- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The cyclin-dependent kinase inhibitor p27Kip1 (p27) has been involved in promoting autophagy and survival in conditions of metabolic stress. While the signaling cascade upstream of
p27 leading to its cytoplasmic localization and autophagy induction has been extensively studied, how p27 stimulates the autophagic process remains unclear. Here, we investigated the
mechanism by which p27 promotes autophagy upon glucose deprivation. Mouse embryo fibroblasts (MEFs) lacking p27 exhibit a decreased autophagy flux compared to wild-type cells and this is
correlated with an abnormal distribution of autophagosomes. Indeed, while autophagosomes are mainly located in the perinuclear area in wild-type cells, they are distributed throughout the
cytoplasm in p27-null MEFs. Autophagosome trafficking towards the perinuclear area, where most lysosomes reside, is critical for autophagosome–lysosome fusion and cargo degradation. Vesicle
trafficking is mediated by motor proteins, themselves recruited preferentially to acetylated microtubules, and autophagy flux is directly correlated to microtubule acetylation levels. p27−/−
MEFs exhibit a marked reduction in microtubule acetylation levels and restoring microtubule acetylation in these cells, either by re-expressing p27 or with deacetylase inhibitors, restores
perinuclear positioning of autophagosomes and autophagy flux. Finally, we find that p27 promotes microtubule acetylation by binding to and stabilizing α-tubulin acetyltransferase (ATAT1) in
glucose-deprived cells. ATAT1 knockdown results in random distribution of autophagosomes in p27+/+ MEFs and impaired autophagy flux, similar to that observed in p27−/− cells. Overall, in
response to glucose starvation, p27 promotes autophagy by facilitating autophagosome trafficking along microtubule tracks by maintaining elevated microtubule acetylation via an
ATAT1-dependent mechanism. SIMILAR CONTENT BEING VIEWED BY OTHERS ER PROTEINS DECIPHER THE TUBULIN CODE TO REGULATE ORGANELLE DISTRIBUTION Article Open access 15 December 2021 ATP-CITRATE
LYASE PROMOTES AXONAL TRANSPORT ACROSS SPECIES Article Open access 07 October 2021 P27 CONTROLS RAGULATOR AND MTOR ACTIVITY IN AMINO ACID-DEPRIVED CELLS TO REGULATE THE AUTOPHAGY–LYSOSOMAL
PATHWAY AND COORDINATE CELL CYCLE AND CELL GROWTH Article 17 August 2020 INTRODUCTION p27Kip1 (p27) is a cell cycle inhibitor that restrains cell proliferation by binding to cyclin/CDK
complexes and inhibiting their activity1. In this way, p27 acts as a tumor suppressor and loss of p27 expression in mice promotes tumor development2,3. In many cancers, oncogenic kinases
phosphorylate p27, leading to its cytoplasmic localization, which inactivates its tumor suppressor functions4,5,6,7. Increasing evidence indicates that cytoplasmic localization of p27
confers resistance to anticancer therapies and negatively affects cancer patient prognosis7,8. p27 may sustain cancer progression via several mechanisms, including the regulation of cell
migration and invasion9,10,11,12, stemness13,14, and autophagy15,16,17,18,19,20,21. Macroautophagy (hereafter autophagy) is a physiological self-digestion process in which intracellular
components are sequestrated within double-membrane autophagosomes and delivered to lysosomes for degradation22. In normal conditions, autophagy occurs at basal levels to keep cells healthy
by preventing the accumulation of damaged organelles and proteins. During stress, such as starvation, hypoxia, ionizing radiations, or chemotherapeutic treatment, autophagy levels
dramatically increase, which is considered as a self-defense mechanism22. In the context of cancer, autophagy is thought to initially prevent tumor development by participating in the
maintenance of cytoplasm quality and genome integrity. In contrast, in established tumors, autophagy sustains proliferation and survival by generating energy and substrates for anabolic
reactions23. Upon energetic stress, 5’-AMP-activated protein kinase (AMPK) phosphorylates the ATG1/ULK1 kinase to initiate autophagy. Then, a subset of ATG (autophagy-related) proteins is
recruited to the site of autophagosome formation on the endoplasmic reticulum membrane. ATG proteins orchestrate all steps of autophagy and are used as markers to monitor this process.
Transcriptional regulation and post-translational modifications of ATG proteins play a crucial role in the control of autophagy24. Additionally, other factors such as cytoskeleton and
membrane dynamics regulate autophagy under basal and stress conditions25. Membranes play an important role in the early steps of autophagy as a source of lipid for expanding autophagosomes.
On the other hand, the actin, intermediate filament, and microtubule (MT) cytoskeleton have all been reported to contribute to both early and late stages of autophagy26. During autophagosome
formation, MT act as a scaffold for the assembly of the autophagy machinery and for signaling proteins that regulate the extent of the autophagic response25,26. Once formed, autophagosomes
are transported along MT tracks towards the minus-end of MTs and the microtubule-organizing center (MTOC), where most lysosomes reside26,27,28. This retrograde transport is mediated by
dyneins27,28. However, kinesin-dependent plus-end movement also appears to be needed, as autophagosomes accumulate and fail to fuse with lysosomes in KIF5B-depleted cells29. This is in
agreement with other reports indicating that autophagosomes move bidirectionally along MTs to finally accumulate in the vicinity of the MTOC30. MT acetylation was shown to promote the
recruitment of motor proteins to MT and intracellular trafficking25,31,32, as well as autophagosome–lysosome fusion and autophagy33,34,35,36. Nevertheless, how MT acetylation is controlled
during autophagy is still unclear. p27 was recently implicated in the promotion of basal and stress-induced autophagy15,16,17,18,19,20,21. Following glucose and/or serum deprivation, LKB1
activates AMPK, which in turn phosphorylates p27 on T198, T170 and/or S83, causing its cytoplasmic retention and promoting autophagy17,37. Expression of a p27 mutant localizing in the
cytoplasm was sufficient to promote autophagy17. Upon glucose deprivation, p27-mediated autophagy confers resistance to apoptosis, suggesting that p27 plays an important role in the cellular
response to energetic stress17. While the pro-autophagic role of p27 is well established, little is known about the molecular mechanism by which p27 favors autophagy upon starvation. Here,
we investigated how p27 promotes autophagy in response to glucose starvation. Our data indicate that cytoplasmic p27 facilitates autophagy by promoting the transport of autophagosomes
towards the MTOC and their fusion with lysosomes. At the molecular level, p27 promotes MT acetylation by binding to and stabilizing ATAT1, an enzyme responsible for MT acetylation.
Glucose-deprived p27 knockout cells exhibit an autophagosome positioning defect, impaired autophagy flux, and low MT acetylation levels due to reduced ATAT1 expression. Increasing MT
acetylation by treatment with HDAC inhibitors or re-expression of p27 rescues the autophagy defect of p27-null MEFs. Taken together, these data reveal the molecular mechanism by which p27
promotes autophagy in response to glucose starvation. RESULTS P27 PROMOTES AUTOPHAGY DURING GLUCOSE DEPRIVATION p27 was previously reported to promote autophagy and cell survival in response
to glucose and/or serum deprivation17. To investigate the mechanism underlying the role of p27 in autophagy, p27+/+ and p27−/− mouse embryo fibroblasts (MEFs) were glucose-deprived and
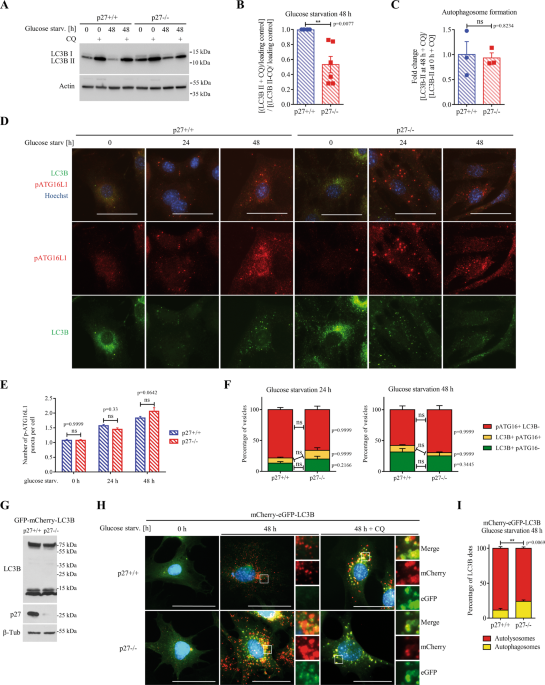
autophagy was evaluated by monitoring ATG8/MAP1LC3/LC3B (hereafter LC3B) levels. Since autophagy is a highly dynamic process and LC3B undergoes rounds of synthesis and degradation, LC3B-II
levels were measured in the presence and absence of chloroquine (CQ), which blocks lysosomal degradation, to determine autophagy flux, represented by the ratio between LC3B-II levels in the
presence and absence of CQ38. Upon glucose deprivation, autophagic flux was significantly lower in p27−/− MEFs compared to wild-type cells (Fig. 1A, B). The overall expression of LC3B may
also be used as a marker of autophagy flux38. Monitoring of turboGFP (tGFP) fluorescence signal in cells expressing tGFP-LC3B with an IncuCyte showed an accumulation of LC3B signal in p27−/−
MEFs during glucose deprivation (Fig. S1A, B), suggesting reduced LC3B turnover and impaired autophagy flux. p62 is known as a selective substrate of autophagy and as such, its accumulation
during starvation is a good indicator of impaired autophagy flux38. After 48 h of glucose starvation, p62 levels, monitored by immunofluorescence (Fig. S1C, D) and immunoblot (Fig. S1E, F),
were lower in p27+/+ MEFs compared to p27−/− cells, supporting the idea that p27 promotes autophagy and that autophagy flux is partially impaired in the absence of p27. A series of
experiments was then performed to determine which step of the autophagic process is affected by p27. First, autophagosome formation was assessed by measuring LC3B-II levels at 0 h and 48 h
of glucose starvation in the presence of CQ to block lysosomal proteolysis39 and similar levels were detected in p27+/+ and p27−/− MEFs (Fig. 1C), indicating that p27 does not affect
autophagosome biogenesis governed by ATG proteins. This was confirmed by monitoring ATG16L1 phosphorylation on S278 and LC3B signal that allows distinguishing between newly formed
autophagophores (p-ATG16L1+, LC3B−), expanding autophagosomes (p-ATG16L1+, LC3B+) and sealed/mature autophagosomes (p-ATG16L1-, LC3B+), which did not show any significant difference between
p27+/+ and p27−/− cells (Fig. 1D–F)40. Similarly, the knockout of p27 did not affect the number and nature of ATG5-positive vesicles (Fig. S1A–C), which represent early autophagic
structures41,42, suggesting that p27 is dispensable for early autophagy events. To distinguish between autophagosomes and autolysosomes, we used MEFs stably expressing tandem fluorescent
mCherry-eGFP-LC3B (Fig. 1G), in which autophagosomes appear as yellow puncta due to mCherry and GFP fluorescence, while autolysosomes appear in red as a result of GFP fluorescence quenching
in the acidic environment of the lysosomal lumen43. Upon glucose deprivation, p27−/− MEFs exhibited less red autolysosome puncta than p27+/+ cells, suggesting defective autophagosome
maturation in the absence of p27 (Fig. 1H, I). Glucose deprivation leads to AMPK activation and the subsequent phosphorylation of p27, causing its cytoplasmic retention and
stabilization17,20,37,44. In agreement with these reports, AMPK was activated in glucose-deprived MEFs (Fig. 2A) and p27 levels slightly increased, although not significantly (Fig. 2B, C).
Similarly, monitoring p27 subcellular localization by immunofluorescence showed a progressive relocalization of p27 in the cytoplasm and an increase in the cytoplasmic to nuclear ratio of
p27 after 48 h of glucose starvation (Fig. 2D, E). Thus, prolonged glucose deprivation induces p27 relocalization to the cytoplasm, which promotes autophagy. P27 REGULATES AUTOPHAGOSOME
POSITIONING IN GLUCOSE-DEPRIVED CELLS The delivery of autophagosomes to the perinuclear area is crucial for their fusion with lysosomes and for cargo degradation25,30,33. Remarkably, the
distribution of LC3B+ vesicles was different in p27−/− cells compared to p27+/+ MEFs (Fig. 3A). While LC3B+ vesicles were predominantly juxtanuclear in over 90% of p27+/+ MEFs, it was the
case in only about a third of p27−/− cells (Fig. 3B). Instead, in p27-null MEFs, LC3B+ vesicles were distributed throughout the cytoplasm (Fig. 3A). To confirm the effect of p27 on LC3B+
vesicle positioning, p27 was re-expressed in p27−/− MEFs (Fig. 3C) and the presence of p27 restored a predominantly perinuclear localization of LC3B+ vesicles in glucose-starved cells (Fig.
3D, E). Taken together, these data suggest that p27 regulates autophagosome localization during glucose deprivation and that defective autophagosome movement in p27−/− cells impairs
autophagosome–lysosome fusion. We recently found that in response to amino acid deprivation, p27 promotes autophagy by interfering with the assembly of the Ragulator complex, thereby
participating in mTOR inhibition21. The lysosome-associated multiprotein complex BORC mediates MT-dependent trafficking of autophagosomes and lysosomes during autophagy and
autophagosome–lysosome fusion45,46. Importantly, upon nutrient depletion, Ragulator inhibits BORC and promotes the juxtanuclear positioning of vesicles, thus facilitating
autophagosome–lysosome fusion47,48. We therefore tested whether p27-mediated inhibition of Ragulator on lysosomes may contribute to the regulation of vesicle positioning in glucose-deprived
cells. First, inhibition of mTOR signaling was confirmed in glucose deprived p27+/+ and p27−/− MEFs (Fig. S3A, B). In these conditions, p27−/− cells did not maintain elevated mTOR activity
as observed under amino acid starvation21. In fact, p27+/+ cells display increased p70 S6K1 phosphorylation upon glucose withdrawal compared to p27−/− cells, which is associated with
autophagy-related mTOR re-activation49, as indicated by the inhibition of mTORC1 activity following CQ treatment in p27+/+ cells (Fig. S3, A, B). Analysis of LC3B+ vesicle localization in
glucose-starved p27+/+ and p27−/− MEFs transfected with LAMTOR1 siRNA (Figure S3C) revealed that LAMTOR1 silencing increased the peripheral distribution of autophagosomes in p27+/+ cells, as
previously reported47,48, but had no effect on autophagosome localization in p27−/− cells (Fig. S3D, E). These results indicate that p27 regulates autophagosome positioning in a
LAMTOR1-independent manner in glucose-deprived cells. MICROTUBULE ACETYLATION DETERMINES AUTOPHAGOSOME POSITIONING AND AUTOPHAGY FLUX Autophagosome trafficking along MT tracks is mediated by
motor proteins27,28,29. The recruitment of motor proteins to MT is controlled by MT stability and their post-translational modifications, such as acetylation at lysine 40 (K40)31,32,50. To
test the importance of the MT network in glucose deprivation-induced autophagy, the correlation between MT acetylation levels, autophagosome positioning and autophagy flux was determined in
p27+/+ MEFs (Fig. 4). Cells were immunostained for LC3B and acetylated-K40 α-tubulin (Ac-α-tubulin). Cells were classified into three categories in function of their MT acetylation status:
low MT acetylation, perinuclear MT acetylation and hyperacetylated MT network (Fig. 4A). Then, the distribution and levels of LC3B+ vesicles were analyzed within each cell population (Fig.
4A, B). The same experiments were performed in the presence of 50 µM CQ to block autophagy and estimate the impact of MT acetylation on autophagy flux (Fig. 4A, C). In cells with MT
acetylation in the perinuclear area, LC3B+ vesicles were also perinuclear and CQ caused a strong accumulation of these vesicles, suggesting that these cells have high autophagy flux (Fig.
4). This is consistent with the idea that autophagosome–lysosome fusion occurs mostly in the proximity of the MTOC30,45. Cells with hyperacetylated tubulin had a low abundance of LC3B+
vesicles and their number dramatically increased upon CQ addition, indicative of a high autophagy flux (Fig. 4). Importantly, LC3B puncta appeared predominantly in the perinuclear zone
following CQ treatment, reinforcing the idea that autophagosomes are delivered in the proximity of the centrosome for efficient fusion with lysosomes. In contrast, cells with low MT
acetylation levels also exhibited low numbers of LC3B+ vesicles, which only slightly increased (mostly in the peripheral area) upon CQ addition, suggesting a low autophagy flux in these
cells (Fig. 4). Taken together, these data suggest that MT acetylation favors autophagosome trafficking towards the perinuclear zone and autophagy flux in glucose-deprived cells. Thus, MT
acetylation does not appear to be necessary for autophagosome–lysosome fusion but increases its efficiency, as previously described51. P27 CONTROLS AUTOPHAGOSOME POSITIONING BY INCREASING
MICROTUBULE ACETYLATION Several reports have described the regulation of MT dynamics by p27, either through direct binding to MT or via the regulation of other proteins11,50,52,53. Although
different mechanisms have been involved, the overall effect of p27 is to stabilize microtubules. Measurement of α-tubulin acetylation at K40 by immunofluorescence (Fig. 5A, B) and immunoblot
(Fig. 5C) indicated that p27 promotes MT acetylation, as previously reported11,50, under both basal and glucose starvation conditions (24 h and 48 h). Furthermore, re-expression of p27 in
p27−/− cells was sufficient to increase MT acetylation (Fig. 5D–F) and to restore the perinuclear localization of autophagosomes in glucose starvation conditions compared to empty vector
infected p27−/− cells (Fig. 5G). Similar results were obtained in glucose-starved primary p27+/+ and p27−/− MEFs in the presence of CQ, with p27+/+ cells exhibiting elevated MT acetylation
levels and perinuclear autophagosome positioning, while p27−/− MEFs had reduced MT acetylation levels and autophagosomes were distributed throughout the cytoplasm (Figure S4). Thus, p27
regulates MT acetylation and impaired LC3B+ vesicle trafficking in glucose starved MEFs appears to be a consequence of lower tubulin acetylation levels in these cells. To determine the
importance of MT acetylation in p27-mediated regulation of glucose starvation-induced autophagy, p27+/+ and p27−/− MEFs were treated with the histone deacetylase (HDAC) inhibitor
Trichostatin A (TSA). TSA was shown to prevent α-tubulin deacetylation by blocking HDAC6 activity54. TSA treatment dramatically increased MT acetylation (Fig. 6A–C), as expected, and induced
the accumulation of LC3B+ vesicle around the nucleus in p27−/− cells (Fig. 6A, D). In addition, while TSA treatment did not affect autophagy rate in full medium (Fig. S5A, B, D, G),
suggesting that microtubule acetylation is dispensable for basal autophagy, TSA rescued impaired autophagosome maturation and autophagy flux in p27−/− cells in glucose-deprivation conditions
(Fig. S5A, C, E, H), without significant effect in p27+/+ MEFs in which tubulin acetylation levels are already elevated. On the other hand, TSA did not rescue the autophagy defect of p27−/−
MEFs upon amino acid starvation (Fig. S5F, I), although it increased autophagy flux in p27+/+ cells, albeit not significantly. Consistent with the positive effect of TSA on autophagosome
maturation, we observed a marked reduction in p62 levels in p27−/− MEFs, indicative of a restored autophagy flux (Fig. S6). Taken together, these data suggest that p27 regulates autophagy
flux via a MT acetylation-dependent mechanism in glucose-deprived cells. P27 INCREASES MICROTUBULE ACETYLATION VIA STABILIZATION OF ATAT1 p27 was recently reported to interact with the MT
acetylating enzyme α-tubulin acetyltranferase (ATAT1) and to increase its stability, which was important in regulating axonal trafficking in cortical and motor neurons50. We confirmed the
physical interaction between p27 and ATAT1 by co-immunoprecipitation (co-IP) experiments (Fig. 7A). Co-transfection of increasing amounts of p27 increased ATAT1 protein levels (Fig. 7B–D),
consistent with p27 stabilizing ATAT150. This was confirmed by cycloheximide treatment to block protein translation and determine the half-life of ATAT1 by measuring ATAT-GFP fluorescence
with an IncuCyte in the presence or absence of p27 expression in HEK 293 cells (Fig. 7E, F). While ATAT1 had a half-life of about 4 h in the absence of p27, it increased to approximately 16
h upon p27 overexpression, as previously reported50. Since ATAT1 regulates MT-dependent axonal transport during neurogenesis50, we tested if the same mechanism was implicated in the
regulation of autophagosome trafficking in glucose starved MEFs. Autophagosome positioning was analyzed in glucose-deprived p27+/+ and p27−/− MEFs transfected with ATAT1 siRNA. Since we do
not have a functional antibody against endogenous ATAT1, siRNA efficiency was verified against a myc-tagged ATAT1 (Fig. 7G). ATAT1 knockdown dramatically reduced MT acetylation (Fig. 7H, I),
as expected, and significantly decreased the localization of LC3B+ vesicles in the perinuclear area in p27+/+ MEFs, whereas it did not affect the already randomly distributed LC3B+ vesicles
in p27−/− cells (Fig. 7H, J). The depletion of ATAT1 in MEFs, validated by monitoring MT acetylation levels (Fig. 8D), resulted in reduced autolysosome formation in p27+/+ cells without
affecting p27−/− cells (Fig. 8A, B), in which autophagosome maturation is already impaired. ATAT1 knockdown did not significantly affect autophagosome maturation in amino acid-deprived cells
(Fig. 8A, C), consistent with the idea that the regulation of autophagy in response to glucose or amino acid starvation by p27 is mediated by distinct mechanisms (Figure S3 and21).
Collectively, these results indicate that in glucose-deprived cells, p27 promotes autophagy by controlling autophagosome positioning via ATAT1-dependent MT acetylation. DISCUSSION Several
lines of evidence indicate that cellular energy status controls p27 levels and localization17,20,37,44,55,56,57. In response to glucose starvation, a fraction of p27 relocalizes to the
cytoplasm and this is accompanied by a mild increase in p27 levels17,20,37,44,56. However, some reports observed p27 degradation following its cytoplasmic relocalization in glucose-deprived
cells55,57. These differences may reflect the use of distinct cellular models and that starvation was performed in low glucose conditions55,57, instead of glucose-free medium and dialyzed
serum as in the present study. The cytoplasmic accumulation of p27 in glucose-deprived cells may seem surprising. Given its role as a cyclin/CDK inhibitor in the nucleus, one would expect
the maintenance of p27 nuclear localization in conditions of metabolic stress to enforce cell cycle arrest, as observed in serum-deprived cells58. Nevertheless, the remaining nuclear p27
likely still contributes to G1 cell cycle arrest upon glucose deprivation17,56. Thus, both cytoplasmic and nuclear p27 appear to cooperate to adapt the cellular response to extracellular
conditions by causing cell cycle arrest and promoting autophagy to cope with metabolic stress. Indeed, cells lacking p27 cannot face this challenge and undergo apoptosis17,18,21.
Interestingly, lysosomal activity and autophagy were recently found to control the depth of the quiescence state59 and an attractive hypothesis is that p27 could contribute to establishing
quiescence by inhibiting cyclin-CDKs and maintaining cells in a shallow, easily reversible quiescence state by promoting autophagy. Glucose withdrawal was shown to induce autophagy in a
p27-dependent manner17. Our results confirm this pro-autophagic function and indicate a role for p27 in autophagic vesicle positioning. In wild-type cells, autophagosomes are located mostly
in the perinuclear zone of the cell, whereas in p27−/− cells, LC3B+ vacuoles were distributed throughout the cytoplasm. During autophagy, autophagosomes form randomly in the cell periphery
and then move along MT towards the centrosome where they fuse with lysosomes25,30,33,34. Thus, the distribution of autophagosomes in the cell is crucial for their maturation and autophagy
flux. p27 was shown to stabilize MT by several mechanisms and promote their acetylation by stabilizing ATAT111,50,52. p27 promotes MT stability either through direct binding to MT52 or by
its interaction with MAPs, such as Stathmin11,57, a MT destabilizing protein whose activity is inhibited by p27, or PRC153, a MAP that bundles antiparallel MT. Since the effect of p27 on MT
requires its cytoplasmic localization, one would expect MT acetylation to increase in glucose-deprived cells, in which there are more p27 in the cytoplasm. MT acetylation in basal and
starvation conditions is more abundant in p27+/+ than in p27−/− MEFs, but it tends to decrease during starvation, unlike what was reported previously60. These differences are probably due to
differences in cellular models or starvation protocol. Geeraert et al. used EBSS starvation (low glucose, no amino acids, no serum) for 2 h60, whereas in this study, MEFs were glucose
starved in the presence of dialyzed serum and amino acids for 48 h. Although MT acetylation on K40 is considered as a hallmark of MT stability, MT acetylation is a consequence and not a
cause of MT stability61. MT acetylation was shown to facilitate autophagy25,34,36,60. To determine if p27-mediated autophagy was dependent on MT acetylation, MT stability, or both, we used
the HDAC inhibitor TSA that increases MT acetylation without affecting their stability61. Treatment of glucose-deprived p27−/− MEFs with TSA rescued their autophagosome positioning defect
and restored autophagy flux, indicating that MT acetylation, and not their stability, is required for retrograde transport of autophagosomes, consistent with previous studies36,62,63,64.
Intriguingly, genetic depletion of HDAC6 inhibits autophagosome maturation and fusion under basal conditions but not upon starvation62,65. However, mice lacking HDAC6 are viable and develop
normally66, whereas autophagy-deficient mice die perinatally67, indicating that HDAC6 is dispensable for autophagy. Nevertheless, given the multiple functions of HDACs in the cell, results
obtained with pharmacological HDAC inhibitors should be interpreted with caution. Indeed, HDAC inhibition not only affects MT acetylation but also that of many genes and proteins, including
ULK1, ATG5, ATG7, and LC3B, which may either promote or inhibit autophagy68,69,70. Although most of these proteins are targeted by sirtuin deacetylases (class III HDACs), which are not
affected by TSA69,71, ATG7 is acetylated by HDACs sensitive to TSA72. Importantly, nuclear HDACs may epigenetically regulate the expression of autophagy-related genes and deacetylation of
histone H3 and H4 promotes autophagy73,74. Thus, TSA may affect the autophagy machinery in different ways, in addition to its effect on MT. To specifically investigate the importance of MT
acetylation on autophagy, we used siRNA against ATAT1, the enzyme responsible for K40 acetylation of α-tubulin75,76. MT acetylation increases the recruitment of motor proteins to MT tracks,
thereby affecting intracellular trafficking31,32,36. p27 binds to and stabilizes ATAT150 and ATAT1 silencing disrupts the perinuclear localization of autophagosomes in cells expressing p27
and impairs their autophagy flux, suggesting that p27 regulates autophagy via ATAT1. Despite many studies investigating starvation-induced autophagy, few of them have addressed the fact that
autophagy mechanisms differ in function of the type of metabolic stress. Investigation of the mechanism of amino acid deprivation-induced autophagy revealed the regulation of mTORC1
signaling and the Ragulator complex by p2721. In contrast, our results indicate that in glucose-deprived cells, p27 promotes autophagy in a Ragulator-independent manner. Furthermore, neither
TSA treatment nor ATAT1 silencing significantly affects amino acid starvation-induced autophagy, confirming that distinct molecular mechanisms are at play in the regulation of autophagy in
response to amino acid or glucose starvation. This is consistent with previous studies showing that signaling pathways leading to autophagosome formation are different in response to amino
acid and glucose withdrawal. While the former induces autophagy via the canonical pathway mediated by PI(3)P and ULK1/2, the latter generates PI(5)P to induce ATG recruitment to
pre-autophagosomal sites in an ULK-independent manner77,78. Further studies have shown that both types of starvation trigger TFEB nuclear localization and autophagy induction, but TFEB is
regulated by different upstream kinases. In amino acid-deprived cells, mTORC1 inhibition prevents the direct phosphorylation of TFEB that maintains its cytoplasmic localization79. In
contrast, in glucose-deprived cells, mTORC2 activates AKT, which in turn inhibits GSK3β activity, preventing TFEB phosphorylation80. Finally, our results indicate that basal autophagy is not
affected by p27 status or MT acetylation levels. Previous studies have shown differences between basal and stress-induced autophagy, notably the site of autophagosome formation81, upstream
signaling governing autophagy initiation82, and factors controlling autophagosome maturation65. Altogether, it appears that distinct metabolic pathways are interconnected with autophagy to
adapt autophagy rate to environmental nutrient availability. The regulation of autophagic vesicle trafficking by cytoplasmic p27 may have therapeutic relevance. In neuronal cells, axonal
transport of autophagosomes is important for cargo degradation and autophagy defects lead to neurodegenerative diseases36. In line with this, p27 has been involved in neural
development50,52,83. In cancer, autophagy may either prevent or promote tumorigenesis22,23. Similarly, p27 may act as a tumor suppressor or oncogene and its cytoplasmic localization appears
to promote cancer development and progression and is associated with poor prognosis7,8,10. Since p27-mediated autophagy confers resistance to metabolic stress caused by glucose starvation17,
it could be one of the mechanisms by which p27 contributes to tumor progression. Therefore, targeting oncogenic kinases causing p27 cytoplasmic localization, preventing microtubule
acetylation, or inhibiting autophagy could overcome chemoresistance of tumors with cytoplasmic p27. MATERIALS AND METHODS ANTIBODIES, REAGENTS, AND PLASMIDS Mouse anti p27 (F8, sc-1641), p27
(SX53G8.5, sc-53871), and rabbit anti p27 (C19, sc-528), Myc (A14, sc-789), p62/SQSTM1 (H290, sc-25575) antibodies were purchased from Santa Cruz Biotechnology. Mouse anti p27 (610242) and
Grb2 (610112) antibodies were purchased from BD-Transduction Laboratories. Mouse anti β-actin (A2228), and β-tubulin (T4026), α-tubulin-FITC (F2168), Acetyl-K40 α-tubulin (T6793) antibodies
were purchased from Sigma-Aldrich. Rabbit anti LC3A/B (#4108), AMPK (#8532), p-AMPK (T172) (#2535), phospho-p70 S6K1 (Thr389) (#9234), p70 S6K1 (#27087) and LAMTOR1 (#8975) antibodies were
purchased from Cell Signalling Technology. Rabbit anti ATG5 (GTX113309) antibodies were purchased from Genetex. Rabbit anti phospho-Ser278 ATG16L1 (ab195242) antibodies were purchased from
Abcam. Mouse anti LC3B (0231-100/LC3-5F10) was purchased from NanoTools. Secondary antibodies against Ig conjugated to horseradish peroxydase or Cyanine-2 and -3 were purchased from Jackson
ImmunoResearch. siRNA control (sc-108727), siRNA against mouse LAMTOR1 (sc-37007) and ATAT1 (sc-108799) were purchased from Santa Cruz Biotechnology. Chloroquine diphosphate (C6628) and
Trichostatin A (T1952) were purchased from Sigma-Aldrich. p27 constructs and p27 point mutants and deletion mutants in pCS2 + , pcDNA3.1+Hygro (Invitrogen), pQCXIP (Clontech), pBabe puro,
pWZL-Blast were described previously9,84. pEF5B-FRT-GFP-ATAT1 was a gift from Maxence Nachury (Addgene #27099)76. GFP-ATAT1 was subcloned into pQCXIP. pBabe-puro-mCherry-eGFP-LC3B was a gift
from Jayanta Debnath (Addgene #22418)85. All plasmids were verified by DNA sequencing. CELL CULTURE AND TRANSFECTIONS All cells were incubated at 37 °C and 5% CO2 in DMEM (D6429, Sigma),
4.5 g/l glucose supplemented with 10% fetal bovine serum [FBS], 0.1 mM nonessential amino acids and 2 µg/ml penicillin–streptomycin. Primary MEFs were isolated from p27+/+ or p27−/− embryos
and immortalized by retroviral infection with a vector encoding the human papilloma virus E6 protein and hygromycin resistance, as previously9,12. Two days after infection, cells were
selected with appropriate antibiotics at following concentrations: 2 µg/mL puromycin, 250 µg/mL hygromycin, 16 µg/mL of blasticidin. Cells were kept under selection at all times. HEK 293
cells were authenticated by short tandem repeat profiling. All cells were mycoplasma free as estimated by routine DAPI staining. For glucose deprivation, cells were rinsed twice with PBS and
once with glucose-free DMEM (D5030, Sigma-Aldrich) supplemented with 0.1 mM nonessential amino acids, 2 µg/ml penicillin–streptomycin, 2mM L-glutamine and 10% dialyzed FBS. FBS was dialyzed
at 4 °C in 3,500 MW cut-off dialysis tubing (SpectrumLabs, 132111) against PBS for 6 h and overnight. Pharmacological inhibitors were used at the following concentrations: Chloroquine 50 µM
for 2 h (immunofluorescence) or 24 h (immunoblotting) in LC3B turnover assays as indicated in figure legend; TSA 200 nM for 1 h. Control cells were treated with the same volume of
corresponding vehicle. For co-immunoprecipitations, HEK 293 cells were transfected by the calcium phosphate method and collected 24 h post-transfection. siRNAs were transfected for 48 h
before starvation using Interferin (Polyplus transfection) according to manufacturer’s instructions. IMMUNOFLUORESCENCE MEFs were seeded on coverslips and grown to 50-60% confluence. Cells
were rinsed and starved for 24–48 h prior to fixation with 1% PFA for 3 min at room temperature followed by 100% methanol for 5 min at −20 °C. Coverslips were washed three times 5 min with
PBS and blocked for 20 min with blocking solution (PBS, 3% BSA, 0.05% Tween 20 and 0.08% sodium azide) at room temperature. Then, coverslips were incubated with primary antibodies diluted in
blocking solution for 1 h at 37 °C, washed three times 5 min in PBS, and incubated with Cy2, Cy3 or Cy5-conjugated secondary antibodies for 30 min at 37 °C. Next, coverslips were washed
three times 5 min in PBS, with the first wash containing 0.1 μg/mL Hoechst H33342. Where indicated, coverslips were incubated with α-tubulin-FITC antibody prior to H33342 staining.
Coverslips were mounted on glass slides using gelvatol (10% polyvinyl alcohol (w/v), 20% glycerol (v/v), 70 mM Tris pH 8). Image acquisition was performed on a Nikon 90i Eclipse microscope
with DS-Qi2 HQ camera and the NIS Element BR software. NIS, ZEN 3.1 Blue and ImageJ 1.51 W were used for quantifications. To measure fluorescent intensity of specific cellular compartments,
regions of interest (ROI) were delineated and the signal was analyzed within the ROI. To determine the positioning of autophagosome, LC3B integrated density within perinuclear ROI was
expressed as percentage of whole cell LC3B integrated density. Cells with more than 50% of LC3B signal in perinuclear ROI were counted as cells with predominantly perinuclear autophagosomes.
To estimate autophagy flux by IF, the average LC3B fluorescence intensity was determined in cells treated with CQ. Cells with mean fluorescence intensity higher than this average value were
considered having high autophagy flux, while cells with mean fluorescence intensity lower than this value were considered as having low autophagy flux. Similarly, acetylated tubulin
fluorescence intensity was evaluated by comparing to a threshold corresponding to average microtubule acetylation in p27+/+ cells. Cells with MT acetylation below or above this value were
considered as cells with low microtubule acetylation or hyperacetylation, respectively. Cells in which MT hyperacetylation was observed only in the perinuclear zone were considered as cells
with perinuclear MT acetylation. The numbers of LC3B, p-ATG16L1 and ATG5 puncta were determined using Find maxima function in ImageJ. IMMUNOPRECIPITATION Cells were lysed in IP buffer (150
mM NaCl, 50 mM HEPES pH 7.5, 1% NP-40, 1 mM EDTA, 2.5 mM EGTA, 0.1% Tween20 and 10% glycerol, complemented with 1 mM DTT, 10 mM β-glycerophosphate, 10 mM NaF, 10 mM sodium orthovanadate, and
10 µg/ml Aprotinin, 10 µg/ml Bestatin, 10 µg/ml Leupeptin and 10 µg/ml Pepstatin A). Lysates were sonicated for 10 s, centrifuged for 5 min at 12,500 rpm and supernatants were collected.
Protein concentrations were determined by Bradford assay and 500 µg of proteins were incubated with 12 µl protein-A sepharose beads (IPA300, Repligen) and 3 µg of indicated antibodies.
Antibody-beads complexes were rinsed 4 times in IP buffer and eluted in 10 µl 4X Laemmli buffer (8%SDS, 40% Glycerol, 240 mM Tris-HCl pH 6.8, 400 mM DTT). Samples were boiled 3 min at 96 °C
prior to loading onto SDS-PAGE gel. Immunoblotting of 1/10th of input was used to determine amounts of respective proteins in lysates and for loading control. IMMUNOBLOT Cells were lysed in
2X Laemmli buffer, sonicated twice for 15 s and boiled 3 min at 96 °C. Proteins were quantified by micro BCA assay (Thermo Scientific Pierce, 23235) and 20–30 µg of proteins were loaded on
SDS-PAGE gel following the addition of DTT at 200 mM final concentration. After electrophoresis, proteins were transferred to polyvinylidene difluoride membrane (Immobilon-P, Millipore).
Membranes were blocked with PBS-T (PBS, 0.1% Tween-20), 5% non-fat dry milk prior to incubation with primary antibodies overnight at 4 °C. The following day, membranes were rinsed thrice
with PBS-T and incubated with HRP-conjugated secondary antibody for 4–6 h at room temperature. Signal was visualized with enhanced chemiluminescence detection reagents (Millipore, BioRad,
and Ozyme) on autoradiographic films (Blue Devil) or with a Fusion Solo S (Vilber) digital acquisition system. Densitometry analyses were performed with the ImageJ software. Proteins of
interests were normalized to loading control (β-Actin, β-Tubulin, or Grb2) and phospho-protein signals were normalized to total levels of the corresponding protein. To determine autophagy
flux, the ratio of LC3B-II/loading control in the presence of CQ by that in the absence of CQ was calculated. AUTOPHAGY TURBOGFP-LC3B INCUCYTE ASSAY Cells expressing turboGFP-LC3B were
seeded (5000 cells/well in 96-well plates) and grown overnight in MEM (Sigma, 56419 C) medium supplemented with 4.5 g/l glucose, 10% FBS, 0.1 mM nonessential amino acids and 2 µg/ml
penicillin–streptomycin before starvation as described above. Images were acquired with an Incucyte FLR system equipped with a 20x objective and tGFP intensity was measured as a ratio
between fluorescence object confluence (green fluorescence) and confluence (phase contrast) using the Incucyte software. Values were normalized to fluorescence values in the first scan (0
h), which was acquired prior to starvation. DETERMINATION OF ATAT1 HALF-LIFE HEK 293 cells were transfected with ATAT1-GFP and/or p27 encoding plasmids for 48 h and treated with 50 µg/ml
cycloheximide (CHX) for 16 h. GFP fluorescence intensity was monitored using the IncuCyte at intervals of 4 h. The fluorescence signal was normalized to cell confluence at each time point
using the Incucyte software. STATISTICAL ANALYSES Data were analyzed using the Graphpad Prism 6.0 software. Statistical significance between three groups or more were determined using ANOVA
test followed by Bonferroni test for multiple comparisons. Difference between two groups was evaluated using unpaired two-tailed _t_ test with Welch’s correction. Data are presented as mean
± SEM. Symbols used are ns: _p_ > 0.05; *: _p_ ≤ 0.05; **: _p_ ≤ 0.01; ***: _p_ ≤ 0.001; ****: _p_ ≤ 0.0001. DATA AVAILABILITY The datasets used and/or analyzed during the current study
are included with this published article as supplementary information files or are available from the corresponding author on reasonable request. REFERENCES * Besson, A., Dowdy, S. F. &
Roberts, J. M. CDK Inhibitors: cell cycle regulators and beyond. _Developmental Cell_ 14, 159–169 (2008). Article CAS PubMed Google Scholar * Fero, M. L., Randel, E., Gurley, K. E.,
Roberts, J. M. & Kemp, C. J. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. _Nature_ 396, 177–180 (1998). Article CAS PubMed PubMed Central Google Scholar *
Fero, M. L. et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. _Cell_ 85, 733–744 (1996). Article CAS
PubMed Google Scholar * Min, Y. H. et al. Cytoplasmic mislocalization of p27Kip1 protein is associated with constitutive phosphorylation of Akt or protein kinase B and poor prognosis in
acute myelogenous leukemia. _Cancer Res._ 64, 5225–5231 (2004). Article CAS PubMed Google Scholar * Liang, J. et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes
p27-mediated G1 arrest. _Nat. Med._ 8, 1153–1160 (2002). Article CAS PubMed Google Scholar * Fujita, N., Sato, S., Katayama, K. & Tsuruo, T. Akt-dependent phosphorylation of p27Kip1
promotes binding to 14-3-3 and cytoplasmic localization. _J. Biol. Chem._ 277, 28706–28713 (2002). Article CAS PubMed Google Scholar * Wander, S. A., Zhao, D. & Slingerland, J. M.
p27: a barometer of signaling deregulation and potential predictor of response to targeted therapies. _Clin. Cancer Res._ 17, 12–18 (2011). Article CAS PubMed Google Scholar * Chu, I.
M., Hengst, L. & Slingerland, J. M. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. _Nat. Rev. Cancer_ 8, 253–267 (2008). Article CAS
PubMed Google Scholar * Besson, A., Gurian-West, M., Schmidt, A., Hall, A. & Roberts, J. M. p27Kip1 modulates cell migration through the regulation of RhoA activation. _Genes Dev._ 18,
862–876 (2004). Article CAS PubMed PubMed Central Google Scholar * Besson, A., Assoian, R. K. & Roberts, J. M. Regulation of the cytoskeleton: an oncogenic function for CDK
inhibitors? _Nat. Rev. Cancer_ 4, 948–955 (2004). Article CAS PubMed Google Scholar * Baldassarre, G. et al. p27Kip1-stathmin interaction influences sarcoma cell migration and invasion.
_Cancer cell_ 7, 51–63 (2005). Article CAS PubMed Google Scholar * Jeannot P. et al. p27(Kip1) promotes invadopodia turnover and invasion through the regulation of the PAK1/Cortactin
pathway. _Elife_ 6 https://doi.org/10.7554/eLife.22207 (2017) * Besson, A. et al. Discovery of an oncogenic activity in p27Kip1 that causes stem cell expansion and a multiple tumor
phenotype. _Genes Dev._ 21, 1731–1746 (2007). Article CAS PubMed PubMed Central Google Scholar * Choudhury, S. et al. Molecular profiling of human mammary gland links breast cancer risk
to a p27(+) cell population with progenitor characteristics. _Cell Stem Cell_ 13, 117–130 (2013). Article CAS PubMed PubMed Central Google Scholar * Sun, X. et al. p27 Protein protects
metabolically stressed cardiomyocytes from apoptosis by promoting autophagy. _J. Biol. Chem._ 289, 16924–16935 (2014). Article CAS PubMed PubMed Central Google Scholar * Su, M. et al.
MicroRNA-221 inhibits autophagy and promotes heart failure by modulating the p27/CDK2/mTOR axis. _Cell death Differ._ 22, 986–999 (2015). Article CAS PubMed Google Scholar * Liang, J. et
al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. _Nat. Cell Biol._ 9, 218–224 (2007). Article CAS
PubMed Google Scholar * Campos, T. et al. Rheb promotes cancer cell survival through p27Kip1-dependent activation of autophagy. _Mol. Carcinog._ 55, 220–229 (2016). Article CAS PubMed
Google Scholar * Campos, T. et al. Rapamycin requires AMPK activity and p27 expression for promoting autophagy-dependent Tsc2-null cell survival. _Biochim. Biophys. Acta_ 1863, 1200–1207
(2016). Article CAS PubMed Google Scholar * White, J. P. et al. The AMPK/p27(Kip1) axis regulates autophagy/apoptosis decisions in aged skeletal muscle stem cells. _Stem Cell Rep._ 11,
425–39 (2018). Article CAS Google Scholar * Nowosad, A. et al. p27 controls Ragulator and mTOR activity in amino acid-deprived cells to regulate the autophagy-lysosomal pathway and
coordinate cell cycle and cell growth. _Nat. Cell Biol._ 22, 1076–90 (2020). Article CAS PubMed Google Scholar * Levine, B. & Kroemer, G. Autophagy in the pathogenesis of disease.
_Cell_ 132, 27–42 (2008). Article CAS PubMed PubMed Central Google Scholar * White, E. The role for autophagy in cancer. _J. Clin. Invest._ 125, 42–46 (2015). Article PubMed PubMed
Central Google Scholar * Botti-Millet, J., Nascimbeni, A. C., Dupont, N., Morel, E. & Codogno, P. Fine-tuning autophagy: from transcriptional to posttranslational regulation. _Am. J.
Physiol. Cell Physiol._ 311, C351–C362 (2016). Article PubMed Google Scholar * Mackeh, R., Perdiz, D., Lorin, S., Codogno, P. & Pous, C. Autophagy and microtubules—new story, old
players. _J. Cell Sci._ 126, 1071–1080 (2013). Article CAS PubMed Google Scholar * Kast, D. J. & Dominguez, R. The cytoskeleton-autophagy connection. _Curr. Biol._ 27, R318–R26
(2017). Article CAS PubMed PubMed Central Google Scholar * Kimura, S., Noda, T. & Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with
lysosomes. _Cell Struct. Funct._ 33, 109–122 (2008). Article CAS PubMed Google Scholar * Ravikumar, B. et al. Dynein mutations impair autophagic clearance of aggregate-prone proteins.
_Nat. Genet._ 37, 771–776 (2005). Article CAS PubMed Google Scholar * Cardoso, C. M. et al. Depletion of kinesin 5B affects lysosomal distribution and stability and induces peri-nuclear
accumulation of autophagosomes in cancer cells. _PloS ONE_ 4, e4424 (2009). Article PubMed PubMed Central CAS Google Scholar * Jahreiss, L., Menzies, F. M. & Rubinsztein, D. C. The
itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. _Traffic_ 9, 574–587 (2008). Article CAS PubMed PubMed Central Google Scholar * Reed, N. A.
et al. Microtubule acetylation promotes kinesin-1 binding and transport. _Curr. Biol._ 16, 2166–2172 (2006). Article CAS PubMed Google Scholar * Dompierre, J. P. et al. Histone
deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. _J. Neurosci._ 27, 3571–3583 (2007). Article CAS PubMed PubMed
Central Google Scholar * Kochl, R., Hu, X. W., Chan, E. Y. & Tooze, S. A. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. _Traffic_ 7,
129–145 (2006). Article CAS PubMed Google Scholar * Xie, R., Nguyen, S., McKeehan, W. L. & Liu, L. Acetylated microtubules are required for fusion of autophagosomes with lysosomes.
_BMC Cell Biol._ 11, 89 (2010). Article PubMed PubMed Central CAS Google Scholar * McLendon, P. M. et al. Tubulin hyperacetylation is adaptive in cardiac proteotoxicity by promoting
autophagy. _Proc. Natl Acad. Sci. USA_ 111, E5178–E5186 (2014). Article CAS PubMed PubMed Central Google Scholar * Esteves, A. R. et al. Acetylation as a major determinant to
microtubule-dependent autophagy: relevance to Alzheimer’s and Parkinson disease pathology. _Biochim. Biophys. Acta Mol. Basis Dis._ 1865, 2008–23 (2019). Article CAS PubMed Google Scholar
* Short, J. D. et al. AMP-activated protein kinase signaling results in cytoplasmic sequestration of p27. _Cancer Res._ 68, 6496–6506 (2008). Article CAS PubMed PubMed Central Google
Scholar * Mizushima, N., Yoshimori, T. & Levine, B. Methods in mammalian autophagy research. _Cell_ 140, 313–326 (2010). Article CAS PubMed PubMed Central Google Scholar *
Pampliega, O. et al. Functional interaction between autophagy and ciliogenesis. _Nature_ 502, 194–200 (2013). Article CAS PubMed PubMed Central Google Scholar * Tian, W. et al. An
antibody for analysis of autophagy induction. _Nat. Methods_ 17, 232–239 (2020). Article CAS PubMed Google Scholar * Kishi-Itakura, C., Koyama-Honda, I., Itakura, E. & Mizushima, N.
Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. _J. Cell Sci._ 127, 4089–4102 (2014). Article CAS PubMed Google Scholar * Noda, N. N.,
Fujioka, Y., Hanada, T., Ohsumi, Y. & Inagaki, F. Structure of the Atg12-Atg5 conjugate reveals a platform for stimulating Atg8-PE conjugation. _EMBO Rep._ 14, 206–211 (2013). Article
CAS PubMed Google Scholar * Kimura, S., Noda, T. & Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3.
_Autophagy_ 3, 452–460 (2007). Article CAS PubMed Google Scholar * Short, J. D. et al. AMPK-mediated phosphorylation of murine p27 at T197 promotes binding of 14-3-3 proteins and
increases p27 stability. _Mol. Carcinog._ 49, 429–439 (2010). Article CAS PubMed Google Scholar * Korolchuk, V. I. et al. Lysosomal positioning coordinates cellular nutrient responses.
_Nat. Cell Biol._ 13, 453–460 (2011). Article CAS PubMed PubMed Central Google Scholar * Jia, R., Guardia, C. M., Pu, J., Chen, Y. & Bonifacino, J. S. BORC coordinates encounter and
fusion of lysosomes with autophagosomes. _Autophagy_ 13, 1648–63 (2017). Article CAS PubMed PubMed Central Google Scholar * Filipek, P. A. et al. LAMTOR/Ragulator is a negative
regulator of Arl8b- and BORC-dependent late endosomal positioning. _J. Cell Biol._ 216, 4199–215 (2017). Article CAS PubMed PubMed Central Google Scholar * Pu, J., Keren-Kaplan, T.
& Bonifacino, J. S. A Ragulator-BORC interaction controls lysosome positioning in response to amino acid availability. _J. Cell Biol._ 216, 4183–97 (2017). Article CAS PubMed PubMed
Central Google Scholar * Yu, L. et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. _Nature_ 465, 942–946 (2010). Article CAS PubMed PubMed Central Google
Scholar * Morelli, G. et al. p27(Kip1) modulates axonal transport by regulating alpha-tubulin acetyltransferase 1 stability. _Cell Rep._ 23, 2429–42 (2018). Article CAS PubMed Google
Scholar * Fass, E., Shvets, E., Degani, I., Hirschberg, K. & Elazar, Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with
lysosomes. _J. Biol. Chem._ 281, 36303–36316 (2006). Article CAS PubMed Google Scholar * Godin Juliette, D. et al. p27Kip1 is a microtubule-associated protein that promotes microtubule
polymerization during neuron migration. _Dev. Cell_ 23, 729–744 (2012). Article CAS PubMed Google Scholar * Perchey, R. T. et al. p27(Kip1) regulates the microtubule bundling activity of
PRC1. _Biochim. Biophys. Acta Mol. Cell Res._ 1865, 1630–1639 (2018). Article CAS PubMed Google Scholar * Hubbert, C. et al. HDAC6 is a microtubule-associated deacetylase. _Nature_ 417,
455–458 (2002). Article CAS PubMed Google Scholar * Nagano, Y. et al. Siah1/SIP regulates p27(kip1) stability and cell migration under metabolic stress. _Cell Cycle_ 10, 2592–2602
(2011). Article CAS PubMed PubMed Central Google Scholar * Dalvai, M., Schubart, K., Besson, A. & Matthias, P. Oct1 is required for mTOR-induced G1 cell cycle arrest via the control
of p27(Kip1) expression. _Cell Cycle_ 9, 3933–3944 (2010). Article CAS PubMed Google Scholar * Wang, J. et al. Phosphorylation at Ser10 triggered p27 degradation and promoted
gallbladder carcinoma cell migration and invasion by regulating stathmin1 under glucose deficiency. _Cell Signal_ 80, 109923 (2021). Article CAS PubMed Google Scholar * Coats, S.,
Flanagan, W. M., Nourse, J. & Roberts, J. M. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. _Science_ 272, 877–880 (1996). Article CAS PubMed
Google Scholar * Fujimaki, K. et al. Graded regulation of cellular quiescence depth between proliferation and senescence by a lysosomal dimmer switch. _Proc. Natl Acad. Sci. USA_ 116,
22624–34 (2019). Article CAS PubMed PubMed Central Google Scholar * Geeraert, C. et al. Starvation-induced hyperacetylation of tubulin is required for the stimulation of autophagy by
nutrient deprivation. _J. Biol. Chem._ 285, 24184–24194 (2010). Article CAS PubMed PubMed Central Google Scholar * Palazzo, A., Ackerman, B. & Gundersen, G. G. Cell biology: tubulin
acetylation and cell motility. _Nature_ 421, 230 (2003). Article CAS PubMed Google Scholar * Iwata, A., Riley, B. E., Johnston, J. A. & Kopito, R. R. HDAC6 and microtubules are
required for autophagic degradation of aggregated huntingtin. _J. Biol. Chem._ 280, 40282–40292 (2005). Article CAS PubMed Google Scholar * Guedes-Dias, P. et al. HDAC6 inhibition
induces mitochondrial fusion, autophagic flux and reduces diffuse mutant huntingtin in striatal neurons. _Biochim. Biophys. Acta_ 1852, 2484–2493 (2015). Article CAS PubMed Google Scholar
* Gilardini Montani, M. S. et al. Histone deacetylase inhibitors VPA and TSA induce apoptosis and autophagy in pancreatic cancer cells. _Cell Oncol. (Dordr.)_ 40, 167–80 (2017). Article
CAS Google Scholar * Lee, J.-Y. et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. _EMBO J._ 29, 969–980 (2010). Article CAS
PubMed PubMed Central Google Scholar * Zhang, Y. et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. _Mol. Cell. Biol._ 28,
1688–1701 (2008). Article CAS PubMed PubMed Central Google Scholar * Kuma, A., Komatsu, M. & Mizushima, N. Autophagy-monitoring and autophagy-deficient mice. _Autophagy_ 13, 1619–28
(2017). Article CAS PubMed PubMed Central Google Scholar * Sebti, S. et al. BAT3 modulates p300-dependent acetylation of p53 and autophagy-related protein 7 (ATG7) during autophagy.
_Proc. Natl Acad. Sci. USA_ 111, 4115–4120 (2014). Article CAS PubMed PubMed Central Google Scholar * Huang, R. et al. Deacetylation of nuclear LC3 drives autophagy initiation under
starvation. _Mol. Cell_ 57, 456–466 (2015). Article CAS PubMed Google Scholar * Lin, S. Y. et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy.
_Science_ 336, 477–481 (2012). Article CAS PubMed Google Scholar * Ng, F. & Tang, B. L. Sirtuins’ modulation of autophagy. _J. Cell Physiol._ 228, 2262–2270 (2013). Article CAS
PubMed Google Scholar * Stankov, M. V. et al. Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. _Leukemia_ 28, 577–588 (2014). Article CAS
PubMed Google Scholar * Fullgrabe, J. et al. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. _Nature_ 500, 468–471 (2013). Article PubMed PubMed
Central CAS Google Scholar * He, M. et al. MoSnt2-dependent deacetylation of histone H3 mediates MoTor-dependent autophagy and plant infection by the rice blast fungus Magnaporthe oryzae.
_Autophagy_ 14, 1543–61 (2018). Article CAS PubMed PubMed Central Google Scholar * Akella, J. S. et al. MEC-17 is an alpha-tubulin acetyltransferase. _Nature_ 467, 218–222 (2010).
Article CAS PubMed PubMed Central Google Scholar * Shida, T., Cueva, J. G., Xu, Z., Goodman, M. B. & Nachury, M. V. The major alpha-tubulin K40 acetyltransferase alphaTAT1 promotes
rapid ciliogenesis and efficient mechanosensation. _Proc. Natl Acad. Sci. USA_ 107, 21517–21522 (2010). Article CAS PubMed PubMed Central Google Scholar * Vicinanza, M. et al. PI(5)P
regulates autophagosome biogenesis. _Mol. Cell_ 57, 219–234 (2015). Article CAS PubMed PubMed Central Google Scholar * Cheong, H., Lindsten, T., Wu, J., Lu, C. & Thompson, C. B.
Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. _Proc. Natl Acad. Sci. USA_ 108, 11121–11126 (2011). Article CAS PubMed PubMed Central Google Scholar * Pena-Llopis, S. et
al. Regulation of TFEB and V-ATPases by mTORC1. _EMBO J._ 30, 3242–3258 (2011). Article CAS PubMed PubMed Central Google Scholar * Li, L. et al. A TFEB nuclear export signal integrates
amino acid supply and glucose availability. _Nat. Commun._ 9, 2685 (2018). Article PubMed PubMed Central CAS Google Scholar * Musiwaro, P., Smith, M., Manifava, M., Walker, S. A. &
Ktistakis, N. T. Characteristics and requirements of basal autophagy in HEK 293 cells. _Autophagy_ 9, 1407–1417 (2013). Article CAS PubMed Google Scholar * Wei, Y. et al. The
stress-responsive kinases MAPKAPK2/MAPKAPK3 activate starvation-induced autophagy through Beclin 1 phosphorylation. _Elife_ 4, e05289 (2015). Article PubMed Central Google Scholar *
Nguyen, L. et al. p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. _Genes Dev._ 20, 1511–1524 (2006). Article CAS PubMed PubMed Central
Google Scholar * Serres, M. P. et al. p27(Kip1) controls cytokinesis via the regulation of citron kinase activation. _J. Clin. Invest._ 122, 844–858 (2012). Article CAS PubMed PubMed
Central Google Scholar * N’Diaye, E. N. et al. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. _EMBO Rep._ 10, 173–179 (2009). Article
PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS The authors thank Raphael Culerrier for technical assistance with cloning. We are grateful to Maxence Nachury
(Stanford University) and Jayanta Debnath (University of California San Francisco) for providing reagents. A.N. was supported by a studentship from the Fondation ARC pour la Recherche sur le
Cancer. S.M. is supported by a grant from the Ligue Nationale Contre le Cancer. This project was supported by funds from the Ligue Nationale Contre le Cancer (# 2FI13911UPQV) and Fondation
ARC pour la Recherche sur le Cancer (PJA 20171206148) to A.B. A.B. is supported by funds from the Fondation pour la Recherche Médicale (FRM Equipe EQU202103012639). FUNDING A.N. was
supported by a studentship from the Fondation ARC pour la Recherche sur le Cancer. S.M. is supported by a grant from the Ligue Nationale Contre le Cancer. This project was supported by funds
from the Ligue Nationale Contre le Cancer (# 2FI13911UPQV) and Fondation ARC pour la Recherche sur le Cancer (PJA 20171206148) to A.B. A.B. is supported by funds from the Fondation pour la
Recherche Médicale (FRM Equipe EQU202103012639). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * MCD, Centre de Biologie Intégrative, Université de Toulouse, CNRS, UPS, 31062, Toulouse, Cedex,
France Ada Nowosad, Justine Creff, Pauline Jeannot, Raphael Culerrier & Arnaud Besson * Institut Necker-Enfants Malades (INEM), INSERM U1151-CNRS UMR 8253, F-75015, Paris, France
Patrice Codogno * Université Paris, F-75005, Paris, France Patrice Codogno * Cancer Research Center of Toulouse (CRCT), INSERM U1037, CNRS ERL5294, University of Toulouse, Toulouse, France
Stephane Manenti * GIGA-Stem Cells, University of Liège, CHU Sart Tilman, Liège, Belgium Laurent Nguyen Authors * Ada Nowosad View author publications You can also search for this author
inPubMed Google Scholar * Justine Creff View author publications You can also search for this author inPubMed Google Scholar * Pauline Jeannot View author publications You can also search
for this author inPubMed Google Scholar * Raphael Culerrier View author publications You can also search for this author inPubMed Google Scholar * Patrice Codogno View author publications
You can also search for this author inPubMed Google Scholar * Stephane Manenti View author publications You can also search for this author inPubMed Google Scholar * Laurent Nguyen View
author publications You can also search for this author inPubMed Google Scholar * Arnaud Besson View author publications You can also search for this author inPubMed Google Scholar
CONTRIBUTIONS A.N., J.C., P.J., and A.B. performed experiments. R.C. contributed tools for experiments. A.N., J.C., P.J., P.C., S.M., L.N., and A.B. designed experiments and analyzed the
data. A.N. and A.B. wrote the paper with contributions from all authors. CORRESPONDING AUTHOR Correspondence to Arnaud Besson. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare no
competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Edited by
M. Hamasaki SUPPLEMENTARY INFORMATION FIGURE S1 FIGURE S2 FIGURE S3 FIGURE S4 FIGURE S5 FIGURE S6 DATASET FIG. 1 DATASET FIG. 2 DATASET FIG. 3 DATASET FIG. 4 DATASET FIG. 5 DATASET FIG. 6
DATASET FIG. 7 DATASET FIG. 8 DATASET FIG. S1 DATASET FIG. S2 DATASET FIG. S3 DATASET FIG. S4 DATASET FIG. S5 DATASET FIG. S6 RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed
under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article
are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Nowosad, A., Creff, J., Jeannot, P. _et al._ p27 controls autophagic
vesicle trafficking in glucose-deprived cells via the regulation of ATAT1-mediated microtubule acetylation. _Cell Death Dis_ 12, 481 (2021). https://doi.org/10.1038/s41419-021-03759-9
Download citation * Received: 02 March 2020 * Revised: 15 April 2021 * Accepted: 26 April 2021 * Published: 13 May 2021 * DOI: https://doi.org/10.1038/s41419-021-03759-9 SHARE THIS ARTICLE
Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided
by the Springer Nature SharedIt content-sharing initiative








