- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Despite remarkable efficacy, targeted treatments often yield a subpopulation of residual tumor cells in part due to non-genetic adaptions. Previous mechanistic understanding on the emergence
of these drug-tolerant persisters (DTPs) has been limited to epigenetic and transcriptional reprogramming. Here, by comprehensively interrogating therapy-induced early dynamic protein
changes in diverse oncogene-addicted non-small cell lung cancer models, we identified adaptive MCL1 increase as a new and universal mechanism to confer apoptotic evasion and DTP formation.
In detail, acute MAPK signaling disruption in the presence of genotype-based tyrosine kinase inhibitors (TKIs) prompted mitochondrial accumulation of pro-apoptotic BH3-only protein BIM,
which sequestered MCL1 away from MULE-mediated degradation. A small-molecule combination screen uncovered that PI3K-mTOR pathway blockade prohibited MCL1 upregulation. Biochemical and
immunocytochemical evidence indicated that mTOR complex 2 (mTORC2) bound and phosphorylated MCL1, facilitating its interaction with BIM. As a result, short-term polytherapy combining
antineoplastic TKIs with PI3K, mTOR or MCL1 inhibitors sufficed to prevent DTP development and promote cancer eradication. Collectively, these findings support that upfront and transient
targeting of BIM-dependent, mTORC2-regulated adaptive MCL1 preservation holds enormous promise to improve the therapeutic index of molecular targeted agents.
The identification of driver aberrations underlying drug sensitivity to selective kinase inhibitors has revolutionized clinical management of oncogene-addicted non-small cell lung cancer
(NSCLC) over the past two decades [1]. More than 50% of lung adenocarcinomas, the major NSCLC histotype, harbor actionable molecular targets, including EGFR mutations, ALK/ROS1/RET/NTRK
rearrangements, and MET exon 14 skipping variants, among others, and therefore are amenable to genotype-matched therapeutics [2]. Despite pervasive and dramatic initial efficacy, targeted
treatments rarely induce complete tumor eradication, but rather retain minimal residual disease (MRD) which ultimately results in cancer relapse and represents the key obstacle to cure
[3,4,5]. Prior studies reveal that a fraction of malignant cells acquire the ability to evade death programs and enter a drug-tolerant persister (DTP) state to serve as a reservoir for the
emergence of acquired resistance [6,7,8,9], thus establishing the therapeutic potential of MRD-directed regimens. Nonetheless, the molecular basis underpinning cell survival from lethal drug
exposure remains to be fully elucidated.
At present, two non-mutually exclusive mechanisms have been uncovered to govern the formation of DTP phenotype. On one hand, the intrinsic genetic heterogeneity in a cancer cell population
can allow selection and enrichment of preexisting refractory subclones through a Darwinian evolutionary process [8]. On the other hand, mounting evidence implicates non-mutational adaption
of tumor cells as an alternative route toward drug tolerance [10,11,12,13]. The adaptive responses may involve a multitude of intracellular events at different layers, e.g., epigenetic
dysregulation consisting of aberrant histone code [6, 14, 15], transcriptional rewiring that activates the targeted or bypass pathways [16,17,18,19], as well as the less explored alterations
at protein levels. While the importance of epigenetic modifiers and transcriptional feedback loops are exemplified by a large body of recent literature, emerging roles for
post-translational regulation in shaping DTP dynamics have not been comprehensively investigated.
In this study, using a range of representative lung adenocarcinoma models, we unveiled a previously unrecognized interplay between the BCL2 family proteins in promoting DTP development.
Specifically, a rapid increase of MCL1 abundance was triggered by diverse targeted drugs, engendering adaptive resistance to treatment-induced programmed cell death. Mechanistically, BIM
accumulation upon MAPK pathway inhibition and its sequestration of MCL1 attenuated MULE-mediated protein degradation. Of interest, the BIM-dependent MCL1 preservation was associated with
MCL1 phosphorylation by mTOR complex 2 (mTORC2). Based on these findings, we proposed a rational strategy of transient polytherapy to prevent the appearance of DTP cells and potentially
enhance the therapeutic index of current molecular agents.
We reasoned that specific regulation at the protein level, in addition to epigenetic and transcriptional programs, might contribute to tumor cell survival from initial drug treatment. To
test this hypothesis, A549 (harboring KRASG12S), PC9 (harboring EGFRE746-A750 del) and NCI-H3122 (harboring EML4-ALK fusion gene) were selected and treated with genotype-matched tyrosine
kinase inhibitors (TKIs), i.e., the MEK inhibitor trametinib, the EGFR inhibitor erlotinib and the ALK inhibitor crizotinib, respectively. We first considered possible roles for incomplete
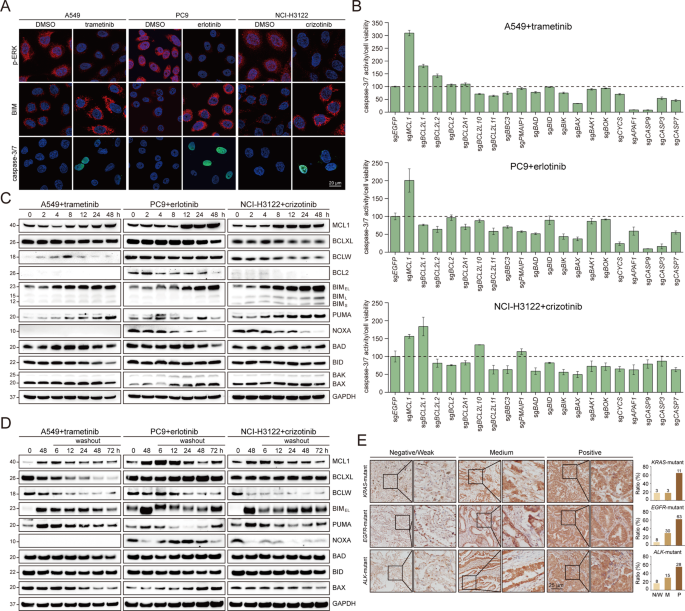
suppression of neoplastic signaling or inadequate induction of cell death in conferring the DTP status. As assessed by immunofluorescence staining (Fig. 1A), 48-hour TKI administration
uniformly diminished ERK phosphorylation in all cells, indicating efficient inhibition of oncogenic signals. Likewise, a dramatic accumulation of BIM protein, reportedly essential for
antitumor effects of targeted therapies [20, 21], was invariably observed at the single-cell level, implying potent engagement of intrinsic apoptotic machinery. However, caspase-3/7 activity
was not detected within every cell, suggesting occasionally compromised capacity to stimulate subsequent activation of caspase cascade. These data pointed to a model that the mitochondrial
apoptotic pathway did not properly proceed in some cases, and prompted us to perform an unbiased CRISPR (clustered regularly interspaced short palindromic repeats) screen on a focused list
of key mediators. As expected, individual knockout of pro-apoptotic BIM (encoded by BCL2L11), cell death effector BAX, or multiple downstream factors, attenuated TKI-induced caspase-3/7
activation. On the contrary, MCL1 depletion consistently potentiated the onset of caspase-dependent apoptosis in response to small-molecule inhibitors (Fig. 1B). In the meantime, we examined
dynamic changes of putative apoptogenic regulators, in particular the BCL2 family members [22], and found that MCL1 protein was notably upregulated with prolonged drug exposure (Fig. 1C).
Intriguingly, elevated MCL1 gradually declined following compound withdrawal, demonstrating the reversibility of this phenomenon (Fig. 1D). Of clinical relevance, although it was not
feasible to monitor MCL1 levels immediately after treatment, we noted that in a large cohort of lung adenocarcinoma with distinct genetic alterations, MCL1 exhibited prominent expression, as
evidenced by positive immunohistochemical staining in 64.7% of KRAS-mutant, 62.4% of EGFR-altered and 54.9% of ALK-rearranged tumor specimens (Fig. 1E) using a validated antibody
(Supplementary Fig. 1A). These observations were extended to a panel of lung cancer cell lines encompassing different oncogenic drivers and expressing various amounts of BCL2 family proteins
(Supplementary Fig. 1B). MCL1 induction by relevant TKIs was confirmed in diverse models harboring EGFR, ALK, MET, HER2, KRAS or BRAF aberrations (Supplementary Fig. 1C). Taken together, we
identified adaptive MCL1 upregulation as a new candidate mechanism by which oncogene-addicted tumor cells could survive the profound antagonistic effects of molecular targeted therapies.
A Representative immunofluorescent images of phosphorylated ERK (p-ERK, T202/Y204), BIM protein and caspase-3/7 activatable nuclear dye in A549, PC9 and NCI-H3122 cells treated with DMSO or
indicated inhibitors (trametinib: 1 µM; erlotinib: 1 µM; crizotinib: 3 µM). Scale bar, 20 µm. B A549, PC9 and NCI-H3122 cells were infected with lentiviral sgRNAs targeting selected genes in
the mitochondrial apoptotic pathway, and treated with indicated inhibitors for 72 h (trametinib: 1 µM; erlotinib: 1 µM; crizotinib: 3 µM). Caspase-3/7 activity and cell viability were
measured by Caspase-Glo 3/7 and CellTiter-Glo assays, respectively. Results were presented as the ratio of caspase-3/7 activity/cell viability. Each column represented the mean value of
three biological replicates, and error bars indicated standard deviation. C A549, PC9 and NCI-H3122 cells were treated with indicated inhibitors over a time course (trametinib: 1 µM;
erlotinib: 1 µM; crizotinib: 3 µM), and cell lysates were analyzed by immunoblotting. D A549, PC9 and NCI-H3122 cells were treated with indicated inhibitors for 48 hours (trametinib: 1 µM;
erlotinib: 1 µM; crizotinib: 3 µM), followed by drug withdrawal lasting various durations. Cell lysates were analyzed by immunoblotting. E Immunohistochemical staining of MCL1 in a cohort of
KRAS-mutant (17 samples), EGFR-altered (101 samples) or ALK-rearranged (51 samples) lung adenocarcinoma. Representative images and quantification for negative/weak, medium or positive
staining were shown.
To validate the essential role of MCL1 in the setting of DTP formation, MCL1 was knocked out using two different single-guide RNAs (sgRNAs) in A549, PC9 and NCI-H3122 cells. Genetic MCL1
deletion did not significantly affect cell viability at baseline, but evidently sensitized lung cancer to TKI treatment and resulted in fewer surviving DTP cells (Fig. 2A). Consistently,
MCL1 loss led to a marked decrease of IC50 (compound concentration causing 50% inhibition) in all three cell lines (Fig. 2B). Western blot analysis showed that 48-hour drug exposure yielded
more cleaved caspase-3, PARP and GSDME in MCL1-depleted cells, demonstrating enhanced apoptosis and pyroptosis (Fig. 2C). In line with these in vitro findings, MCL1 ablation in conjunction
with small-molecule inhibitors efficaciously blocked tumor growth in mouse xenograft models (Fig. 2D). To complement the genetic data, we exploited a pharmacological tool, S63845, a BH3
mimetic with exceptional selectivity and potency against MCL1 [23]. When combined with targeted agents, S63845 effectively activated both apoptotic and pyroptotic cell death, as indicated by
caspase-3, PARP and GSDME cleavage (Fig. 2E), as well as cytochrome c leakage from mitochondria into cytosol (Supplementary Fig. 2A). We also performed dose matrix assays to assess the
effects of drug combination using the Bliss independence model. The heatmaps for Bliss synergy scores illustrated that each TKI and S63845 synergistically reduced tumor cell viability (Fig.
2F), which was further verified by crystal violet staining (Fig. 2G) and IC50 measurements (Supplementary Fig. 2B). Similar results were obtained in a spectrum of oncogene-driven cell lines
treated with S63845 and corresponding targeted inhibitors (Supplementary Fig. 2C). Therefore, genetic or pharmacological MCL1 inhibition impeded DTP generation and enhanced tumor response to
molecular therapeutics.
A The MCL1 gene was knocked out in A549, PC9 and NCI-H3122 cells using the CRISPR-Cas9 system, and MCL1 protein was analyzed by immunoblotting. Drug response (trametinib: 1 µM; erlotinib: 1
µM; crizotinib: 3 µM) upon MCL1 depletion was assayed by phase-contrast microscopy or crystal violet staining, and quantified as shown in bar graphs. *P


:max_bytes(150000):strip_icc():focal(999x0:1001x2)/shawn-mendes-fan-selfie-1-91322-75e4e820aedf48809901c5f76ad838c3.jpg)


