Santa Cruz
sc-17764, 1:1000 Cell Signaling #42406), anti-MITOK (1:1000 Sigma HPA010980), anti-GRP75 (1:1000, Santa Cruz sc-133137), anti-IMMT (1:1000, Proteintech 10179-1-AP), anti-COXIV (1:1000, Cell
Signaling #4844), anti-HSP60 (1:1000, Santa Cruz sc-59567), anti-TFAM (1:1000, Abnova H00007019-B01P), anti-Parkin (1:1000, Santa Cruz sc-32282), anti-polyubiquitin (1:1000, Enzo
BLM-PW8810-0400), anti-pS6 (Ser 240/244; 1:1000, Cell Signalling #5364), anti-pGSK3β (Ser9, 1:1000, Cell Signalling #9336), anti-p4EBP1 (Thr 37/46, 1:1000, Cell Signalling #2855), anti-Flag
(1:1000 Cell Signalling #14793). Secondary HRP-conjugated antibodies were purchased from Bio-Rad and used at 1:5000 dilution. To perform the polyubiquitin WB on the subcellular fractions all
the procedures were done in the presence of proteasome inhibitors MG132 (10 µM, Merck) and PR619 (1 µM, Merck). Densitometric analysis was conducted with the Fiji distribution of ImageJ
[26]. Initially, the signal intensity ratio between the protein of interest and the protein used as loading control was quantified. Subsequently, the fold change of the treated muscle versus
its contralateral counterpart was calculated and plotted. IMMUNOFLUORESCENCE For immunohistochemistry analysis and fibre size measurements, AAV9-MITOK injected muscle cryosections were
fixed with 4% FA for 20 minutes, quenched with 50 mM NH4Cl in phosphate-buffer saline (PBS) and blocked in PBS containing 0.5% BSA and 10% goat serum for 20 min. Sections were then incubated
with primary antibodies for 1 h at 37 °C. The following antibodies were used: anti-flag (1:100, Cell Signaling) and anti-p62 (1:100, Sigma). Alexa Fluor 488- or 555-conjugated secondary
antibodies (Thermo Fisher Scientific) were used. For the detection of sarcolemma, WGA antibody (1:1000) was used, while Hoechst was used to mark nuclei. Fibre size measurements were
performed with the Fiji distribution of ImageJ [26]. HEMATOXYLIN AND EOSIN STAINING Hematoxylin and Eosin (H&E) staining was performed on 20 µm-thick cryosections of TA and EDL muscles.
The staining was carried out using the Rapid Frozen Sections H&E staining Kit (Bio-Optica). SIRIUS RED STAINING Sirius Red staining was performed on 20 µm-thick cryosections of EDL
muscles. Sections were incubated overnight at RT in a Bouin Solution (saturated picric acid, 37% formaldehyde, 5% acetic acid). After washing in running water, sections were incubated in a
solution of 0.1% Direct Red 80 in saturated picric acid for 1 h at RT. After washing with 2% acetic acid, sections were dehydrated in sequence with 40-50% ethanol, 70% ethanol, 96% ethanol,
100% ethanol, 1:1 ethanol-xylene (100%), 100% xylene. Slides were mounted with Eukitt (Sigma). SDH STAINING 20 µm-thick cryosections of TA, EDL and soleus muscles were stained with SDH
incubating solution, prepared by the addition to SDH stock solution (0.2 M Na+-succinate, 0.2 M Phosphate buffer pH 7.4) of nitroblue tetrazolium (final concentration 1 mg/mL), for 20
minutes at 37 °C. Sections were rinsed three times in water and assembled with Mowiol. ELECTRON MICROSCOPY EDL muscles were fixed with a fixative solution (3.5% glutaraldehyde in 0.1 M
NaCaCo buffer, pH 7.2) at RT. Small bundles of fibres were post-fixed in 2% OsO4 in the same buffer for 2 h and then block-stained in aqueous saturated uranyl acetate. After dehydration,
specimens were embedded in an epoxy resin (Epon 812). Ultrathin sections (approximately 30-40 nm) were stained in 4% uranyl acetate and lead citrate. STATISTICAL ANALYSIS OF DATA The sample
size was determined based on similar experiments. Depending on the number of expected comparisons per experimental group, the type I error α was set between 0.55% and 5% bilaterally. The
required number of animals is the minimum number needed to achieve a test power of 80%. Statistical data are expressed as mean ± SD unless otherwise specified. For comparison between two
independent groups, unpaired t tests were used. Adjusted *_p_ < 0.05, **_p_ < 0.01, and ***_p_ < 0.001. For animal studies, randomization was applied, no blinding was done. DATA
AVAILABILITY The datasets generated and analysed during the current study are available from the corresponding author on reasonable request. REFERENCES * Checchetto V, Leanza L, De Stefani
D, Rizzuto R, Gulbins E, Szabo I. Mitochondrial K+ channels and their implications for disease mechanisms. Pharm Ther. 2021;227:107874. Article CAS Google Scholar * Malinska D, Mirandola
SR, Kunz WS. Mitochondrial potassium channels and reactive oxygen species. FEBS Lett. 2010;584:2043–8. Article CAS PubMed Google Scholar * Kowaltowski AJ, Seetharaman S, Paucek P, Garlid
KD. Bioenergetic consequences of opening the ATP-sensitive K(+) channel of heart mitochondria. Am J Physiol Heart Circ Physiol. 2001;280:H649–57. Article CAS PubMed Google Scholar *
Minners J, Van Den Bos EJ, Yellon DM, Schwalb H, Opie LH, Sack MN. Dinitrophenol, cyclosporin A, and trimetazidine modulate preconditioning in the isolated rat heart: Support for a
mitochondrial role in cardioprotection. Cardiovasc Res. 2000;47:68–73. Article CAS PubMed Google Scholar * Paggio A, Checchetto V, Campo A, Menabò R, Di Marco G, Di Lisa F, et al.
Identification of an ATP-sensitive potassium channel in mitochondria. Nature. 2019;572:609–13. Article CAS PubMed PubMed Central Google Scholar * Debska G, Kicinska A, Skalska J,
Szewczyk A, May R, Elger CE, et al. Opening of potassium channels modulates mitochondrial function in rat skeletal muscle. Biochim et Biophys Acta (BBA) - Bioenerg. 2002;1556:97–105. Article
CAS Google Scholar * Montoya-Pérez R, Saavedra-Molina A, Trujillo X, Huerta M, Andrade F, Sánchez-Pastor E, et al. Inhibition of oxygen consumption in skeletal muscle-derived
mitochondria by pinacidil, diazoxide, and glibenclamide, but not by 5-hydroxydecanoate. J Bioenerg Biomembr. 2010;42:21–27. Article PubMed Google Scholar * García MC, Hernández A, Sánchez
JA. Role of mitochondrial ATP-sensitive potassium channels on fatigue in mouse muscle fibers. Biochem Biophys Res Commun. 2009;385:28–32. Article PubMed Google Scholar * Sánchez-Duarte
E, Trujillo X, Huerta M, Ortiz-Mesina M, Cortés-Rojo C, Manzo-Ávalos S, et al. Mitochondrial KATP channels in skeletal muscle: are protein kinases C and G, and nitric oxide synthase involved
in the fatigue process? Open Access Anim Physiol. 2012;4:21–28. Google Scholar * Luévano-Martínez LA, Kowaltowski AJ. Phosphatidylglycerol-derived phospholipids have a universal,
domain-crossing role in stress responses. Arch Biochem Biophys. 2015;585:90–97. Article PubMed Google Scholar * Romanello V, Sandri M. The connection between the dynamic remodeling of the
mitochondrial network and the regulation of muscle mass. Cell Mol Life Sci. 2020;78:1305–28. Article PubMed PubMed Central Google Scholar * Sartori R, Romanello V, Sandri M. Mechanisms
of muscle atrophy and hypertrophy: implications in health and disease. Nat Commun 2021 12:1. 2021;12:1–12. Google Scholar * Ebert SM, Monteys AM, Fox DK, Bongers KS, Shields BE, Malmberg
SE, et al. The Transcription Factor ATF4 Promotes Skeletal Myofiber Atrophy during Fasting. Mol Endocrinol. 2010;24:790–9. Article CAS PubMed PubMed Central Google Scholar * Ebert SM,
Dyle MC, Kunkel SD, Bullard SA, Bongers KS, Fox DK, et al. Stress-induced Skeletal Muscle Gadd45a Expression Reprograms Myonuclei and Causes Muscle Atrophy. J Biol Chem. 2012;287:27290–301.
Article CAS PubMed PubMed Central Google Scholar * Bongers KS, Fox DK, Kunkel SD, Stebounova LV, Murry DJ, Pufall MA, et al. Spermine oxidase maintains basal skeletal muscle gene
expression and fiber size and is strongly repressed by conditions that cause skeletal muscle atrophy. Am J Physiol Endocrinol Metab. 2015;308:E144–E158. Article CAS PubMed Google Scholar
* Fox DK, Ebert SM, Bongers KS, Dyle MC, Bullard SA, Dierdorff JM, et al. p53 and ATF4 mediate distinct and additive pathways to skeletal muscle atrophy during limb immobilization. Am J
Physiol Endocrinol Metab. 2014;307:E245–E261. Article CAS PubMed PubMed Central Google Scholar * Thada V, Greenberg RA. Unpaved roads: How the DNA damage response navigates endogenous
genotoxins. DNA Repair (Amst). 2022;118:103383. Article CAS PubMed PubMed Central Google Scholar * Fennel ZJ, Amorim FT, Deyhle MR, Hafen PS, Mermier CM. The heat shock connection:
skeletal muscle hypertrophy and atrophy. Am J Physiol Regul Integr Comp Physiol. 2022;323:R133–R148. Article CAS PubMed Google Scholar * Cramer AAW, Prasad V, Eftestøl E, Song T, Hansson
KA, Dugdale HF, et al. Nuclear numbers in syncytial muscle fibers promote size but limit the development of larger myonuclear domains. Nat Commun. 2020;11:6287. Article CAS PubMed PubMed
Central Google Scholar * Hansson KA, Eftestøl E, Bruusgaard JC, Juvkam I, Cramer AW, Malthe-Sørenssen A, et al. Myonuclear content regulates cell size with similar scaling properties in
mice and humans. Nat Commun. 2020;11:6288. Article CAS PubMed PubMed Central Google Scholar * Vega-Moreno J, Tirado-Cortes A, Álvarez R, Irles C, Mas-Oliva J, Ortega A. Cholesterol
Depletion Uncouples β-dystroglycans from Discrete Sarcolemmal Domains, Reducing the Mechanical Activity of Skeletal Muscle. Cell Physiol Biochem. 2012;29:905–18. Article CAS PubMed Google
Scholar * Kohin S, Stary CM, Howlett RA, Hogan MC. Preconditioning improves function and recovery of single muscle fibers during severe hypoxia and reoxygenation. Am J Physiol-Cell
Physiol. 2001;281:C142–C146. Article CAS PubMed Google Scholar * Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, et al. Autophagy is required to maintain muscle mass. Cell
Metab. 2009;10:507–15. Article CAS PubMed Google Scholar * Ebert SM, Bullard SA, Basisty N, Marcotte GR, Skopec ZP, Dierdorff JM, et al. Activating transcription factor 4 (ATF4)
promotes skeletal muscle atrophy by forming a heterodimer with the transcriptional regulator C/EBPβ. J Biol Chem. 2020;295:2787–803. Article CAS PubMed PubMed Central Google Scholar *
Bongers KS, Fox DK, Ebert SM, Kunkel SD, Dyle MC, Bullard SA, et al. Skeletal muscle denervation causes skeletal muscle atrophy through a pathway that involves both Gadd45a and HDAC4. Am J
Physiol Endocrinol Metab. 2013;305:907–15. Article Google Scholar * Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for
biological-image analysis. Nat Methods. 2012;9:676–82. Article CAS PubMed Google Scholar * Pendin D, Norante R, De Nadai A, Gherardi G, Vajente N, Basso E, et al. A Synthetic Fluorescent
Mitochondria-Targeted Sensor for Ratiometric Imaging of Calcium in Live Cells. Angew Chem Int Ed. 2019;58:9917–22. Article CAS Google Scholar * Blaauw B, Mammucari C, Toniolo L, Agatea
L, Abraham R, Sandri M, et al. Akt activation prevents the force drop induced by eccentric contractions in dystrophin-deficient skeletal muscle. Hum Mol Genet. 2008;17:3686–96. Article CAS
PubMed Google Scholar * Ju JS, Varadhachary AS, Miller SE, Weihl CC. Quantitation of ‘autophagic flux’ in mature skeletal muscle. Autophagy. 2010;6:929–35. Article CAS PubMed PubMed
Central Google Scholar * Rozen S, Skaletsky H Primer3 on the WWW for General Users and for Biologist Programmers. In: _Bioinformatics Methods and Protocols_. Humana Press: New Jersey,
2000, pp 365–86. Download references ACKNOWLEDGEMENTS This research was supported with funding from the Italian Ministry of University and Research (PRIN 2022B32SCL to C.M., PRIN
2020R2BP2E_002 to R.R., PRIN 2017YF9FBS and 2020RRJP5L to D.D.S.), the French Muscular Dystrophy Association (22493 and 24863 to C.M.), the Italian Ministry of Health (RF-2016-02363566 to
R.R.), the CARIPARO foundation (to R.R. and to D.D.S.), the Veneto Region (POR FESR to C.M.), the University of Padova (BIRD161124, STARS mitoPOC to D.D.S., BIRD222201 to G.G), the CARIPLO
foundation (to D.D.S.). The authors are grateful to Federico Caicci for electron microscopy and to Vanina Romanello and Tito Calì for critical discussion. AUTHOR INFORMATION Author notes *
These authors contributed equally: Giulia Di Marco, Gaia Gherardi. AUTHORS AND AFFILIATIONS * Department of Biomedical Sciences, University of Padova, Padova, Italy Giulia Di Marco, Gaia
Gherardi, Agnese De Mario, Ilaria Piazza, Bert Blaauw, Rosario Rizzuto, Diego De Stefani & Cristina Mammucari * Biozentrum, University of Basel, Basel, Switzerland Martina Baraldo *
Department of Pharmaceutical and Pharmacological Sciences, University of Padova, Padova, Italy Andrea Mattarei * Venetian Institute of Molecular Medicine, Padova, Italy Bert Blaauw * Myology
Center (CIR-Myo), University of Padova, Padova, Italy Bert Blaauw, Rosario Rizzuto & Cristina Mammucari Authors * Giulia Di Marco View author publications You can also search for this
author inPubMed Google Scholar * Gaia Gherardi View author publications You can also search for this author inPubMed Google Scholar * Agnese De Mario View author publications You can also
search for this author inPubMed Google Scholar * Ilaria Piazza View author publications You can also search for this author inPubMed Google Scholar * Martina Baraldo View author publications
You can also search for this author inPubMed Google Scholar * Andrea Mattarei View author publications You can also search for this author inPubMed Google Scholar * Bert Blaauw View author
publications You can also search for this author inPubMed Google Scholar * Rosario Rizzuto View author publications You can also search for this author inPubMed Google Scholar * Diego De
Stefani View author publications You can also search for this author inPubMed Google Scholar * Cristina Mammucari View author publications You can also search for this author inPubMed Google
Scholar CONTRIBUTIONS G.D.M., G.G., A.D.M. and I.P. performed experiments, M.B. performed force measurements, A.M. designed and performed synthesis of mt-fura2.3 probe, B.B. supervised
force measurements, R.R., D.D.S. and C.M. conceived the research, C.M. and G.G. wrote the paper, C.M. directed the research. All authors analyzed the data and reviewed the manuscript.
CORRESPONDING AUTHOR Correspondence to Cristina Mammucari. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ETHICS APPROVAL AND CONSENT TO PARTICIPATE Not
applicable. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Edited by Professor
Paolo Pinton SUPPLEMENTARY INFORMATION SUPPLEMENTAL MATERIAL RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License,
which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link
to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless
indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or
exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints
and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Di Marco, G., Gherardi, G., De Mario, A. _et al._ The mitochondrial ATP-dependent potassium channel (mitoKATP) controls skeletal muscle
structure and function. _Cell Death Dis_ 15, 58 (2024). https://doi.org/10.1038/s41419-024-06426-x Download citation * Received: 19 June 2023 * Revised: 20 December 2023 * Accepted: 02
January 2024 * Published: 17 January 2024 * DOI: https://doi.org/10.1038/s41419-024-06426-x SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative