- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The processes leading from disturbed B-cell development to adult B-cell progenitor acute lymphoblastic leukemia (BCP-ALL) remain poorly understood. Here, we describe _Irf4__−/−_
mice as prone to developing BCP-ALL with age. _Irf4__−/−_ preB-I cells exhibited impaired differentiation but enhanced proliferation in response to IL-7, along with reduced retention in the
IL-7 providing bone marrow niche due to decreased CXCL12 responsiveness. Thus selected, preB-I cells acquired _Jak3_ mutations, probably following irregular AID activity, resulting in
malignant transformation. We demonstrate heightened IL-7 sensitivity due to _Jak3_ mutants, devise a model to explain it, and describe structural and functional similarities to _Jak2_
mutations often occurring in human Ph-like ALL. Finally, targeting JAK signaling with Ruxolitinib in vivo prolonged survival of mice bearing established _Irf4__−/−_ leukemia. Intriguingly,
organ infiltration including leukemic meningeosis was selectively reduced without affecting blood blast counts. In this work, we present spontaneous leukemogenesis following IRF4 deficiency
with potential implications for high-risk BCP-ALL in adult humans. SIMILAR CONTENT BEING VIEWED BY OTHERS INTERLEUKIN-7 RECEPTOR Α MUTATIONAL ACTIVATION CAN INITIATE PRECURSOR B-CELL ACUTE
LYMPHOBLASTIC LEUKEMIA Article Open access 14 December 2021 ACTIVATED INTERLEUKIN-7 RECEPTOR SIGNALING DRIVES B-CELL ACUTE LYMPHOBLASTIC LEUKEMIA IN MICE Article 30 June 2021 AN INSTRUCTIVE
ROLE FOR INTERLEUKIN-7 RECEPTOR Α IN THE DEVELOPMENT OF HUMAN B-CELL PRECURSOR LEUKEMIA Article Open access 03 February 2022 INTRODUCTION Two signaling pathways via the Interleukin-7
receptor (IL-7R) and the preB cell receptor (preBCR) ensure an orderly progression of B lymphopoiesis [1,2,3]. ProB cells adhere to bone marrow (BM) stromal cells (SCs) expressing CXCL12 and
VCAM-1 through CXCR4 and VLA-4 respectively, while SC-derived IL-7 induces their proliferation [4]. The formation of the preBCR composed of Igμ protein and the surrogate light chain (ψL),
consisting of λ5 and VPREB, marks the entrance to the preB cell stage. Signaling via the preBCR in turn induces the transcription factor (TF) interferon regulatory factor 4 (IRF4) which is
also critical during T-cell differentiation [5, 6]. In preB cells, IRF4 halts cycling and facilitates recombination of the light chain locus by RAG1/2 [1]. Despite its importance,
_Irf4__−/−_ mice still develop, albeit less, surface (s)Igμ+ mature B cells [7], likely due to a partially redundant function of IRF8. Accordingly, _Irf4,8__−/−_ B progenitors are completely
arrested at the preB cell stage [8]. Disruption of this developmental track can provoke B-cell progenitor acute lymphoblastic leukemia (BCP-ALL). In humans, this disease preferentially
affects children (age 0–19), while most deaths however occur in the adult population [9]. Cases affecting adolescents and young adults (AYA) display a different set of driver mutations
compared to childhood BCP-ALL [10,11,12,13]. Herein, we report that adult _Irf4__−/−_ mice spontaneously develop BCP-ALL with age and delineate the steps from disturbed _Irf4__−/−_ B
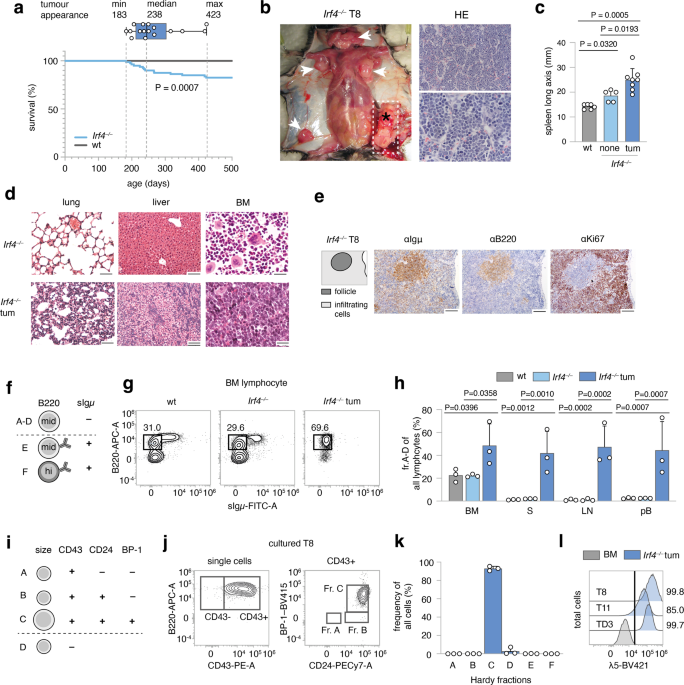
lymphopoiesis to overt leukemia. RESULTS _IRF4_ _−/−_ MICE SPONTANEOUSLY DEVELOP PREB CELL LEUKEMIA Following the serendipitous finding, that some aged _Irf4__−/−_ mice developed tumors and
died, we systematically observed 80 _Irf4__–/–_ mice over time. We detected 14 tumors (incidence 17.5%), that spontaneously appeared in lymph node (LN) areas (mean age: 268d, median: 238d,
Fig. 1a). Tumors were neither detected in mice younger than 150d nor in C57BL/6 wild-type (wt) mice housed in the same room. All tumors (Fig. 1b shows a representative tumor in situ) were
accompanied by lymphadenopathy (arrowheads) and increased spleen size (Fig. 1c). The suspected lymphomatous origin was corroborated microscopically (Fig. 1b, right panels), with infiltration
of mononucleated cells into the BM, lung, and liver (Fig. 1d). Due to the known impaired maturation of _Irf4__−/−_ preB cells [7], spontaneous eruption of preB-leukemia seemed plausible: In
spleen sections (Fig. 1e), infiltrating cells stained positive for both B220 and Igμ (although less than untransformed “follicle” B cells) and Ki67. By flow cytometry, BM samples from tumor
mice harbored an expanded pro/preB cell compartment (Hardy fraction (fr.)A-D [14], B220midsIgμ–) (Fig. 1f, g). Fr.A-D cells were detected also in peripheral lymphoid organs and blood of
tumor mice (Fig. 1h). Following the Hardy classification (Fig. 1i), we determined tumor cells to be fr.C preB cells (B220midsIgμ–CD43+CD24+BP-1+) (Fig. 1j, k, sFig. 1a). In addition, Igμ,
but not Igκ/λ was detected intracellularly (sFig. 1b). Lastly, tumors stained positive for surface λ5, part of the ψL (Fig. 1l). These attributes characterize the disease as preB-I cell
BCP-ALL. To prove clonality, we sequenced the VDJ junctions of the IgH region in three tumors (Supplementary Table 1). Almost all sequences per tumor were identical, demonstrating clonality.
The tumors (three examples) further displayed copy number variations (CNV) (sFig. 1h), targeting differing genomic regions. Finally, transfer of tumor cells robustly elicited leukemia in wt
acceptor mice (sFig. 1d–g) with as little as 500 transferred cells (sFig. 1f, g), indicating bona fide malignancy. B LYMPHOPOIESIS IN _IRF4_ _−/−_ MICE HARBORS A HYPERPROLIFERATIVE PREB-I
CELL COMPARTMENT The uniform appearance of BCP-ALL in _Irf4__−/−_ mice suggested a defined preleukemic pro/preB cell state vulnerable to immortalization. Dimensional reduction of BM samples
stained for B-cell differentiation markers, identified an enlarged fr.C preB cell compartment already in healthy _Irf4__−/−_ mice (Fig. 2a–c, sFig. 2a). This disturbed, but productive B-cell
maturation confirms and extends previous reports [7]. Expression analysis of IL-7Rα and of CD2 (sFig. 2b, c), which accompanies cytosolic Igμ expression [15] further showed an increased
frequency of CD2-/dimIL-7Rα+B220+sIgμ− preB cells in _Irf4__−/−_ mice. Purified BM B220+ cells from _Irf4__−/−_ and wt mice were cultured with IL-7 (Fig. 2d–g) to compare proliferative
capacities. After 6d, _Irf4__−/−_ cells had expanded roughly three-fold, whereas wt cell numbers decreased. Phenotypically, _Irf4__−/−_ cells accumulated at the fr.C stage (Fig. 2e, f) and
expressed surface λ5 (Fig. 2g, h); exactly like _Irf4__−/−_ leukemia. In contrast, wt cells differentiated further, losing surface CD43 (making them fr.D) (Fig. 2e, f) with some cells
expressing sIgμ (fr.E). Thus, IL-7 unmasked the leukemic potential of the fr.C compartment in _Irf4__−/−_ mice with both unchecked proliferation and a reinforced differentiation block.
Notably, IL-7 dependent _Irf4__−/−_ preB-I cell proliferation was blocked by NIBR3049 and Ruxolitinib, inhibitors of the IL-7R downstream actors JAK3 and JAK1 respectively (sFig. 2d). _IRF4_
_−/−_ B-CELL PROGENITORS EXHIBIT REDUCED RETENTION TO THE BM NICHE As overt leukemia is characterized by systemic presence, we tested whether already preleukemic _Irf4__−/−_ B-cell
progenitors would leak from the BM. To reduce the complex Hardy classification, we identified early B-cell progenitors, approximately until the preB-I stage, by B220+CD2–/dim expression
(sFig. 2b). We detected higher frequencies of splenic B220+CD2–/dim cells in _Irf4__−/−_ than in wt mice (Fig. 2h), which accumulated with age. Thus, premature BM evasion adds to the
impaired differentiation and hyperproliferation that characterize _Irf4__−/−_ preleukemia. Potentially, this finding represented a systemic consequence of reduced vicinity to BM niche cells.
We, therefore, analyzed the proximity of _Irf4__−/−_ and wt B220+CD2–/dim cells to IL-7+ BMSCs in situ using _Il-7_eGFP reporter mice (Fig. 2j–m, sFig. 2e, f, Supplementary Movie 1) [16].
In femur cryosections, the B220+CD2–/dim subset (Fig. 2j, arrowheads) but not the whole _Irf4__−/−_ B220+ cell compartment was on average located further away from IL-7+ BMSCs, compared to
wt control (Fig. 2l, m). We excluded differences in IL-7+ BMSC abundance between genotypes (sFig. 2g). B progenitor retention to BM is secured via the interaction of CXCR4 on pro/preB cells
with the IL-7+ BMSC-derived chemokine CXCL12 [17, 18]. Notably, _Irf4__−/−_ pro/preB cells expressed markedly lower levels of CXCR4 compared to wt cells (Fig. 2n). Chemokine migration assays
with Hardy fr.A-D cells showed that _Irf4__−/−_ cells indeed migrated significantly less towards CXCL12 (Fig. 2o). Thus, reduced CXCR4-CXCL12 interaction likely induces the systemic seeding
of _Irf4__−/−_ B progeny. Inversely, direct cell interactions are likely not responsible, because _Irf4__−/−_ and wt fr.A-D cells adhered equally to monolayers of OP-9 cells in vitro (sFig.
2h). THE IL-7-JAK-STAT-AXIS IS RECURRENTLY ALTERED IN _IRF4_ _−/−_ LEUKEMIA Most likely, a second, acquired genetic alteration was necessary for bona fide leukemia development and arose
with low frequency per time, explaining the affected age and relatively low penetrance. Importantly, IL-7 deprivation of BM-evaded pro/preB cells should create strong survival stress and
potential selection pressure for bona fide leukemogenesis. To identify somatically acquired mutations, we performed whole-exome sequencing (WES) of three independent tumors (T8, T10, T11)
compared to sorted B220+sIgµ− cells from _Irf4__−/−_ BM. Comparisons of the single nucleotide variants (SNVs) between the three samples identified nine genes affected in all three tumor
samples (Fig. 3a). Out of these, SNVs in four genes (_Rrs1, Jak3, AW82073,_ and _Duxf3_) showed alternate base frequencies close to 0.5 or 1 (Fig. 3b), suggesting that they could be present
on one or both alleles of all leukemic cells. Although we did not exclude the oncogenic potential of the other three genes, we focused on _Jak3_, because it is associated with IL-7R
signaling. We detected _Jak3_ mutations also in other tumors TD1, TD2, TD3, and T14 by Sanger- and RNA-sequencing (Fig. 3c, Supplementary Table 2). Thus, seven out of seven tested tumors
carried _Jak3_ mutations (“JAK3mut”). All mutations targeted either the active kinase domain or the pseudokinase domain regulating JAK3 activity. Some of these SNVs have been described
before [19]. Further, using two different classifiers, no gene fusions could be detected (see methods). Analysis of typical BCP-ALL genes [20] identified some respective mutations at low
frequencies, indicative of subclonal events (Fig. 3d). Among these, mutations in _Jak1_, the partner to JAK3 in IL-7R signaling, were detected in both T8 and T11. To analyze the role of the
JAK3mut, we transduced _Irf4__−/−_ preB-I cell cultures with retroviruses (RVs) encoding no or wt JAK3 or the JAK3mut R653H and T844M. Culturing transduced cells in the presence of αIL-7 to
test for IL-7 independency unexpectedly resulted in cell death after a few days with no benefit for cells expressing JAK3mut (sFig. 3a, b). To test if JAK3mut would confer advantages with
limited IL-7, RV-infected _Irf4__−/−_ preB-I cell cultures were exposed to decreasing IL-7 concentrations (Fig. 3e–g). At 0.1 and 0.01 ng/ml IL-7, both JAK3mut-, but not JAK3wt-RV led to the
outgrowth of transduced over untransduced cells after 6d of culture (Fig. 3f, g). Thus, JAK3mut confer IL-7-hypersensitivity, but not -independency. Accordingly, ex vivo cultured
_Jak3_-mutated T8 and T11 cells also still depended on IL-7 (sFig. 3c, d). However, the tumor cells exhibited increased proliferation (sFig. 3e) and λ5 surface retention (sFig. f, g) in
decreased IL-7 concentrations when compared to wt or _Irf4__−/−_ preB cell culture. _AICDA_ IS UPREGULATED IN _IRF4_ _−/−_ PREB CELLS BY LPS AND DEPRIVATION OF IL-7 Because six out of seven
_Jak3_ mutations were C to T base exchanges (Table 2), we suspected a specific mutagenic agent. DNA-editing enzymes including the APOBEC family member AID can deamidate cytosines, e.g.,
during somatic hypermutation [21, 22]. Repair mechanisms most often ultimately cause C to T conversions [23, 24]. Notably, AID is induced in wt preB cells by IL-7 withdrawal and LPS
stimulation and acts as a facilitator of human BCP-ALL [25]. Therefore, we compared _Aicda_ expression in sorted _Irf4__−/−_ and wt fr.A-D cells to that of wt mesenteric (m)LN- and CD4+
TH1-cells as controls and to individual leukemia samples. While mLN cells highly expressed _Aicda_, fr.A-D preB- and leukemia cells, but not TH1-cells, also expressed readily detectable
amounts (Fig. 3h). Furthermore, like their wt counterpart [25], in vitro expanded _Irf4__–/–_ preB-I cells upregulated _Aicda_ further under LPS treatment and during IL-7 withdrawal (Fig.
3i). This finding can explain how BM evasion and exposure to pathogens might cooperatively initiate mutagenic processes via AID in vulnerable _Irf4__−/−_ preB-I cells. To test if T8
exhibited signs of previous AID activity on a global level, we analyzed C:T/G:A-transition frequencies in WES of T8, as well as BM-sorted _Irf4__−/−_ and wt fr.A-D cells compared with
matched tail-tip samples. Indeed, we found a marked preponderance of C:T/G:A-transitions in T8, when filtering on putative somatic core SNVs (Fig. 3j). _JAK3_ MUTATIONS IN MICE MIRROR _JAK2_
MUTATIONS IN HUMAN PH-LIKE ALL Next, we compared _Irf4__−/−_ leukemia to the complex landscape of human BCP-ALL subtypes (reviewed in refs. [11, 26, 27]), using a published human BCP-ALL
cohort for which a random forest classifier had been established (Methods for details) [28]. Only mildly (potentially due to the interspecies comparison) elevated prediction scores were
generated for Ph+, Ph-like, KMT2a- and DUX4-rearranged human BCP-ALL (sFig. 4a, b). Since all of these except Ph-like are defined by specific gene rearrangements, that we had not detected in
_Irf4__−/−_ mouse leukemia, we excluded them as comparable candidates. Ph-like ALL harbors recurrent genetic alterations in signaling molecules, especially in CRLF2 and JAK2 [20]. While
BCP-ALL overall preferentially affects children, the incidence of the Ph-like subtype increases from 10% in children to above 25% in AYA and adults [20, 29], reminiscent of the older age of
_Irf4__−/−_ leukemic mice. Furthermore, a published dataset of 154 Ph-like BCP-ALL cases exhibited 10-fold reduced _IRF4_ transcripts, when compared to other BCP-ALL subtypes [20]. While in
human Ph-like ALL, _Jak2_ is commonly mutated, we report recurrent _Jak3_ mutations in _Irf4__−/−_ mice. As both proteins are part of distinct but similar signaling complexes in B-cell
progenitors (Fig. 4a), we investigated structural and functional similarities between the specific _Jak3_ and _Jak2_ mutations. Comparisons of amino-acid sequences revealed high protein-wide
interspecies and intermolecular similarities for both proteins (Fig. 4b). Mapping the two amino acids R653 and T844 (mutated in _Irf4__−/−_ mice) onto JAK3 structure predictions, generated
by the alpha-fold algorithm [30], revealed that the two amino acids are in direct contact at an interface of JH1-JH2 domains (Fig. 4c). This interface specifically is highly conserved in
JAK2 compared to JAK3 (Fig. 4d, f, sFig. 4c, d). Intriguingly, R683 (corresponding to R653 in JAK3) is by far the most commonly mutated amino acid in JAK2 in Ph-like ALL, while mutations
targeting T875 (corresponding to T844) also have been described [31]. These findings suggest that mutations in human JAK2 and mouse JAK3 affect a highly similar functional hotspot. As
mentioned above, JAK2 and JAK3 are part of distinct, but similar receptors: JAK3 binds the common γ-chain involved in IL-7 signaling, while JAK2 associates with CRLF2 involved in TSLP
signaling. Both signals involve the IL-7Rα chain and the same downstream pathways (STATs, PI3K) [32]. Therefore, the alternative presence of JAK3/JAK2 mutations between mouse and human
BCP-ALL might reflect different cytokine preferences. Human proB/preB cells proliferate in response to both TSLP and IL-7 [33]. However, in _Irf4__−/−_ BM cells IL-7, but not TSLP induced
robust proliferation (Fig. 4g) as well as high frequencies and absolute counts of CD43+ (Fig. 4h) and λ5+ preB cells (Fig. 4i, j). IRF4 RE-EXPRESSION LEADS TO CELL DEATH AND DIFFERENTIATION
As _Irf4_ deletion was a prerequisite for leukemia in our model, we examined the effect of forced IRF4 re-expression, using RVs coding for GFP alone (EV-RV) or plus IRF4 (IRF4-RV). When
re-introducing IRF4 into T8 or T11, GFP+ IRF4-expressing-, but not GFP+ control cells gradually disappeared over time (Fig. 5a). AnnexinV/PI stainings confirmed apoptosis (not shown).
Further, we noted the loss of surface λ5-expression induced by IRF4-RV (Fig. 5b, c). Comparing the transcriptomes of still viable cells 24 h after transduction revealed strong induction of
“apoptotic process” and “innate immune response” gene ontology (GO) gene-sets (gs) (Fig. 5d). Markov clustering of GO gs affected by IRF4 re-expression (Fig. 5e, f) further identified
several coregulated B-cell differentiation gs (Fig. 5f), with downregulated ψL components _Igll1_, _Vpreb1,_ and _Vpreb2_, but upregulated differentiation genes including _Igμ_, _Igκ,_ and
_Blnk_ (Fig. 5g, h, sFig. 5a, b). Similar results were obtained for T11 (sFig. 5c, d). Therefore, fully transformed leukemia remained targetable by IRF4 re-expression. SMALL COMPOUND AGENTS
AFFECTING _IRF4_ _−/−_ LEUKEMIA _CELLS_ IN VITRO Next, we screened a collection of kinase inhibitors for their capacity to kill _Irf4__−/−_ leukemia cells in vitro. We included NIBR3049
targeting JAK3, Ruxolitinib, an inhibitor of JAK1/2 (downstream of JAK3), and Dexamethasone, a cornerstone for treating lymphomatous malignancies. Furthermore, we included inhibitors of NFκB
(IKK, TAK1), JNK, MEK, ERK, PP2A, GFI1, FAK, and the Bruton tyrosine kinase (BTK) acting downstream of the BCR. A variety of these substances potently killed tumor cells (sFig. 6a),
implying the involvement of multiple pathways in leukemia cell survival. The efficacy of Ruxolitinib and NIBR3049 corroborated our results concerning _Jak3_ driver mutations. Furthermore,
inhibitors of GFI1 and PP2A, as well as NFκB and JNK, were potent. In contrast, inhibiting BTK, MEK and ERK had no impact. IN VIVO THERAPY OF ESTABLISHED _IRF4_ _−/−_ B-ALL Next, we
implemented JAK inhibition as in vivo treatment for _Irf4__−/−_ leukemia. We began induction therapy with Dexamethasone around day 12 after adoptively transferring 3 × 105 T8.1 cells i.p.
into wt mice (Fig. 6a), when overt leukemia was noted in peripheral blood (pB) (Fig. 6b “pre”). After 7d of treatment, leukemic cell numbers in pB were robustly reduced (Fig. 6b “post”),
although few cells reproducibly remained detectable (Fig. 6c). Maintenance therapy was continued with Ruxolitinib or vehicle control by oral gavage twice daily for the following 12 days
(Fig. 6a, c). Importantly, the half-life of Ruxolitinib in mice is only 0.8 h (“Australian Public Assessment Report for Ruxolitinib”, Australian Government), implying that any observed in
vivo effectiveness might be underestimated. Despite maintenance therapy, leukemic cells in pB reappeared, with no significant difference between treatment groups (Fig. 6c). However,
treatment with Ruxolitinib resulted in a clear survival benefit (Fig. 6d) and marked improvement of a prominent neurological symptom: in sham-treated animals, temporary limpness of the tail
and hind legs occurred seconds after gavage, which we quantified using a newly established scoring system (ranging from 0 to 3, see Methods). Mechanistically, ultrasound imaging revealed an
echogenic paravertebral mass (sFig. 7a, b) in score 3, but not score 0 mice. By histology, score 3 correlated with severe infiltration of blasts into the spinal canal (X in Fig. 6f),
extending into spinal nerve roots (arrowhead in Fig. 6f). Therefore, paraparesis likely represented a manifestation of mouse leukemic meningeosis, exacerbated by gavage-induced increases in
intraabdominal pressure. Paraparesis was reproducibly relieved during Ruxolitinib treatment (Fig. 6e), correlating with the suppression of perimyelon infiltration that ensued in
vehicle-treated mice after the end of induction therapy (Fig. 6f, h). In contrast, the severely impaired hematopoiesis in sham-treated mice, indicated by low CAE+ cell frequencies, was not
significantly ameliorated by Ruxolitinib (Fig. 6i, j). These findings raised the possibility that Ruxolitinib preferentially targets infiltration of solid organs rather than BM or pB.
Accordingly, Ruxolitinib fully blocked the liver infiltration as observed in sham-treated mice (Fig. 6k, l). As tissue infiltration is regulated by homing receptors, we treated T8.1 and T8.2
cells with Ruxolitinib in vitro and recorded the expression of CD29 (integrin β1), which pairs with various integrin alpha chains involved in cell- and tissue adhesion [34, 35]. Notably, on
T8.1 and T8.2, Ruxolitinib reduced CD29 expression dose-dependently (sFig. 6c–e) while it even slightly increased expression of MHC I molecules (H2Db, H2Kb), stained as a specificity
control. DISCUSSION The herein described spontaneous leukemogenesis in _Irf4__−/−_ mouse stresses the particular vulnerability of preB-I cells. Our data provide insights for (a) conditions
promoting leukemogenesis, (b) functional consequences of _Jak_ mutations, (c) parallels of mouse and human BCP-ALL and (d) potential in vivo treatment: (a) We provide evidence for a two-hit
leukemogenesis model: The first hit (_Irf4_ loss) resulted in reduced differentiation, IL-7-dependent hyperproliferation, and impaired retention to the BM niche (Fig. 7). A second hit
(targeting _Jak3_ in our model) created a dominant survival signal, probably founding overt preB-I leukemia. The induction of BCP-ALL in _Irf4__−/−_ mice are similar to _Ikzf1_ and _Pax5_
mutated mouse models [36,37,38], implying similarities between these TF-alterations. Probably, one shared mechanism is the differentiative impairment. Importantly, for _Irf4__−/−_ fr.A-D
cells we even detect slightly higher levels of _Pax5_ compared to wt fr.A-D cells (sFig. 8), ruling out that the findings in _Irf4__−/−_ mice merely mirror those of _Pax5_ deficiency. The
reverse remains conceivable; that _Ikzf1_ and _Pax5_ mutations converge in lowering IRF4 expression. In addition to mice mutated in _Pax5_ or _Ikzf1_, _Irf4/Irf8__−/−_, and _Irf4/Spi1__−/−_
mice have been shown to develop leukemia early in life at a high incidence [39, 40]. Contrasting these studies, we report that a single deficiency for IRF4 fully suffices for leukemogenesis.
We excluded secondary alterations in _Irf8, Spi1_ in our model: we found unchanged expression and gene sequence of IRF8 (not shown) and normal amounts of _Spi1_ transcripts (sFig. 8a) in
_Irf4__−/−_ fr.A-D cells. The single IRF4 deficiency models potential clonal initiating events better than _Irf4/Irf8__−/−_ or _Irf4/Spi1__−/−_ mice, because _Irf4__−/−_ mice harbor
productive B-cell development. We newly describe that a preleukemic alteration can lead to reduced BM retention, presenting a tentative explanation for the induction of mutagenic signals, as
deprivation from IL-7 and exposure to bacterial compounds can cooperatively induce the mutagenic agent AID [25]. (b) Why do _Jak3_ mutations only lead to enhanced sensitivity to, but not
complete independence of IL-7? Analysis of JAK3 and JAK2 structure implied that mutations of R683/R653 and T875/T844 might decrease JH1-JH2 interaction strength. This would imply reduced
auto-inhibition as the GOF mechanism—in line with findings for the JAK family member TYK2 [41]. This alone cannot explain cytokine independency, owing to the receptor biology: The two
preassembled receptor chains keep JAKs intracellularly separated [42]. Ligand binding is needed for a conformational change that brings JAKs into the proximity needed for
cross-phosphorylation. To explain our observations, we propose an oncogene model with two equilibria (Fig. 8a, b): the first is determined by cytokine concentration and dictates the
probability of receptor conformation change (Fig. 8a). The second, independent equilibrium (Fig. 8b), is determined by the interaction strength at the JH1-JH2 interface and dictates the
probability of JH1 and JH2 dissociation. Only the combination of the “bound” and “active” state (Fig. 8c) would result in the elicitation of a signal (Fig. 8c, green frame). In this model,
JAK mutations would only affect the second equilibrium (Fig. 8d, red arrows). Sporadic ligand binding would still be needed for elicitation of signaling. The model stringently predicts the
better exploitation of low cytokine concentrations for JAKmut that we observed in vitro. Figure 8d depicts theoretical probabilities of receptor states in the presence (top row) or absence
(bottom row) of JAK mutations. Our findings that JAK3mut confer heightened cytokine sensitivity, but not -independence, is in contrast to what has been found for JAK2-R683G mutants expressed
in the commonly used BaF3 cell line [43]. However, BaF3 cells depend on IL-3 and not IL-7Rα cytokines (i.e., IL-7 or TSLP). Therefore, it remains conceivable, that the IL-3 receptor
provides a different physiology, which may deviate from the IL-7R physiology in primary B progenitors. (c) The finding that _Irf4__−/−_ preB-I cells respond preferentially to IL-7 over TSLP
presents a possible explanation, why mouse models of BCP-ALL acquire _Jak3_ mutations, and human Ph-like ALL typically harbors _Jak2_ mutations. Our comparison of JAK structure predictions
yielded corresponding mutations likely to elicit similar downstream effects. (d) Lastly, our in vivo experiments reinforce Ruxolitinib as a potential treatment for JAK-driven BCP-ALL. The
compound represents an important therapeutic agent in myeloproliferative disease and is already studied for the treatment of Ph-like-ALL [44, 45]. We describe a preferential effect of
Ruxolitinib on CNS- and organ infiltration, potentially due to reductions in integrin expression on leukemia cells. These effects are of potential translational importance because current
CNS-targeted therapies for ALL remain toxic. METHODS MICE C57Bl/6 mice were purchased from Charles River, Sulzfeld, Germany. _Irf4__−/−_ mice [7] and _Il-7__eGFP_ mice [46] (provided by Koji
Tokoyoda, DRFZ Berlin) were bred on the C57Bl/6 background and housed in the animal facility of the Biomedical Research Center at the University of Marburg, Germany. If not stated
otherwise, all mice used in the presented experiments were 8–12 weeks old and sex-matched. TUMOR CELL LINES AND CELL CULTURE Stable tumor cell lines T8.1, T8.2, and T11 were established from
primary _Irf4__–/–_ leukemia cells (derived from primary tumor 8, i.e., T8, or tumor 11 (T11)) by culturing them on a monolayer of irradiated (30 Gy) ST2 stromal cells [47] grown to
confluency in Opti-MEM medium (31985070, ThermoFisher Scientific) supplied with 1% cell culture supernatant from JIL-7.6 J558 cells [48] (a gift from Fritz Melchers, Berlin) as a source of
IL-7. After several passages, T8 and T11 cells grew independently of ST2 cells. For in vitro inhibitor experiments, 2.5 × 105 T8.1 or T8.2 cells (or T11 cells) were cultured in 500 µL RPMI
medium in 48 well plates in the presence of the indicated concentrations of inhibitors. To determine the percentage of viable cells, samples were stained using Annexin V and propidium iodide
(PI) (see below) after 48 h. Substances used include Defactinib (S7654, Selleckchem), Oxocaenol (O9890, Sigma), GANT61 (Sigma, G9048), SP203580 (EI-286-0001, Enzo), SP600125 (EI-305-0010,
Enzo), PD98059, Promega), Ibrutinib (S2680, Selleckchem), BAY11-7082 (ALX-270-219, Alexis), Dexamethasone (PZN 08704491, mibe GmbH) and Ocadaic acid (O4511, Sigma). MURINE PRO/PREB CELL
CULTURES Femur and tibia bones from 8 to 12 weeks old mice were explanted and cleaned from adherent tissues. Cells were extracted via centrifugation at 11 × 103 RPM for 10 s. Total BM cells
were enriched for B220+ (sIgµ−) B lineage cells using an in-house magnetic-activated cell sorting protocol. Briefly, whole bone marrow cells were stained with a mix of FITC-conjugated
antibodies to (Igµ), CD11b, B220, Ter119, CD49b, CD4, and CD8 (all from eBioscience), followed by incubation with an anti FITC/streptavidin/biotin/magnetic bead complex (Miltenyi Biotec) and
magnetic sorting using a microcentrifugation tube stand (Miltenyi Biotec) [49]. Sorting efficiency, as confirmed by flow cytometry, routinely exceeded 90%. Cells were seeded at a density of
1 × 105 cells per well in 200 µL RPMI complete (96-well plates, Greiner). Pro/preB cell cultures were propagated with 10 ng/mL rmIL-7 (217-17, Peprotech) in RPMI-1640 medium complete
(R8758, Sigma-Aldrich, supplemented with: 10% FCS (Sigma-Aldrich), 2 mM l-glutamine (Biochrom), 50 µM β-mercaptoethanole (Sigma-Aldrich), 0.03/0.05 g per 500 mL Penicillin G/Streptomycin
Sulfate, 1% non-essential amino acids (PAA Laboratories)). In some experiments, pro/preB cells (1.25 × 106/mL medium) were treated for 24 h with LPS (Sigma, 1 µg/ml), anti-IL-7 (BioXCell, 10
µg/ml), rmIL-7, or respective combinations, before generating mRNA for qRT-PCR. TRANSWELL MIGRATION ASSAY AND OP-9 ADHESION ASSAY For the transwell migration assays, Hardy fr.A-D cells were
magnetically sorted from BM of wt and _Irf4__−/−_ mice as described above (with addition of FITC-conjugated anti-Igµ antibody), and 2 × 105 cells in RPMI (without additives, FCS-free)
containing 10 ng/mL rmIL-7 seeded in 50 µL in the top chamber of 96-well 5 µm pore uncoated 96-well transwell plates (HTS transwell® Corning). The bottom chamber was flooded with 200 µL RPMI
containing indicated concentrations of rmCXCL12 (Peprotech). After 16 h, inserts were removed, cells in the bottom chamber were collected, counted, and analyzed for B220 surface expression
using flow cytometry. The fraction of migrated cells was calculated as n(migrated) × freqB220(migrated)/n(input) × freqB220(input). Normalization to B220+ cells reduced interexperimental
differences due to differences in cell purity after magnetic selection. For OP-9 adhesion assays, 5 × 103 OP-9 cells (a gift from Hyun-Dong Chang, DRZF Berlin) were seeded in 96-well
microtiter plates 24 h before the assay. On the day of the assay, fr.A-D cells were purified as above and 2 × 105 fr.A-D cells were seeded on top of OP-9 monolayers in RPMI complete + 10
ng/mL rmIL-7. Plates were centrifuged briefly to accelerate cell descension. After 1 h, suspended cells were collected in the supernatant and by washing OP-9 monolayers two times with PBS.
FLOW CYTOMETRY AND CELL SORTING For surface staining of B lineage markers, cells were harvested, resuspended in PBS/1% FCS and stained with anti-B220 (RA3-6B2, Biolegend), anti-Igµ (II/41,
BD Bioscience), anti-CD43 (RM2-5, Biolegend), anti-CD24 (M1/69, invitrogen), anti-BP-1 (BP-1, BD Bioscience), anti-CD2 (RM2-5, Biolegend), anti-CXCR4 (L276F12, Biolegend), anti-CD127
(=IL-7Rα) (A7R34, BD Bioscience), anti-CD179b (=λ5) (LM34, BD Bioscience) as indicated (20 min at room temperature in the dark). All antibodies were employed at a dilution of 1:500.
Fluorescence was recorded using either a FACS Aria III (BD) or an Attune NxT (Thermo-Fisher) analyzer. Data analysis was performed using the FlowJo V10 software (BD). For dimensional
reduction, we used the t-Distributed Stochastic Neighbor Embedding (tSNE) [50] algorithm built into FlowJo V10. Epitopes on BM cells from _Irf4__−/−_ and wt control mice used for dimensional
reduction analysis comprised B220, sIgµ, CD43, CD24, BP-1. For RNA and WES analyses, BM cells were surface labeled for B220 and sIgµ expression, and B220+sIgµ− cells were sorted using a
FACS Aria III (BD Bioscience). Sorting efficiency was routinely above 95%. To determine cell viability, AnnexinV/PI staining was performed using 5 µL AnnexinV (640905, Biolegend) per 500 µL
HBSS. After 20 min of incubation at room temperature in the dark, 1 µL PI (421301, Biolegend) was added, and cells were immediately measured. CNVS ANALYSIS CNVs were analyzed in tumor
samples 8, 10, and 14 and compared to _Irf4__−/−_ normal tail tissue. Whole DNA was extracted from 5 × 106 cells per sample using the Macherey-Nagel NucleoSpin Tissue kit (REF 740952.50)
according to the manufacturer’s protocol. Library preparation was performed using the Illumina Nextera DNA kit according to the manufacturer’s instructions. Sequencing was performed on an
Illumina-HiSeq-1500 platform in rapid-run mode at the Genomics Core Facility of Philipps-University Marburg. Fastq quality control was performed using custom scripts. Raw sequenced reads
were aligned to the Ensembl Mus musculus reference (revision 79) using Bowtie2 (version 2.0.0) [51] with standard parameterization. Analysis of CNVs was performed using the cn.mops (Copy
Number estimation by a Mixture Of PoissonS) package (version 1.18.1) [52] with the following parametrization: prior impact = 1, lower threshold −0.9, upper threshold = 0.5 minimum width = 4.
Window length was set to 10000 and the algorithm was run in unpaired mode. BM CRYOSECTIONS AND ANALYSIS OF B PROGENITOR VICINITY TO IL-7+ BMSCS Mouse femora from _Irf4__−/−_ or _wt
il-7__eGFP_ reporter mice were explanted, cleaned from soft tissues, and fixated overnight in 4% PFA PBS (Alfa Aesar). Samples were then dehydrated by incubation in 30% sucrose in PBS for 24
h. Dried and dehydrated femora were snap-frozen in cryomolds® (Tissue-Tek) using O.C.T freezing medium (Tissue-Tek) by being placed in a beaker of Hexan, surrounded by a beaker of Acetone
and dry ice. Samples were stored at −20 °C until processing. Cryosections of 7 µm were generated with a Leica cryostat (DB80 LX microtome blades, Leica) using Kawamoto tape [53]
(Section-lab) as described before [54]. Cryosections were stained with antibodies against B220 (RA3-6B2, Biolegend), CD2 (14-0021-85, eBioscience, conjugated to AF555 using lightning-Link
kit, Abcam), GFP (Rockland goat polyclonal anti-GFP, 600-101-215) with secondary rabbit anti-goat F(ab’)2 AF488 (thermo-scientific A21222). Samples were then mounted in DAPI ProLong Gold
Antifade (ThermoFisher Scientific). Images were recorded using a Leica confocal (SP8i) microscope. Image analysis was performed in IMARIS (version 9.7.2). HISTOLOGICAL ANALYSES Tissue
samples were immediately fixed in 4% PFA PBS solution. Histological analysis was performed on 3 µm thick sections from paraffin-embedded tissue as described previously [55]. Briefly,
rehydrated paraffin sections were first blocked with 0.3% H2O2 and goat normal serum. For immunohistochemical (IHC) stainings, rat antibodies against CD45R/B220 (clone RA3-6B2, BD) and KI67
(clone TEC-3, Dako) were then incubated on the tissue slices and the bound antibody was detected with biotinylated goat anti-rat IgG (Southern Biotechnology). Bound antibody was visualized
with the Vectastain-kit (Vector Laboratories) according to the manufacturer’s protocol. Hematoxylin-Eosin (HE) stainings were performed according to standard procedures. Cells of the
granulocytic lineage were stained on paraffin-embedded tissues with the Naphthol AS-D Chloracetate (Specific Esterase, CAE) Kit (Ref: 91C-1KT, Sigma-Aldrich) according to the
manufacturer's protocol. In the in vivo therapeutic experiments, we calculated the narrowing of the spinal cord using the equation _A_t/(_A_sca–_A_sp), where _A_sca is the area of the
spinal canal, _A_t that of the tumor, and _A_sp that of the spinal cord area. Two different cross-sections per animal were examined. The infiltration of the liver was calculated by dividing
the tumor area in the liver by the whole area of the liver section. Three whole liver sections were analyzed per animal. All measurements were performed using Fijii [56]. WHOLE-EXOME
SEQUENCING AND BIOSTATISTICAL ANALYSIS To determine SNV within leukemia samples, genomic (g)DNA was extracted both from primary _Irf4__−/−_ tumors as well as FACS-sorted control B220+sIgµ−
BM fr.A-D cells using the High Pure PCR Template Preparation kit from Roche (11796828001). The integrity of the resultant gDNA was confirmed in a 2% Agarose gel. Macrogen in Seoul performed
SureSelect All Exon V6 library preparation and sequenced exons on a NovaSeq platform producing 2 × 150 bp reads at a coverage of 100× (50× on-target coverage). Fastq quality control was
performed using FASTQC (version 0.11.9). Raw sequenced reads were aligned to the Ensembl Mus musculus reference (revision 96) using STAR (version 2.6.1d) using default parametrization.
Soft-clipped aligned reads were then subjected to variant calling analysis. Position-wise pile-up files were generated using samtools (version 1.9) with the mpileup option and a pile-up
quality threshold of 15, both for single sample and matched variant calling. Subsequently, variant calling was performed for SNP and InDel detection using VarScan2 (version 2.3.9) on single
samples with the following parametrization: sampling depth = 100,000, minimum variant frequency = 0.05, minimum coverage = 8, minimum variant reads = 2, minimum average read quality = 15 and
a _p_ value threshold was set to 0.05. Only primary alignments were considered, the strand filter was enabled, and duplicates were removed. As a comparison, matched tumor-normal variant
calling was performed with VarScan as well using an identical parameter setting with the somatic _p_ value threshold set to 0.05. For Fig. 3n raw sequenced reads were aligned to the Ensembl
Mus musculus reference (revision 96) using Burrows-Wheeler Aligner (BWA version 0.7.17) using default parametrization [57]. Prior to variant calling, aligned reads were filtered using a
custom filter that excludes reads with more than three mismatches, more than two indels, or a mapping quality below 20 using pysam (version 0.16.0.1). Duplicates were marked and removed
using Picard (GATK version 4.1.6.0) [58]. Filtered aligned reads were then subjected to variant calling analysis. Position-wise pile-up files were generated using samtools (version 1.9) with
the mpileup option and a minimal base quality threshold of 20. Subsequently, variant calling performed for SNP detection using VarScan2 (version 2.4.4) using matched tumor-normal (somatic)
mode with the following parametrization: sampling depth = 100,000, minimum variant frequency = 0.2, minimum coverage = 8, minimum variant supporting reads = 5, minimum average read quality =
20 and a somatic _p_ value threshold was set to 0.05. Only primary alignments were considered, and the strand filter was enabled. SNP calls were filtered to high confidence somatic
mutations using VarScan’s somaticFilter method, SNPs with a variant allele frequency above 0 in the matched reference sample were excluded. SANGER SEQUENCING AND POLYMERASE CHAIN REACTION
SNVs in the JAK3 gene were confirmed by Sanger sequencing of PCR fragments spanning the _Jak3_ pseudokinase and kinase region (primers used for PCR amplification and Sanger Sequencing: mJAK3
for, mJAK3 rev s. Supplemental Data). Sequencing services were provided by Microsynth Seqlab. To determine the clonality of tumor cells, the VµH region was amplified by PCR. Amplicons were
run on an agarose gel and extracted using the QIAquick Gel Extraction Kit (Qiagen). DNA fragments were then cloned into the vector pJet1.2 (Thermo Scientific) and transformed into DH10B E.
coli. The indicated numbers of clones (Fig. 1g) for each PCR amplicon were sequenced and aligned with software from IMGT/V-quest [59]. RETROVIRAL TRANSDUCTION OF _JAK3-_MUTANTS AND IL-7
INDEPENDENCY ASSAY The coding sequence of murine _Jak3_ was amplified from pCineo-Jak3 (a gift from Olli Silvennoinen from Tampere-university in Finland) and cloned into the pMSCV-Thy1.1
expression plasmid using _BamHI_ and _SalI_ restriction digestion. Site-directed mutagenesis was performed following the manufacturer’s protocol using the Quick-Change II site-directed
mutagenesis kit (Agilent Technologies; primers employed are listed in the Supplemental Materials). Viral supernatant from mutated pMSCV-Thy1.1-Jak3 constructs was produced as described
previously [49]. For viral transduction, 5 × 105 IL-7 dependent primary _Irf4__−/−_ preB-I cell cultures were resuspended in 400 µL RPMI medium (D5030, Sigma-Aldrich) with 600 µL viral
supernatant and 1.5 µL polybrene and spun in culture plates at 2700 rpm for 90 min at 37 °C. Cells were then replenished with a conditioned medium and rested for 24 h. Transduction
efficiency was measured by flow cytometry using surface staining for Thy1.1 (OX-70, Biolegend). For the IL-7 independency assay (Fig. 3b), transduced cells were split and cultured with
either recombinant murine (rm)IL-7 or 10 µg/ml neutralizing anti-IL-7 antibody (BE0048, Bio X Cell). RNA-SEQUENCING AND BIOSTATISTICAL ANALYSIS RNA extraction from primary tumor samples and
FACS-sorted B220+ sIgµ− pro/preB cells was performed using Trizol extraction. Quality control was performed using the Bioanalyzer RNA 6000 NanoChip (Agilent Technologies). Library
preparation was performed at the Institute for Immunology, University Medical Center of the Johannes Gutenberg-University Mainz using the NEBNext Ultra Library Prep kit (New England
Biolabs). For deep sequencing, the Illumina-HiSeq- 4000 platform was used (Beijing Genomic Institute). Quality control on the sequencing data were performed with the FastQC tool (version
0.11.2, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). RNA-sequencing reads were aligned to the ENSEMBL Mus_musculus.GRCm38 reference genome. The corresponding annotation
(ENSEMBL v76) was also retrieved from ENSEMBL FTP website. The STAR aligner (version 2.4.0j) was used to perform mapping to the reference genome. Alignments were processed with the
featureCounts function [60] of the Rsubread package, using the annotation file also used for supporting the alignment. Exploratory Data Analysis was performed with the pcaExplorer package
[61]. Differential expression analysis was performed with DESeq2 package [62], setting the false discovery rate (FDR) cutoff to 0.1. DESeq2 datasets were analyzed using the GeneTonic [63]
and pcaExplorer packages. To assess the possible occurrence of gene fusions, we applied two different methods, Star-Fusion (version 1.10.1) and Arriba (version 2.1.0). For STAR-Fusion,
required meta reference files were created from the Ensembl Mus musculus reverence (revision 100) as recommended in the STAR-Fusion manual. In case of Arriba, we used the mm10 + GENCODEM25
assembly. In each case, we used the dockerized versions of the tools. Raw fastq files were used as an input for both tools. Subsequently, raw reads were mapped using the recommended
alternative STAR settings recommended in the tools manual to leverage chimeric reads from the alignments. Default filters as recommended by the STAR-Fusion and Arriba manuals were applied to
limit the false-positive rate. For the same reason, known blacklisted regions as provided by the Arriba release were excluded from the analysis. BCP-ALL SUBTYPE PREDICTIONS USING RANDOM
FOREST CLASSIFIER Human genes (GRCh38.p13, v104) with annotated orthologous genes in mice were extracted from ensembl database using the BiomaRt online tool. Gene counts from RNA-sequencing
of a previously published human BCP-ALL cohort [28] and of murine tumor samples were subsetted to include only human-mouse orthologous genes. The resulting gene counts were normalized by
variant stabilization transformation using the R package DESeq2 version 1.32.0. Allocation of the murine tumor samples to human BCP-ALL molecular subtypes was performed based on gene
expression using a random forest machine learning algorithm (R package caret version 6.0-88) trained on the human cohort. Predictions were plotted using R package pheatmap version 1.0.12.
Differential gene expression was analyzed in R package DESeq2 and resulting gene lists ranked by log2-fold-change were analyzed in GSEA version 4.1.0. JAK STRUCTURE AND SEQUENCE ANALYSIS
Mouse JAK2 (AF-Q62120) and JAK3 (AF-Q62137) structure predictions were acquired from the AlphaFold protein structure database [64] and visualized in UCSF ChimeraX (version 1.2.5) [65].
Multiple sequence alignments were performed using the EMBL-EBI Clustal Omega tool. QUANTITATIVE REAL-TIME (QRT-)PCR Total RNA was extracted both from primary _Irf4__−/−_ tumors as well as
FACS-sorted control B220+sIgµ− BM fr.A-D cells of either _Irf4__−/−_ or wt animals using the Gdansk extractme kit (EM09.1) according to the manufacturer’s protocol. cDNA was prepared from
whole RNA samples using the RevertAid cDNA kit from Thermo Fisher (K1621). qRT-PCR for _Aicda_, _Spi1,_ and _Pax5_ was performed using the SybrGreen MasterMix reagent (4385612,
AppliedBiosystems) in a StepOnePlus cycler (AppliedBiosystems). Data presented as percentage of HPRT using the formula _x_ = 1/2(cyclesAicda − cyclesHPRT) × 100. IN VIVO THERAPEUTIC STUDIES
AND ULTRASOUND IMAGING Mice were injected with 3 × 105 T8.1 cells intraperitoneally and monitored daily for clinical symptoms. When mice began showing signs of general morbidity, leukemia
was confirmed by FACS analysis of tail vein blood for B220+ sIgµ− blast cells. When blast cells in pB reached 25 (mean 50)%, therapy was initiated with oral Dexamethasone (Jenapharm) at 6
mg/L supplied ad libitum in the drinking water for seven days. Maintenance therapy comprised either Ruxolitinib-phosphate (S5243, Sellekchem) 1 mg (in 2% DMSO, 30% PEG300 in H2O, as proposed
by the manufacturer), Defactinib (S7654, Sellekchem) 1.2 mg (in 5% DMSO, 50% PEG300, 5% Tween 80 in H2O, as proposed by the manufacturer) or vehicle control (5% DMSO, 50% PEG300, 5% Tween
80 in H2O) administered twice daily via oral gavage. During the course of the disease, this treatment led to paraparesis of the hind legs and tail. A clinical scoring system was established
according to the extent of paraparesis and mice were scored daily accordingly: Scores 0–3: (0) no paraparesis, (1) paraparesis induced by treatment intervention, resolves within 30 s, (2)
paraparesis induced by treatment intervention, does not resolve within 30 s, (3) persistent paraparesis, independent of treatment intervention. Score 3 prompted sacrification of affected
mice. High-resolution ultrasound imaging was performed using a Visual Sonics Vevo 2100 System (FUJIFILM VisualSonics, Toronto, Canada) with microscan transducer MS-550-D, 22–55 MHz (FUJIFILM
VisualSonics, Toronto, Canada) as described previously [66]. STATISTICAL ANALYSIS Statistical analysis was performed using the GraphPad 9.0 software. Data are commonly presented as mean ±
SD. Prior to significance testing, normal distribution and homogeneity of variances were confirmed by Shapiro–Wilk test and Brown–Forsythe testing. Statistical significance when comparing
two normally distributed groups was evaluated using two-tailed unpaired _t_ tests. In case of significant differences in variances between groups, Welch’s correction was applied to account
for non-norminal distribution of data. When comparing multiple groups, a one-way or two-way analysis of variance was performed, depending on the number of variables that differed between
compared groups. This was followed by a Tukey’s Sidak, or Dunnett’s _post hoc_ test, as indicated in figure legends. An alpha level of _P_ < 0.05 was employed for significance testing. In
the in vivo experiment, all animals were included in the analyses. REPORTING SUMMARY Further information on research design is available in the Nature Research Reporting Summary linked to
this article. DATA AVAILABILITY The RNAseq datasets generated during the current study have been deposited in the Gene Expression Omnibus (GEO) archive and are available under the accession
number GSE192424. The WES datasets can be accessed under PRJNA706650 in the sequencing read archive (SRA). REFERENCES * Johnson K, Hashimshony T, Sawai CM, Pongubala JMR, Skok JA, Aifantis
I, et al. Regulation of immunoglobulin light-chain recombination by the transcription factor IRF-4 and the attenuation of interleukin-7 signaling. Immunity 2008;28:335–45. Article CAS
PubMed Google Scholar * Geier JK, Schlissel MS. Pre-BCR signals and the control of Ig gene rearrangements. Semin Immunol 2006;18:31–9. Article CAS PubMed Google Scholar * Herzog S,
Reth M, Jumaa H. Regulation of B-cell proliferation and differentiation by pre-B-cell receptor signalling. Nat Rev Immunol 2009;9:195–205. Article CAS PubMed Google Scholar * Fistonich
C, Zehentmeier S, Bednarski JJ, Miao R, Schjerven H, Sleckman BP, et al. Cell circuits between B cell progenitors and IL-7+ mesenchymal progenitor cells control B cell development cell
circuits control B cell development. J Exp Med2018;215:2586–99. Article CAS PubMed PubMed Central Google Scholar * Huber M, Lohoff M. IRF4 at the crossroads of effector T-cell fate
decision. Eur J Immunol 2014;44:1886–95. Article CAS PubMed Google Scholar * Lohoff M, Mak TW. Roles of interferon-regulatory factors in T-helper-cell differentiation. Nat Rev Immunol
2005;5:125–35. Article CAS PubMed Google Scholar * Mittrücker H-W, Matsuyama T, Grossman A, Kündig TM, Potter J, Shahinian A, et al. Requirement for the transcription factor LSIRF/IRF4
for mature B and T lymphocyte function. Science 1997;275:540–3. Article PubMed Google Scholar * Lu R, Medina KL, Lancki DW, Singh H. IRF-4,8 orchestrate the pre-B-to-B transition in
lymphocyte development. Gene Dev 2003;17:1703–8. Article CAS PubMed PubMed Central Google Scholar * Katz AJ, Chia VM, Schoonen WM, Kelsh MA. Acute lymphoblastic leukemia: an assessment
of international incidence, survival, and disease burden. Cancer Cause Control 2015;26:1627–42. Article Google Scholar * Tasian SK, Hurtz C, Wertheim GB, Bailey NG, Lim MS, Harvey RC, et
al. High incidence of Philadelphia chromosome-like acute lymphoblastic leukemia in older adults with B-ALL. Leukemia 2016;31:981–4. Article PubMed PubMed Central Google Scholar * Roberts
KG. Genetics and prognosis of ALL in children vs adults. Hematol Am Soc Hematol Educ Program 2018;2018:137–45. Article Google Scholar * Mullighan CG. Genomic characterization of childhood
acute lymphoblastic leukemia. Semin Hematol 2013;50:314–24. Article CAS PubMed Google Scholar * Liu Y-F, Wang B-Y, Zhang W-N, Huang J-Y, Li B-S, Zhang M, et al. Genomic profiling of
adult and pediatric b-cell acute lymphoblastic leukemia. Ebiomedicine 2016;8:173–83. Article PubMed PubMed Central Google Scholar * Hardy RR, Carmack CE, Shinton SA, Kemp JD, Hayakawa K.
Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J Exp Med 1991;173:1213–25. Article CAS PubMed Google Scholar * Sen J, Arceci RJ, Jones
W, Burakoff SJ. Expression and ontogeny of murine CD2. Eur J Immunol 1989;19:1297–302. Article CAS PubMed Google Scholar * Zehentmeier S, Pereira JP. Cell circuits and niches controlling
B cell development. Immunol Rev 2019;289:142–57. Article CAS PubMed PubMed Central Google Scholar * Tokoyoda K, Egawa T, Sugiyama T, Choi B-I, Nagasawa T. Cellular niches controlling B
lymphocyte behavior within bone marrow during development. Immunity 2004;20:707–18. Article CAS PubMed Google Scholar * Ma Q, Jones D, Springer TA. The chemokine receptor CXCR4 is
required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity 1999;10:463–71. Article CAS PubMed Google Scholar * Batista CR, Lim
M, Laramée A-S, Abu-Sardanah F, Xu LS, Hossain R, et al. Driver mutations in Janus kinases in a mouse model of B-cell leukemia induced by deletion of PU.1 and Spi-B. Blood Adv
2018;2:2798–810. Article CAS PubMed PubMed Central Google Scholar * Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang Y-L, Pei D, et al. Targetable kinase-activating lesions in Ph-like
acute lymphoblastic leukemia. N. Engl J Med 2014;371:1005–15. Article PubMed PubMed Central Google Scholar * Refsland EW, Harris RS. The APOBEC3 family of retroelement restriction
factors. Curr Top Microbiol 2013;371:1–27. CAS Google Scholar * Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody
diversification. Nature 2002;418:99–104. Article CAS PubMed Google Scholar * Noia JD, Neuberger MS. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA
glycosylase. Nature 2002;419:43–8. Article PubMed Google Scholar * Wilson TM, Vaisman A, Martomo SA, Sullivan P, Lan L, Hanaoka F, et al. MSH2–MSH6 stimulates DNA polymerase η, suggesting
a role for A:T mutations in antibody genes. J Exp Med 2005;201:637–45. Article CAS PubMed PubMed Central Google Scholar * Swaminathan S, Klemm L, Park E, Papaemmanuil E, Ford A, Kweon
S-M, et al. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat Immunol 2015;16:766–74. Article CAS PubMed PubMed Central Google Scholar * Malard F, Mohty M.
Acute lymphoblastic leukaemia. Lancet 2020;395:1146–62. Article CAS PubMed Google Scholar * Mullighan CG. How advanced are we in targeting novel subtypes of ALL? Best Pract Res Clin
Haematol 2019;32:101095. Article PubMed PubMed Central Google Scholar * Bastian L, Schroeder MP, Eckert C, Schlee C, Tanchez JO, Kämpf S, et al. PAX5 biallelic genomic alterations define
a novel subgroup of B-cell precursor acute lymphoblastic leukemia. Leukemia 2019;33:1895–909. Article CAS PubMed Google Scholar * Jain N, Roberts KG, Jabbour E, Patel K, Eterovic AK,
Chen K, et al. Ph-like acute lymphoblastic leukemia: a high-risk subtype in adults. Blood 2017;129:572–81. Article CAS PubMed PubMed Central Google Scholar * Jumper J, Evans R, Pritzel
A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021;596:583–9. Article CAS PubMed PubMed Central Google Scholar *
Herold T, Schneider S, Metzeler K, Neumann M, Hartmann L, Roberts KG, et al. Philadelphia chromosome-like acute lymphoblastic leukemia in adults have frequent IGH-CRLF2 and JAK2 mutations,
persistence of minimal residual disease and poor prognosis. Haematologica. 2016;102:130–8. * Corfe SA, Paige CJ. The many roles of IL-7 in B cell development; mediator of survival,
proliferation and differentiation. Semin Immunol 2012;24:198–208. Article CAS PubMed Google Scholar * Milford TM, Su RJ, Francis OL, Baez I, Martinez SR, Coats JS, et al. TSLP or IL-7
provide an IL-7Rα signal that is critical for human B lymphopoiesis. Eur J Immunol 2016;46:2155–61. Article CAS PubMed PubMed Central Google Scholar * Härzschel A, Zucchetto A, Gattei
V, Hartmann TN. VLA-4 expression and activation in B cell malignancies: functional and clinical aspects. Int J Mol Sci 2020;21:2206. Article PubMed Central Google Scholar * Springer TA.
Adhesion receptors of the immune system. Nature 1990;346:425–34. Article CAS PubMed Google Scholar * Tijchon E, Havinga J, Leeuwen FNvan, Scheijen B. B-lineage transcription factors and
cooperating gene lesions required for leukemia development. Leukemia 2013;27:541–52. Article CAS PubMed Google Scholar * Martin-Lorenzo A, Hauer J, Vicente-Duenas C, Auer F,
Gonzalez-Herrero I, Garcia-Ramirez I, et al. Infection exposure is a causal factor in B-cell precursor acute lymphoblastic leukemia as a result of Pax5-inherited susceptibility. Cancer
Discov 2015;5:1328–43. Article CAS PubMed Google Scholar * Schjerven H, Ayongaba EF, Aghajanirefah A, McLaughlin J, Cheng D, Geng H, et al. Genetic analysis of Ikaros target genes and
tumor suppressor function in BCR-ABL1+ pre–B ALLIkaros tumor suppressor function in Ph+ pre–B ALL. J Exp Med 2017;214:793–814. Article CAS PubMed PubMed Central Google Scholar * Pang
SHM, Minnich M, Gangatirkar P, Zheng Z, Ebert A, Song G, et al. PU.1 cooperates with IRF4 and IRF8 to suppress pre-B-cell leukemia. Leukemia 2016;30:1375–87. Article CAS PubMed PubMed
Central Google Scholar * Jo S-H, Schatz JH, Acquaviva J, Singh H, Ren R. Cooperation between deficiencies of IRF-4 and IRF-8 promotes both myeloid and lymphoid tumorigenesis. Blood
2010;116:2759–67. Article CAS PubMed PubMed Central Google Scholar * Lupardus PJ, Ultsch M, Wallweber H, Kohli PB, Johnson AR, Eigenbrot C. Structure of the pseudokinase–kinase domains
from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc Natl Acad Sci 2014;111:8025–30. Article PubMed PubMed Central Google Scholar * McElroy CA,
Holland PJ, Zhao P, Lim J-M, Wells L, Eisenstein E, et al. Structural reorganization of the interleukin-7 signaling complex. Proc Natl Acad Sci 2012;109:2503–8. Article CAS PubMed PubMed
Central Google Scholar * Hertzberg L, Vendramini E, Ganmore I, Cazzaniga G, Schmitz M, Chalker J, et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which
aberrant expression of CRLF2 is associated with mutated JAK2: a report from the International BFM Study Group. Blood 2010;115:1006–17. Article CAS PubMed Google Scholar * Tasian SK,
Assad A, Hunter DS, Du Y, Loh ML. A Phase 2 study of ruxolitinib with chemotherapy in children with philadelphia chromosome-like acute lymphoblastic leukemia (INCB18424-269/AALL1521):
dose-finding results from the part 1 safety phase. Blood 2018;132:555–555. Article Google Scholar * Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind,
placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl J Med 2012;366:799–807. Article CAS PubMed PubMed Central Google Scholar * Hara T, Shitara S, Imai K, Miyachi H,
Kitano S, Yao H, et al. Identification of IL-7–producing cells in primary and secondary lymphoid organs using IL-7–GFP knock-in mice. J Immunol 2012;189:1577–84. Article CAS PubMed Google
Scholar * Ogawa M, Nishikawa S, Ikuta K, Yamamura F, Naito M, Takahashi K, et al. B cell ontogeny in murine embryo studied by a culture system with the monolayer of a stromal cell clone,
ST2: B cell progenitor develops first in the embryonal body rather than in the yolk sac. Embo J 1988;7:1337–43. Article CAS PubMed PubMed Central Google Scholar * Ceredig R, Boekel
Eten, Rolink A, Melchers F, Andersson J. Fetal liver organ cultures allow the proliferative expansion of pre-B receptor-expressing pre-B-II cells and the differentiation of immature and
mature B cells in vitro. Int Immunol 1998;10:49–59. Article CAS PubMed Google Scholar * Kang CH, Hartmann E, Menke L, Staudenraus D, Abass E-F, Raifer H, et al. A hyperactive mutant of
interferon-regulatory factor 4. Eur J Immunol 2018;49:812–5. Article PubMed Google Scholar * Maaten LVD, Hinton G. Visualizing data using t-SNE. J Mach Learn Res 2008;9:2579–625. Google
Scholar * Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–9. Article CAS PubMed PubMed Central Google Scholar * Klambauer G, Schwarzbauer K,
Mayr A, Clevert D-A, Mitterecker A, Bodenhofer U, et al. cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery
rate. Nucleic Acids Res 2012;40:e69. Article CAS PubMed PubMed Central Google Scholar * Kawamoto T. Use of a new adhesive film for the preparation of multi-purpose fresh-frozen sections
from hard tissues, whole-animals, insects and plants. Arch Histol Cytol 2003;66:123–43. Article PubMed Google Scholar * Zehentmeier S, Roth K, Cseresnyes Z, Sercan Ö, Horn K, Niesner RA,
et al. Static and dynamic components synergize to form a stable survival niche for bone marrow plasma cells. Eur J Immunol 2014;44:2306–17. Article CAS PubMed Google Scholar *
Canene-Adams K. Preparation of formalin-fixed paraffin-embedded tissue for immunohistochemistry. Methods Enzymol 2013;533:225–33. Article CAS PubMed Google Scholar * Schindelin J,
Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012;9:676–82. Article CAS PubMed Google
Scholar * Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009;25:1754–60. Article CAS PubMed PubMed Central Google Scholar *
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res
2010;20:1297–303. Article CAS PubMed PubMed Central Google Scholar * Brochet X, Lefranc M-P, Giudicelli V. IMGT/V-QUEST: the highly customized and integrated system for IG and TR
standardized V-J and V-D-J sequence analysis. Nucleic Acids Res 2008;36:W503–8. Article CAS PubMed PubMed Central Google Scholar * Liao Y, Smyth GK, Shi W. featureCounts: an efficient
general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30:923–30. Article CAS PubMed Google Scholar * Marini F, Binder H. pcaExplorer: an
R/Bioconductor package for interacting with RNA-seq principal components. BMC Bioinformatics 2019;20:331. Article PubMed PubMed Central Google Scholar * Love MI, Huber W, Anders S.
Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. Article PubMed PubMed Central Google Scholar * Marini F, Ludt A, Linke J,
Strauch K. GeneTonic: an R/Bioconductor package for streamlining the interpretation of RNA-seq data. _Biorxiv_. 2021; https://www.biorxiv.org/content/10.1101/2021.05.19.444862v1. * Varadi M,
Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with
high-accuracy models. Nucleic Acids Res. 2021;50:D439–D444. * Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, et al. UCSF ChimeraX: structure visualization for researchers,
educators, and developers. Protein Sci 2021;30:70–82. Article CAS PubMed Google Scholar * Buchholz SM, Goetze RG, Singh SK, Ammer-Herrmenau C, Richards FM, Jodrell DI, et al. Depletion
of macrophages improves therapeutic response to gemcitabine in murine pancreas cancer. Cancers 2020;12:1978. Article CAS PubMed Central Google Scholar Download references
ACKNOWLEDGEMENTS The authors want to thank Koji Tokoyoda (DRFZ, Berlin) for supplying _Il-7__eGFP_ mice for breeding, Olli Silvennoinen (Tampere-university, Finland) for supplying us with
the JAK3 construct, and Fritz Melchers (DRFZ Berlin) for the JIL-7.6 J558 and ST2 cells. Further, Hyun-Dong Chang and Anja Hauser (both DRFZ, Berlin) for supplying OP-9 cells and Kawamoto
materials, respectively. FUNDING D.D.G. received personal funding through the German Cancer Aid, Mildred-Scheel doctoral scholarship (70112922). M.L. was funded by the Deutsche
Forschungsgemeinschaft (DFG) (LO 396/8-1) and the Else Kröner-Fresenius-Stiftung. Open Access funding enabled and organized by Projekt DEAL. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS *
Institute for med. Microbiology & Hospital Hygiene, Philipps University Marburg, Marburg, Germany Dennis Das Gupta, Nadine Samel, Maria Bieringer, Daniel Staudenraus, Hartmann Raifer,
Lisa Menke, Lea Hansal, Bärbel Camara & Michael Lohoff * University Hospital Gießen and Marburg, Dept. of Ophthalmology, Philipps University Marburg, Marburg, Germany Christoph Paul *
MVZ for Laboratory Medicine and Microbiology, Koblenz-Mittelrhein, Germany Nadine Samel * Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI), University Medical Center
of the Johannes Gutenberg-University Mainz, Mainz, Germany Federico Marini * Division of Molecular Immunology, Nikolaus-Fiebiger Center, University of Erlangen-Nürnberg, Erlangen, Germany
Edith Roth, Patrick Daum & Hans-Martin Jäck * Institute for Molecular Biology and Tumor Research (IMT), Center for Tumor- and Immunobiology (ZTI), Philipps University Marburg, Marburg,
Germany Michael Wanzel, Marco Mernberger, Andrea Nist, Uta-Maria Bauer & Thorsten Stiewe * Genomics Core Facility, Philipps University Marburg, Marburg, Germany Marco Mernberger &
Thorsten Stiewe * Core Facility for Mouse Pathology and Electron Microscopy, Philipps University Marburg, Marburg, Germany Frederik Helmprobst & Axel Pagenstecher * University Hospital
Gießen and Marburg, and Philipps University, Institute of Neuropathology, Marburg, Germany Frederik Helmprobst & Axel Pagenstecher * University Hospital Gießen and Marburg, and Philipps
University, Clinic for Gastroenterology and Core Facility Small Animal Ultrasound, Marburg, Germany Malte Buchholz * Core facility for Cellular Imaging, Philipps University Marburg, Marburg,
Germany Katrin Roth * Medical Department II, Hematology and Oncology, University Medical Center Schleswig- Holstein, Kiel, Germany Lorenz Bastian, Alina M. Hartmann & Claudia Baldus *
Institute for Frontier Life and Medical Sciences, Kyoto University, Kyoto, Japan Koichi Ikuta * University Hospital Gießen and Marburg, and Philipps University, Dept. Hematology, Oncology
and Immunology, Marburg, Germany Andreas Neubauer & Andreas Burchert * Institute for Immunology, Research Center for Immunotherapy (FZI), University Cancer Center, University Medical
Center of the Johannes Gutenberg-University Mainz, Mainz, Germany Matthias Klein & Tobias Bopp * German Cancer Consortium (DKTK), Mainz, Germany Tobias Bopp Authors * Dennis Das Gupta
View author publications You can also search for this author inPubMed Google Scholar * Christoph Paul View author publications You can also search for this author inPubMed Google Scholar *
Nadine Samel View author publications You can also search for this author inPubMed Google Scholar * Maria Bieringer View author publications You can also search for this author inPubMed
Google Scholar * Daniel Staudenraus View author publications You can also search for this author inPubMed Google Scholar * Federico Marini View author publications You can also search for
this author inPubMed Google Scholar * Hartmann Raifer View author publications You can also search for this author inPubMed Google Scholar * Lisa Menke View author publications You can also
search for this author inPubMed Google Scholar * Lea Hansal View author publications You can also search for this author inPubMed Google Scholar * Bärbel Camara View author publications You
can also search for this author inPubMed Google Scholar * Edith Roth View author publications You can also search for this author inPubMed Google Scholar * Patrick Daum View author
publications You can also search for this author inPubMed Google Scholar * Michael Wanzel View author publications You can also search for this author inPubMed Google Scholar * Marco
Mernberger View author publications You can also search for this author inPubMed Google Scholar * Andrea Nist View author publications You can also search for this author inPubMed Google
Scholar * Uta-Maria Bauer View author publications You can also search for this author inPubMed Google Scholar * Frederik Helmprobst View author publications You can also search for this
author inPubMed Google Scholar * Malte Buchholz View author publications You can also search for this author inPubMed Google Scholar * Katrin Roth View author publications You can also
search for this author inPubMed Google Scholar * Lorenz Bastian View author publications You can also search for this author inPubMed Google Scholar * Alina M. Hartmann View author
publications You can also search for this author inPubMed Google Scholar * Claudia Baldus View author publications You can also search for this author inPubMed Google Scholar * Koichi Ikuta
View author publications You can also search for this author inPubMed Google Scholar * Andreas Neubauer View author publications You can also search for this author inPubMed Google Scholar *
Andreas Burchert View author publications You can also search for this author inPubMed Google Scholar * Hans-Martin Jäck View author publications You can also search for this author
inPubMed Google Scholar * Matthias Klein View author publications You can also search for this author inPubMed Google Scholar * Tobias Bopp View author publications You can also search for
this author inPubMed Google Scholar * Thorsten Stiewe View author publications You can also search for this author inPubMed Google Scholar * Axel Pagenstecher View author publications You
can also search for this author inPubMed Google Scholar * Michael Lohoff View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS DDG, CP, NS, MB,
FH, ML designed experiments; DDG, CP, NS, MB, DS, LM, BC, FH, LH performed experiments; DDG, CP, MB, HR, ER, PD, MW, MM, AN, UMB, FM, FH, MB, HMJ, AN, AB, MK, TB, TS, AP, LB, AH, CB, ML
analyzed data; DDG and ML prepared the manuscript. CORRESPONDING AUTHOR Correspondence to Michael Lohoff. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests.
STUDY APPROVAL All animal experiments were approved by the local government (Regierungspräsidium Gießen, G49/2018, G34/2021) and conducted according to the German animal protection law.
ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Edited by T Mak SUPPLEMENTARY
INFORMATION SUPPLEMENTARY TABLE 1 SUPPLEMENTARY TABLE 2 SUPPLEMENTARY TABLE 3 SUPPLEMENTARY INFORMATION SUPPLEMENTARY MOVIE 1 AUTHOR CONTRIBUTION FORM 1/3 AUTHOR CONTRIBUTION FORM 2/3 AUTHOR
CONTRIBUTION FORM 3/3 REPORTING SUMMARY FORM RIGHTS AND PERMISSIONS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons
licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise
in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the
permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Das Gupta, D., Paul, C., Samel, N. _et al._ IRF4 deficiency vulnerates B-cell progeny for leukemogenesis via somatically acquired _Jak3_
mutations conferring IL-7 hypersensitivity. _Cell Death Differ_ 29, 2163–2176 (2022). https://doi.org/10.1038/s41418-022-01005-z Download citation * Received: 07 February 2022 * Revised: 07
April 2022 * Accepted: 08 April 2022 * Published: 22 April 2022 * Issue Date: November 2022 * DOI: https://doi.org/10.1038/s41418-022-01005-z SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative








