- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Gallbladder cancer (GBC) is a highly malignant bile duct cancer with poor prognosis due to early invasion and metastasis. However, the molecular mechanisms through which GBC cells
interact with the tumor microenvironment (TME) remain poorly understood. Here, we examined the role of the tumor suppressor apoptosis-stimulating of p53 protein 2 (ASPP2) in regulating GBC
invasion and metastasis and macrophage recruitment. The clinicopathological significance of ASPP2 expression was measured by immunohistochemical analysis in 72 patients with GBC.
Lentivirus-mediated knockdown or overexpression of ASPP2 was used to investigate the biological functions and molecular mechanisms of ASPP2 in GBC cells. Our data showed that downregulation
of ASPP2 in patients with GBC was linked to poor prognosis. Knockdown of ASPP2 induced epithelial–mesenchymal transition (EMT) in GBC cells and influenced the TME. Mechanistically, we
further confirmed that ASPP2 affected the expression and protein binding between atypical protein kinase C (aPKC)-ι and glioma-associated oncogene homolog 1 (GLI1). ASPP2 also induced C−C
motif chemokine ligand (CCL) 2, CCL5, and tumor necrosis factor-α secretion by cancer cells, thereby promoting macrophage recruitment. The latter also induced EMT-like changes in GBC.
Furthermore, ASPP2 deficiency regulated GLI1 transcriptional activity via the noncanonical Hedgehog (Hh) pathway and aPKC-ι/GLI1 signaling loop and promoted GLI1 nuclear localization and
binding to the promoters of target genes. Our findings revealed that downregulation of ASPP2 promoted GBC invasion and metastasis through the aPKC-ι/GLI1 pathway and enhanced macrophage
recruitment. Thus, ASPP2/aPKC-ι/GLI1 pathway may be a potential therapeutic target for the treatment of GBC. SIMILAR CONTENT BEING VIEWED BY OTHERS THE KDM6A-SPARCL1 AXIS BLOCKS METASTASIS
AND REGULATES THE TUMOUR MICROENVIRONMENT OF GASTROINTESTINAL STROMAL TUMOURS BY INHIBITING THE NUCLEAR TRANSLOCATION OF P65 Article 08 February 2022 SUPRABASIN PROMOTES GASTRIC CANCER LIVER
METASTASIS VIA HEPATIC STELLATE CELLS-MEDIATED EGF/CCL2/JAK2 INTERCELLULAR SIGNALING PATHWAYS Article 03 April 2025 SLC7A2 DEFICIENCY PROMOTES HEPATOCELLULAR CARCINOMA PROGRESSION BY
ENHANCING RECRUITMENT OF MYELOID-DERIVED SUPPRESSORS CELLS Article Open access 02 June 2021 INTRODUCTION Gallbladder cancer (GBC), a primary malignancy of the biliary tract, is the sixth
most common gastrointestinal cancer and has a 5-year survival rate of 5%1,2. Such poor prognosis is due, in part, to its aberrant anatomical features, aggressive biological behaviors, and
lack of sensitive screening tests for early diagnosis, resulting in loss of the opportunity for early treatment1,3. Although radical resection is the most promising potential curative
approach for patients, less than 10% of patients are considered candidates for resection because of advanced stage disease, and nearly 50% of patients exhibit lymph node metastasis on
initial diagnosis4,5. Metastasis is a highly complex biological process involving a multistep cascade of genetic and epigenetic events. For tumors to metastasize, the cancer cells must
obtain enhanced invasive capacity, and the tumor microenvironment (TME) must be remodeled6. Growing evidence has supported the concept that the epithelial-to-mesenchymal transition (EMT)
plays pleiotropic roles in tumor metastasis7,8. We previously reported that atypical protein kinase C (aPKC)-ι, as an oncogene and key polarization regulator, is positively correlated with
cholangiocarcinoma (CCA) differentiation and invasion9. We also showed that aPKC-ι induced the EMT in CCA cells and stimulates immunosuppression associated with Snail10. However, it is
unknown how GBC cells modulate the TME and what the molecular mechanisms are associated with the interaction between tumor and host cells during the EMT. Apoptosis-stimulating of p53 protein
2 (ASPP2), a haploinsufficient tumor suppressor that was originally identified as an activator of the p53 family, is a member of the ASPP family, together with ASPP1 and iASPP, and has
several shared structural features, including ankyrin repeats, an SH3 domain, and a proline-rich region11,12. Downregulation of ASPP2 is associated with the advanced stages of many human
cancers, such as breast cancer, hepatocellular carcinoma, and pancreatic cancer13,14,15,16. In the nucleus, direct binding with p53 and stimulation of the transactivation of p53 are
downstream events of ASPP2-induced apoptosis17. However, clinical studies have also detected ASPP2 in the cytoplasm of cancer cells18. Recent studies have shown that ASPP2 controls cell
polarity during central nervous system development and is colocalized with the Par3 complex to act as a regulator of cell−cell adhesion19. Of note, ASPP2 deficiency promoted EMT and tumor
metastasis in multiple types of cancer13; however, it remains unknown whether ASPP2 is involved in the regulation of EMT in GBC. Recent studies of the Hedgehog (Hh) pathway have shown that
this pathway is a critical regulator of cancer progression and has fundamental roles in the development and differentiation of tissues and organs during embryonic life20. Aberrant activation
of the Hh pathway results in a wide variety of human cancers, including GBC21. The transcription factor glioma-associated oncogene homolog 1 (GLI1), which is a central player in the Hh
pathway, mediates Hh signaling and acts as a marker of Hh signaling activation by translocation to the nucleus22. Activated GLI proteins translocate into the nucleus and stimulate the
transcription of Hh pathway target genes, including GLI1, protein patched homolog 1 (PTCH1), smoothened (SMO), and many survival-promoting molecules23. In addition to being activated by the
Hh ligand/PTCH1/SMO axis, also known as the canonical Hh pathway, a growing body of evidence suggests that activation of GLI1 in some cancers is not controlled exclusively by Hh signaling
but is also modulated by other pathways, such as AKT, MAPK/ERK, and KRAS pathways in an Hh ligand/PTCH1/SMO axis-independent manner, also known as noncanonical Hh signaling24. Although the
canonical Hh pathway has been extensively studied, it is still unclear how GLI1 is regulated through noncanonical Hh signaling. Therefore, it is important to identify other upstream
regulators of GLI1. In this study, we evaluated the fundamental roles of ASPP2 in tumor progression and the TME via the noncanonical Hh pathway involving aPKC-ι/GLI1 signaling in GBC.
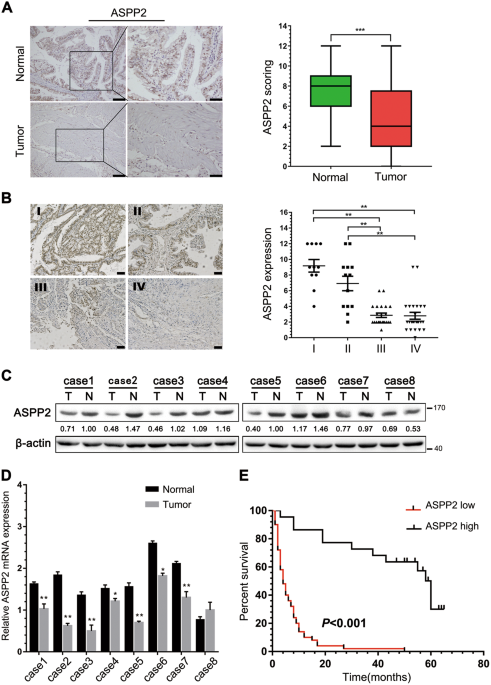
RESULTS ASPP2 WAS DOWNREGULATED AND CORRELATED WITH POOR PROGNOSIS IN PATIENTS WITH GBC To explore the clinical significance of ASPP2 in human GBC, we first examined the expression of ASPP2
by immunohistochemistry (IHC), western blotting, and quantitative real-time polymerase chain reaction (qPCR) in GBC tissues and paired normal gallbladder tissues. IHC analysis showed that
ASPP2 expression was significantly lower in GBC tissues than in 72 pair-matched normal tissues, both in the cytoplasm and nucleus (Fig. 1a). Downregulation of ASPP2 was further confirmed at
the protein and mRNA levels in representative eight paired GBC tissues and normal tissues (Fig. 1c, d). In addition, we investigated whether the expression of ASPP2 was correlated with
clinicopathological characteristics and prognosis in human GBC. Notably, reduced expression of ASPP2 was significantly associated with advanced TNM stage (_χ_2 = 33.513, _P_ _<_ 0.001),
lymph node metastasis (_χ_2 = 19.415, _P_ _<_ 0.001), and poor tumor differentiation (_χ_2 = 25.222, _P_ _<_ 0.001) in GBC (Fig. 1b and Table 1). Kaplan−Meier analysis showed that
patients with low ASPP2 expression exhibited a shorter overall survival (OS) than those with high ASPP2 expression (Fig. 1e). Cox’s multivariate analysis indicated that downregulation of
ASPP2 was an independent prognostic risk factor for OS in patients with GBC (Supplementary Table S3). Moreover, gene set enrichment analysis was used to investigate the correlation between
ASPP2 and several other tumors in the public database _The Cancer Genome Atlas_ (TCGA), and the results showed that ASPP2 was involved in cell adhesion and tight junctions, which are related
to tumor metastasis (Fig. S1). Taken together, these observations suggested that ASPP2 levels were frequently downregulated in human GBC and that ASPP2 may promote the EMT and metastasis.
ASPP2 DEFICIENCY PROMOTED THE PROLIFERATION, MIGRATION, AND INVASION OF GBC CELLS To elucidate whether ASPP2 affected the biological characteristics of GBC, we first evaluated ASPP2
expression in three GBC cell lines (NOZ, OCUG-1, and GBC-SD). We found that ASPP2 expression was decreased in all cell lines, especially in GBC-SD (Fig. S2A). Thus, NOZ and OCUG-1 cells were
chosen for the subsequent studies. Then we stably established ectopic ASPP2 expression or knockdown (KD) GBC cell lines (NOZ and OCUG-1 cells; Fig. 2a). We found that ASPP2 KD induced
EMT-like characteristics with respect to mRNA and protein expression profiles, including downregulation of E-cadherin and upregulation of N-cadherin and vimentin (Fig. 2a and Fig. S2B).
However, ASPP2 KD or overexpression had minimal impact on the expression levels of other ASPP2-induced EMT markers, such as ZEB1 and β-catenin13, in GBC cells (Fig. S2C-D). Cell
proliferation, migration, and invasion were also significantly increased after transfection with lentiviral shRNA targeting endogenous ASPP2. Interestingly, restoration of exogenous ASPP2
expression reversed the effects of ASPP2 KD, suggesting that ASPP2 may act as a switch for the EMT and control the mobility of GBC cells (Fig. 2b–d). In vivo tumorigenicity assays showed
that ASPP2 KD significantly increased the volumes of xenograft tumors compared with those in the blank and negative control groups. Moreover, these effects were reversed by expression of
exogenous ASPP2 (Fig. 2e, f and Fig. S2E-F). Thus, our results indicated that ASPP2 deficiency could facilitate tumor cell proliferation, migration, and invasion in vitro and in vivo. ASPP2
KD PROMOTED THE RECRUITMENT OF MACROPHAGES AND FACILITATED TUMOR LUNG METASTASIS Accumulating evidence has demonstrated that various stromal components, including macrophages, play important
roles in tumor metastasis to the lung and in remodeling of the TME6,25,26. Consistent with this, we found that downregulation of ASPP2 significantly increased lung metastasis, whereas
overexpression of ASPP2 inhibited lung metastasis (Fig. 3a). Next, we used flow cytometry analyses to examine CD11b+F4/80+ macrophages in lung metastases and found that ASPP2 KD enhanced,
whereas ASPP2 overexpression suppressed macrophage recruitment (Fig. 3b), as confirmed by IHC in xenograft tumors (Fig. 3c and Fig. S3B). However, changes in macrophages were not due to
alterations in their origin because the number of macrophages in the spleens and bone marrow of the mice remain unchanged (data not shown). Previous studies have reported that cytokines
played an important role to remodel the TME and promote EMT. The cytokines C−C motif chemokine ligand (CCL) 2 and CCL5, which promote the recruitment of tumor-associated macrophages
(TAMs)27,28, as well as interleukin (IL)-8, which is a ligand for C−X−C motif chemokine receptor (CXCR) 1 and CXCR2, are highly expressed on the surface of M2 macrophages, and TAMs have an
M2-like phenotype29. However, ASPP2-induced changes in cytokines have not yet been extensively investigated. To determine the major cytokines regulated by ASPP2, we examined a series of
cytokines and explored whether ASPP2 could induce cytokine secretion to modulate the TME (Fig. S3A). Interestingly, we found that ASPP2 KD, but not overexpression, increased the expression
of CCL2, CCL5, and tumor necrosis factor (TNF)-α and enhanced their secretion in GBC cells (Fig. 3d; Fig. S3C-E). Furthermore, cell migration assay revealed that ASPP2 KD enhanced, while
ASPP2 overexpression suppressed, the chemoattraction of GBC cells to CD14+ monocytes. Addition of neutralizing antibodies attenuated monocyte migration (Fig. 3e, f). Therefore, the above
results showed that reducing ASPP2 expression promoted the recruitment of macrophages and facilitated tumor lung metastasis. TAM RECRUITMENT WAS NEGATIVELY CORRELATED WITH ASPP2 EXPRESSION
AND INDUCED EMT-LIKE CHANGES Next, we further explored the correlations between TAMs and clinical features in GBC specimens and cells. The expression levels of CD68 and CD163, representative
cell markers of TAMs, were significantly higher in GBC tissues than in pair-matched normal tissues (Fig. 4a). Next, we investigated whether ASPP2 expression was correlated with TAM
recruitment in cancer tissues. The results showed that low ASPP2 expression was associated with increased infiltration of TAMs (Fig. 4b). In addition, we also found EMT-like changes in GBC
samples in which CD68 and CD163 were highly expressed (Fig. 4c). E-cadherin expression was reduced, whereas N-cadherin and vimentin levels were increased in GBC cells following coculture
with macrophages (Fig. 4d). Cell migration was also increased after coculture with the conditioned medium from macrophages (Fig. 4e). Therefore, these observations demonstrated that
downregulation of ASPP2 was associated with TAM recruitment and poor prognosis in patients with GBC. TAM recruitment was negatively correlated with ASPP2 and induced EMT-like changes. ASPP2
REGULATED THE EXPRESSION OF APKC-Ι AND GLI1 Previously, we reported that aPKC-ι is a critical polarization regulator and induces the EMT in CCA30. Interestingly, aPKC-ι also regulates YAP1
nuclear localization and activates the Hh signaling pathway to promote tumor growth31,32. These findings suggest that aPKC-ι may interact with multiple signals involved in tumor progression.
Here, we hypothesized that aPKC-ι may participate in regulation of the EMT via ASPP2. Surprisingly, we found that ASPP2 KD increased the expression of aPKC-ι at both the mRNA and protein
levels. In addition, ASPP2 deficiency significantly increased the expression of GLI1, a major Hh transcriptional regulator, but did not affect YAP1 and its target genes _CTGF_, _CYR61_, and
_ANKRD_ in GBC cell lines. Consistent with this, the expression levels of aPKC-ι and GLI1 were restored by exogenous ASPP2, which was confirmed by qPCR and immunoblotting (Fig. 5a, b). Based
on these findings, we then investigated whether aPKC-ι or GLI1 affected the expression of ASPP2. When endogenous aPKC-ι or GLI1 was knocked down, GLI1 or aPKC-ι expression, but not ASPP2
expression, was significantly decreased (Fig. 5c). ASPP2, aPKC-ι, and GLI1 RNAi led to efficient knockdown of their respective target. ASPP2 RNAi significantly increased aPKC-ι and GLI1
expression. aPKC-ι and GLI1 RNAi also exhibited reduced GLI1 and aPKC-ι expression, but did not affect ASPP2 expression (Fig. 5d). Next, immunoprecipitation experiments were performed to
investigate whether ASPP2 affected binding with aPKC-ι and GLI1. The results showed that aPKC-ι and GLI1 immunoprecipitated with ASPP2. The reciprocal immunoprecipitation confirmed this
interaction. More importantly, ASPP2 KD attenuated the ASPP2−aPKC-ι−GLI1 interaction (Fig. 5e). Furthermore, downregulation of ASPP2 increased, while knockdown aPKC-ι (aPKC-ι-KD) or GLI1
(GLI1-KD) reduced anchorage-independent growth by soft agar growth assays in GBC cells. Co-downregulation of ASPP2 with aPKC-ι or GLI1 reversed this effect (Fig. 5f). Consistently, we found
that aPKC-ι-KD or GLI1-KD suppressed the chemoattraction of GBC cells to CD14+ monocytes, which was abolished by ASPP2 KD (Fig. 5g). Taken together, these findings suggested that ASPP2 may
indirectly activate GLI1 expression, in part through aPKC-ι, thereby regulating the EMT and TME. DEPLETION OF ASPP2 REGULATED GLI1 TRANSCRIPTIONAL ACTIVITY VIA APKC-Ι Although previous
studies have reported that Hh signaling may be a potential therapeutic target for GBC, published preclinical data are limited33. Cyclopamine, an inhibitor of SMO, has been shown to exert few
effects in several cancers, and the mechanism mediating aberrant activation of the Hh pathway in GBC remains unknown. In fact, we found that ASPP2 deficiency or overexpression had little
effect on the expression of PTCH1 and SMO (Fig. S4A-B). Thus, we next examined whether GBCs were characterized by noncanonical, SMO-independent, GLI1 activation. Interestingly, in GBC cells,
cyclopamine did not inhibit ASPP2 KD-induced GLI1 activity (Fig. S4C). Moreover, si-SMO had limited effects on ASPP2 KD-induced GLI1 activity (Fig. 6a and Fig. S4D). In GBC cells, we found
that ASPP2 KD induced GLI1 nuclear localization, which increased with time after lentiviral transfection (Fig. 6b, c and Fig. S4E). Consistent with this, IHC results showed that ASPP2
induced nuclear accumulation of GLI1 in xenograft tumors (Fig. S4F). Because aPKC-ι is a serine/threonine kinase that regulates GLI activity in basal cell carcinoma34, we asked whether
aPKC-ι mediated nuclear localization of GLI1 through phosphorylation. Indeed, we found that phosphorylation of GLI1 had a significant impact on the distribution of GLI1 in the cytoplasm and
nucleus as endogenous or exogenous levels of aPKC-ι increased. In addition, the phosphorylation of GLI1 was largely inhibited by treatment with PSI, an aPKC peptide inhibitor. Of note, in
case of ASPP2 knockdown or aPKC-ι overexpression in NOZ cells, we did not detect aPKC-ι in the nucleus (Fig. 6d). Previous studies reported that 79-KKRALS-84 is highly conserved in GLI1, as
a potential phosphorylation site24. Thus, we hypothesized that Ser84 may be phosphorylated by aPKC-ι. Then, we generated wild-type GLI1 (GLI1-WT) and Ser84 nonphosphorylatable mutant GLI1
(GLI1-S84A). Immunoprecipitation experiments showed that GLI1-WT bound significantly more aPKC-ι than GLI1-S84A (Fig. 6e). As shown in Fig. 1a and Fig S2B, we found that ASPP2 was expressed
in both cytoplasm and nucleus of GBC cells. Intriguingly, recent studies have suggested that the subcellular localization of ASPP2 may have specific functions35. This prompted us to test if
ASPP2 regulated GLI1 in the nucleus. The results showed that ASPP2 deficiency attenuated the ASPP2−GLI1 interaction in the nucleus of NOZ cells (Fig. 6f). Interestingly, consistent with
previously studies, we also found that GLI1 directly bound to the promoter regions of _PRKCI_ (aPKC-ι is encoded by the _PRKCI_ gene) (Fig. 6g). Moreover, chromatin immunoprecipitation
(ChIP) assays confirmed the direct binding of GLI1 to the promoters of _CCL2_, _CCL5_, and _TNFA_ (Fig. 6h). Knockdown ASPP2 may enhance recruitment of TAMs by modulating transcriptional
activity of these target genes. Taken together, these results suggested that ASPP2 regulated GLI1 nuclear translocation through the noncanonical Hh pathway involving aPKC-ι-mediated GLI1
phosphorylation. ASPP2 deficiency decreased the ASPP2−GLI1 interaction and facilitated the binding of GLI1 to the promoter of downstream target genes. aPKC-ι and GLI1 may form a positive
feedback loop to amplify the signal. APKC-Ι AND GLI1 WERE ASSOCIATED WITH POOR PROGNOSIS IN PATIENTS WITH GBC AND MODULATED THE SECRETION OF CHEMOKINES Finally, we further confirmed the
relationships among ASPP2, aPKC-ι, and GLI1 in GBC samples. The results showed that aPKC-ι and GLI1 expression levels were significantly higher in GBC tissues than in pair-matched normal
tissues and were negatively correlated with ASPP2 expression (Fig. 7a–c). The expression levels of aPKC-ι and GLI1 were significantly associated with advanced TNM stage (_χ_2 = 19.965,
15.458, respectively; _P_ _<_ 0.001), lymph node metastasis (_χ_2 = 13.125, 15.044, respectively; _P_ _<_ 0.001), and poor tumor differentiation (_χ_2 = 29.154, 15.273, respectively;
_P_ _<_ 0.001) in GBC (Supplementary Table S2). Multivariate Cox regression analyses indicated that aPKC-ι and GLI1 were independent prognostic factors for OS in patients with GBC
(Supplementary Table S3). Furthermore, we found that the expression of ASPP2 in some tumor tissues was almost equivalent to the normal tissues (Fig. 1c). Then, we further compared ASPP2
expression with GLI1 expression and macrophage infiltration in representative samples. The IHC analysis confirmed that low expression of ASPP2 correlated with increased GLI1 expression and
TAMs infiltration in GBC tissues, while high expression of ASPP2 exhibited opposite results (Fig. S5A-B). Next, we investigated the effects of aPKC-ι and GLI1 on changes in chemokine levels
in the supernatants of GBC cell cultures. Our data suggested that downregulation of aPKC-ι or GLI1 reduced the secretion of CCL2, CCL5, and TNF-α, which further supported our previous
results (Fig. 7d). Taken together, our results strongly suggested that ASPP2 affected GBC invasion and metastasis through the aPKC-ι/GLI1 signaling pathway and remodeling of the TME.
DISCUSSION Tumors develop in a complex environment, which they depend on for invasion and metastasis. The TME is also thought to play an important role in crosstalk with cancer cells36,37.
Although previous studies have shown that ASPP2 is correlated with prognosis in multiple types of cancer, the relationships and molecular mechanisms through which ASPP2 interacts with the
TME remain unknown. Here, we found that downregulation of ASPP2 promoted GBC invasion and metastasis through the aPKC-ι/GLI1 pathway and enhanced macrophage recruitment. Previous studies
have focused on the transactivity effects of p53 through ASPP2 in the nucleus, while accumulating evidence has highlighted the tumor-suppressive role of ASPP2 crosstalk with other
pathways35,38. In this study, our data demonstrated that ASPP2 was frequently downregulated in GBC tissues compared with that in normal tissues. Moreover, multivariate Cox regression
analyses suggested ASPP2 was an independent prognostic factor for the OS of patients with GBC. These clinical data strongly indicated that ASPP2 could be an important prognostic biomarker
for patients with GBC. In this study, we found that ASPP2 may act as a molecular switch for the EMT/MET and control tumor progression. Our findings are supported by other studies
demonstrating that ASPP2 can regulate epithelial plasticity and cell−cell adhesion in vitro and in vivo through different signal pathways13. However, tumor progression is a complex process,
and bidirectional communication between cancer cells and the TME is critical for tumor progression and metastasis25. In these contexts, we hypothesized that ASPP2 may also affect the
microenvironment via a special mechanism. As important regulators of tumorigenesis, macrophages are major components of the TME. Numerous studies have demonstrated that tumor cells can
recruit monocytes to infiltrate the tumor tissue by secreting chemokines and differentiate into TAMs. Interestingly, ASPP2 KD, but not overexpression, increased the expression of _CCL2_,
_CCL5_, and _TNF-α_ and enhanced their secretion, which may promote macrophage recruitment to tumor tissues and support multiple aspects of tumor progression. Our work is consistent with
several previous studies demonstrating that these chemokines can promote the recruitment of TAMs to remodel the TME by the “yin and yang” effect or a positive feedback loop between cancer
cells and macrophages29,39. However, care should be taken when interpreting our findings that macrophages can be induced to TAMs. Although our data showed that ASPP2 KD induced an M2-like
phenotype in macrophages to promote lung metastasis of cancer cells, which was also supported by previous studies, the roles of these cells in metastatic progression cannot be ruled out.
Moreover, it is difficult to simply classify TAMs into M1 or M2 states in the complex TME in vivo, although the cells can be induced into M1 or M2 macrophages in vitro. Indeed, large-scale
transcriptome analysis supported that TAMs have a mixed phenotype of both M1 and M2 macrophages in vivo39,40. In addition, IL-6 and IL-10, which act as cytokines for M2-like macrophages,
were not upregulated following ASPP2 KD in GBC cells. Further studies are needed to determine whether these cytokines induce M2-like macrophages through activation of other immune cells in
the TME, such as Th2 cells, or through a noncanonical pathway. In the present study, we found that ASPP2 regulated the expression of aPKC-ι, which has been shown to affect the nuclear
localization of the transcription factor YAP1 in ovarian cancer. Thus, we assessed whether ASPP2 and aPKC-ι activated a similar signaling pathway in GBC cells. Surprisingly, our data
suggested that ASPP2 and aPKC-ι did not affect the expression of YAP1 and target genes, but regulated GLI1, the major mediator of Hh signaling. Interestingly, our results suggested that GLI1
expression in GBC cells may be driven by an ASPP2/aPKC-ι pathway rather than canonical Hh signaling. It is also noteworthy that ASPP2 was expressed in both cytoplasm and nucleus of GBC
cells. In the cytoplasm, ASPP2 KD promoted GLI1 nuclear translocation via aPKC-ι-mediated GLI1 phosphorylation. In the nucleus, ASPP2 depletion facilitated the dissociation of GLI1 from
ASPP2, which led to activation of GLI1 target genes. aPKC-ι, as a target of GLI1, also activated Hh signaling through a positive feedback loop. However, the nuclear import mechanism of ASPP2
remains enigmatic. In addition, further studies are needed to elucidate the mechanisms mediating the interaction between cytokines secreted by tumor cells and macrophages. In conclusion,
our study highlighted the importance of ASPP2 downregulation in promoting the invasion and metastasis of GBC cells via a noncanonical Hh pathway, i.e., the aPKC-ι/GLI1 signaling pathway,
activating downstream cytokines to recruit macrophages that could then infiltrate into the tumor tissues. Accordingly, our findings expand our knowledge of the functions of ASPP2 in cancer,
including its role as a regulator of the EMT in GBC and as an important upstream factor in the TME. These results provide important insights into the development of new, effective
therapeutic approaches for the treatment of GBC. MATERIALS AND METHODS PATIENTS AND SPECIMENS In total, 72 human GBC tissues and paired normal gallbladder tissues (5 cm distant from tumor)
were collected from patients undergoing resection at the Department of Biliary and Pancreatic Surgery, Tongji Hospital (Wuhan, China) between January 2009 and December 2016. Ethical approval
for the use of human samples was obtained from the Tongji Hospital Research Ethical Committee. None of the patients had received any adjuvant therapy before surgery. All cases were
diagnosed by two independent pathologists. The detailed clinicopathological characteristics of the 72 patients with GBC are listed in Supplementary Table S1. IMMUNOHISTOCHEMISTRY The
expressions of ASPP2, aPKC-ι, GLI1, CD68, and CD163 were detected by immunostaining as previously reported41. The positively stained cells were determined in at least five areas at ×400
magnification and assigned to one of the following categories: score 0 (negative); score 1 (<25% positive cells in a specimen); score 2 (25–50% positive cells in a specimen); score 3
(50–75% positive cells in a specimen); score 4 (>75% positive cells in a specimen). The intensity of immunostaining was scored as follows: 1+ (weak); 2+ (moderate), and 3+ (intense). The
above two values are multiplied to produce a total score. The total score ≤4 was considered as low expression and >4 as high expression42. Evaluation of immunostaining was performed by
three independent experienced investigators who were blinded to the patient conditions. CELL LINES Human GBC cell lines NOZ, GBC-SD, and OCUG-1 were generously provided by Prof. Yingbin Liu,
Xinhua Hospital, Shanghai Jiao Tong University School of Medicine, China. These cells were maintained in William’s medium, RPMI 1640 or Dulbecco’s modified Eagle medium (both from Gibco,
Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco). All cell lines were authenticated, mycoplasma-free and cultured at 37 °C in a humidified incubator containing 5% CO2.
ANIMAL STUDY Six-week-old female BALB/c-nude mice were divided into four groups (_n_ = 5 mice per group) and housed under specific pathogen-free conditions in Central Animal Laboratory,
Tongji Medical College. The mice were injected subcutaneously in the upper back with 5×106 transfected GBC cells. The diameter of tumors and the weight of the mice were measured every 3
days. The volumes of the tumor were calculated using the formula: 1/2 (length × width2). For lung metastasis model, the GBC cells (2×106) were suspended in serum-free medium and injected
into tail vein of mice (_n_ = 3 mice per group). All mice were sacrificed 3 weeks later. The number of lung metastases was counted under a microscope. Animal welfare and experimental
procedures were carried out in accordance with the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines and were approved by the Committee on the Ethics of Animal Experiments
of the Tongji Medical College, HUST. Additional experimental procedures are provided in detail in the Supplementary data. STATISTICAL ANALYSES Statistical analyses were performed using SPSS
22.0 software (IBM SPSS, Armonk, NY, USA) or GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA, USA). The results were presented as the mean ± standard deviation (SD). Quantitative data
were analyzed by two-tailed independent Student’s _t_ tests and analysis of variance. Categorical variables were compared using chi-square tests or Fisher exact tests. Clinical correlations
were analyzed using _χ_2 tests, and survival analysis was conducted by the Kaplan−Meier method with log-rank tests. The univariate and multivariate analyses were assessed using a Cox
proportional hazards model. Differences with _P_ values of less than 0.05 were considered statistically significant. REFERENCES * Hundal, R. & Shaffer, E. A. Gallbladder cancer:
epidemiology and outcome. _Clin. Epidemiol._ 6, 99–109 (2014). PubMed PubMed Central Google Scholar * Nakamura, H. et al. Genomic spectra of biliary tract cancer. _Nat. Genet._ 47,
1003–1010 (2015). Article CAS Google Scholar * Kanthan, R., Senger, J. L., Ahmed, S. & Kanthan, S. C. Gallbladder cancer in the 21st century. _J. Oncol._ 2015, 967472 (2015). Article
Google Scholar * Rakic, M. et al. Gallbladder cancer. _Hepatobiliary Surg. Nutr._ 3, 221–226 (2014). PubMed PubMed Central Google Scholar * Lee, D. G. et al. Loss of NDRG2 promotes
epithelial−mesenchymal transition of gallbladder carcinoma cells through MMP-19-mediated Slug expression. _J. Hepatol._ 63, 1429–1439 (2015). Article CAS Google Scholar * Hsu, D. S. et
al. Acetylation of snail modulates the cytokinome of cancer cells to enhance the recruitment of macrophages. _Cancer Cell_ 26, 534–548 (2014). Article CAS Google Scholar * Thiery, J. P.,
Acloque, H., Huang, R. Y. & Nieto, M. A. Epithelial_mesenchymal transitions in development and disease. _Cell_ 139, 871–890 (2009). Article CAS Google Scholar * Mittal, V. Epithelial
mesenchymal transition in tumor metastasis. _Annu. Rev. Pathol._ 13, 395–412 (2018). Article CAS Google Scholar * Li, Q. et al. Correlation of aPKC-iota and E-cadherin expression with
invasion and prognosis of cholangiocarcinoma. _Hepatobiliary Pancreat. Dis. Int._ 7, 70–75 (2008). PubMed Google Scholar * Qian, Y. et al. aPKC-iota/P-Sp1/Snail signaling induces
epithelial−mesenchymal transition and immunosuppression in cholangiocarcinoma. _Hepatology_ 66, 1165–1182 (2017). Article CAS Google Scholar * Vives, V. et al. ASPP2 is a
haploinsufficient tumor suppressor that cooperates with p53 to suppress tumor growth. _Genes Dev._ 20, 1262–1267 (2006). Article CAS Google Scholar * Bergamaschi, D. et al. ASPP1 and
ASPP2: common activators of p53 family members. _Mol. Cell. Biol._ 24, 1341–1350 (2004). Article CAS Google Scholar * Wang, Y. et al. ASPP2 controls epithelial plasticity and inhibits
metastasis through beta-catenin-dependent regulation of ZEB1. _Nat. Cell Biol._ 16, 1092–1104 (2014). Article CAS Google Scholar * Liu, Z. J. et al. Downregulated mRNA expression of ASPP
and the hypermethylation of the 5’-untranslated region in cancer cell lines retaining wild-type p53. _FEBS Lett._ 579, 1587–1590 (2005). Article CAS Google Scholar * Zhao, J. et al.
Epigenetic silence of ankyrin-repeat-containing, SH3-domain-containing, and proline-rich-region- containing protein 1 (ASPP1) and ASPP2 genes promotes tumor growth in hepatitis B
virus-positive hepatocellular carcinoma. _Hepatology_ 51, 142–153 (2010). Article CAS Google Scholar * Song, B. et al. Downregulation of ASPP2 in pancreatic cancer cells contributes to
increased resistance to gemcitabine through autophagy activation. _Mol. Cancer_ 14, 177 (2015). Article Google Scholar * Trigiante, G. & Lu, X. ASPP [corrected] and cancer. _Nat. Rev.
Cancer_ 6, 217–226 (2006). Article CAS Google Scholar * Ge, W. et al. iASPP is an antioxidative factor and drives cancer growth and drug resistance by competing with Nrf2 for Keap1
binding. _Cancer Cell_ 32, 561–573.e566 (2017). Article CAS Google Scholar * Sottocornola, R. et al. ASPP2 binds Par-3 and controls the polarity and proliferation of neural progenitors
during CNS development. _Dev. Cell_ 19, 126–137 (2010). Article CAS Google Scholar * Blotta, S. et al. Canonical and noncanonical Hedgehog pathway in the pathogenesis of multiple myeloma.
_Blood_ 120, 5002–5013 (2012). Article CAS Google Scholar * Xie, F. et al. Aberrant activation of Sonic hedgehog signaling in chronic cholecystitis and gallbladder carcinoma. _Hum.
Pathol._ 45, 513–521 (2014). Article CAS Google Scholar * Pak, E. & Segal, R. A. Hedgehog signal transduction: key players, oncogenic drivers, and cancer therapy. _Dev. Cell_ 38,
333–344 (2016). Article CAS Google Scholar * Ng, J. M. & Curran, T. The Hedgehog’s tale: developing strategies for targeting cancer. _Nat. Rev. Cancer_ 11, 493–501 (2011). Article
CAS Google Scholar * Wang, Y. et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. _Cancer Cell_ 21, 374–387 (2012). Article CAS Google Scholar * Quail, D. F. & Joyce, J. A.
Microenvironmental regulation of tumor progression and metastasis. _Nat. Med._ 19, 1423–1437 (2013). Article CAS Google Scholar * Su, S. et al. A positive feedback loop between
mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. _Cancer Cell_ 25, 605–620 (2014). Article Google Scholar * Qian, B. Z. et al. CCL2 recruits
inflammatory monocytes to facilitate breast-tumour metastasis. _Nature_ 475, 222–225 (2011). Article CAS Google Scholar * Frankenberger, C. et al. Metastasis suppressors regulate the
tumor microenvironment by blocking recruitment of prometastatic tumor-associated macrophages. _Cancer Res._ 75, 4063–4073 (2015). Article CAS Google Scholar * Biswas, S. K. &
Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. _Nat. Immunol._ 11, 889–896 (2010). Article CAS Google Scholar * Yang, Y. et al.
14-3-3zeta and aPKC-iota synergistically facilitate epithelial-mesenchymal transition of cholangiocarcinoma via GSK-3beta/Snail signaling pathway. _Oncotarget_ 7, 55191–55210 (2016). PubMed
PubMed Central Google Scholar * Wang, Y. et al. PKCι regulates nuclear YAP1 localization and ovarian cancer tumorigenesis. _Oncogene_ 36, 534–545 (2016). Article Google Scholar *
Justilien, V. et al. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. _Cancer Cell_ 25, 139–151 (2014). Article CAS
Google Scholar * Matsushita, S. et al. Hedgehog signaling pathway is a potential therapeutic target for gallbladder cancer. _Cancer Sci._ 105, 272–280 (2014). Article CAS Google Scholar
* Atwood, S. X., Li, M., Alex, L., Tang, J. Y. & Oro, A. E. Gli activation by aPKC iota/lambda regulates basal cell carcinoma growth. _Nature_ 494, 484 (2013). Article CAS Google
Scholar * Lu, M. et al. A code for RanGDP binding in Ankyrin repeats defines a nuclear import pathway. _Cell_ 157, 1130–1145 (2014). Article CAS Google Scholar * Lamouille, S., Xu, J.
& Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. _Nat. Rev. Mol. Cell Biol._ 15, 178–196 (2014). Article CAS Google Scholar * Nieto, M. A., Huang, R. Y.,
Jackson, R. A. & Thiery, J. P. EMT: 2016. _Cell_ 166, 21–45 (2016). Article CAS Google Scholar * Chen, R. et al. Downregulation of ASPP2 improves hepatocellular carcinoma cells
survival via promoting BECN1-dependent autophagy initiation. _Cell Death Dis._ 7, e2512 (2016). Article CAS Google Scholar * Qian, B. Z. & Pollard, J. W. Macrophage diversity enhances
tumor progression and metastasis. _Cell_ 141, 39–51 (2010). Article CAS Google Scholar * Franklin, R. A. et al. The cellular and molecular origin of tumor-associated macrophages.
_Science_ 344, 921 (2014). Article CAS Google Scholar * Du, G. S. et al. Expression of P-aPKC-iota, E-cadherin, and beta-catenin related to invasion and metastasis in hepatocellular
carcinoma. _Ann. Surg. Oncol._ 16, 1578–1586 (2009). Article Google Scholar * Konno, R. et al. Expression of survivin and Bcl-2 in the normal human endometrium. _Mol. Hum. Reprod._ 6,
529–534 (2000). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank Prof. Yingbin Liu and Dr. Shanshan Xiang (Xinhua Hospital, Shanghai Jiao Tong University School of
Medicine) for the generous gift of the GBC cell lines and valuable advice regarding cell culture. This study was financially supported by the National Natural Science Foundation of China
(Nos. 81072000, 81172015, 81572417). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Biliary and Pancreatic Surgery, Affiliated Tongji Hospital, Tongji Medical College, Huazhong
University of Science and Technology, 430030, Wuhan, Hubei, China Li Tian, Zhengdong Deng, Lei Xu, Tao Yang, Wei Yao, Lei Ji, Yun Lu, Jian Zhang & Jianming Wang * Department of
Geriatrics, Affiliated Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 430030, Wuhan, Hubei, China Yan Liu Authors * Li Tian View author publications
You can also search for this author inPubMed Google Scholar * Zhengdong Deng View author publications You can also search for this author inPubMed Google Scholar * Lei Xu View author
publications You can also search for this author inPubMed Google Scholar * Tao Yang View author publications You can also search for this author inPubMed Google Scholar * Wei Yao View author
publications You can also search for this author inPubMed Google Scholar * Lei Ji View author publications You can also search for this author inPubMed Google Scholar * Yun Lu View author
publications You can also search for this author inPubMed Google Scholar * Jian Zhang View author publications You can also search for this author inPubMed Google Scholar * Yan Liu View
author publications You can also search for this author inPubMed Google Scholar * Jianming Wang View author publications You can also search for this author inPubMed Google Scholar
CONTRIBUTIONS L.T.: designed the experiments, carried out most of the experiments, collected and analyzed data, and drafted the manuscript. Z.D. and L.X.: carried out the animal experiment,
collected and analyzed the data. T.Y and W.Y.: provided material and technical support. L.J., Y.Lu, J.Z. and Y.Liu: collected and analyzed the clinical data. W.J.: supervised the project and
designed the experiments, obtained funding, and drafted the manuscript. All authors have contributed to and approved the final manuscript. CORRESPONDING AUTHOR Correspondence to Jianming
Wang. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare that they have no conflict of interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE: Springer Nature remains neutral with
regard to jurisdictional claims in published maps and institutional affiliations. Edited by J-E Ricci ELECTRONIC SUPPLEMENTARY MATERIAL SUPPLEMENTARY EXPERIMENTAL PROCEDURES SUPPLEMENTAL
FIGURE LEGENDS FIGURE S1 FIGURE S2 FIGURE S3 FIGURE S4 FIGURE S5 SUPPLEMENTARY TABLES THE STR AUTHENTICATION OF GBC CELL LINES RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed
under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article
are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Tian, L., Deng, Z., Xu, L. _et al._ Downregulation of ASPP2 promotes
gallbladder cancer metastasis and macrophage recruitment via aPKC-ι/GLI1 pathway. _Cell Death Dis_ 9, 1115 (2018). https://doi.org/10.1038/s41419-018-1145-1 Download citation * Received: 04
July 2018 * Revised: 28 September 2018 * Accepted: 09 October 2018 * Published: 02 November 2018 * DOI: https://doi.org/10.1038/s41419-018-1145-1 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative