- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Mitochondria support multiple cell functions, but an accumulation of dysfunctional or excessive mitochondria is detrimental to cells. We previously demonstrated that a defect in the
autophagic removal of mitochondria, termed mitophagy, leads to the acceleration of apoptosis induced by herpesvirus productive infection. However, the exact molecular mechanisms underlying
activation of mitophagy and regulation of apoptosis remain poorly understood despite the identification of various mitophagy-associated proteins. Here, we report that the mitochondrial
translation elongation factor Tu, a mitophagy-associated protein encoded by the _TUFM_ gene, locates in part on the outer membrane of mitochondria (OMM) where it acts as an inhibitor of
altered mitochondria-induced apoptosis through its autophagic function. Inducible depletion of TUFM potentiated caspase-8-mediated apoptosis in virus-infected cells with accumulation of
altered mitochondria. In addition, TUFM depletion promoted caspase-8 activation induced by treatment with TNF-related apoptosis-inducing ligand in cancer cells, potentially via dysregulation
of mitochondrial dynamics and mitophagy. Importantly, we revealed the existence of and structural requirements for autophagy-competent TUFM on the OMM; the GxxxG motif within the N-terminal
mitochondrial targeting sequences of TUFM was required for self-dimerization and mitophagy. Furthermore, we found that autophagy-competent TUFM was subject to ubiquitin-proteasome-mediated
degradation but stabilized upon mitophagy or autophagy activation. Moreover, overexpression of autophagy-competent TUFM could inhibit caspase-8 activation. These studies extend our knowledge
of mitophagy regulation of apoptosis and could provide a novel strategic basis for targeted therapy of cancer and viral diseases. SIMILAR CONTENT BEING VIEWED BY OTHERS CASPASE-8 IS
REQUIRED FOR HSV-1-INDUCED APOPTOSIS AND PROMOTES EFFECTIVE VIRAL PARTICLE RELEASE VIA AUTOPHAGY INHIBITION Article Open access 24 November 2022 DIRECT CLEAVAGE OF CASPASE-8 BY HERPES
SIMPLEX VIRUS 1 TEGUMENT PROTEIN US11 Article Open access 19 July 2022 THE UBIQUITIN LIGASE TRIM32 PROMOTES THE AUTOPHAGIC RESPONSE TO MYCOBACTERIUM TUBERCULOSIS INFECTION IN MACROPHAGES
Article Open access 05 August 2023 INTRODUCTION Mitochondria are required for a variety of cellular functions. However, an accumulation of dysfunctional mitochondria is harmful to cells,
potentially via increased reactive oxygen species (ROS) and mis-localized mitochondrial DNAs that lead to oxidative stress, hyper inflammatory responses, or amplified apoptosis [1,2,3,4].
The quality control of mitochondria is achieved by balanced actions among mitochondrial biogenesis, dynamics, and mitophagy. Mitophagy is a selective autophagy that removes dysfunctional or
excessive mitochondria [5]. The mitochondria-localized kinase PINK1 and the ubiquitin (Ub) E3 ligase Parkin are the best known mitophagy-associated proteins. Upon the loss of mitochondrial
membrane potential, the PINK1 and Parkin complex induces ubiquitination of mitochondria [6], which is recognized by Ub-binding autophagy receptors, such as p62/SQSTM1, and targeted to
autophagosomes via interaction with autophagy-related protein 8 family proteins, such as LC3 and GABARAP. In addition, ubiquitination-independent mitophagy pathways via mitophagy receptors,
such as NIX/BNIP3L, BNIP3, FUNDC1, and PHB2, have been identified under certain physiological conditions [7,8,9,10]. Mitophagy is also activated by virus infection, where it serves to
counter host antiviral responses, such as type I interferon (IFN) induction and apoptosis, enabling successful virus infection [11,12,13,14,15,16]. We recently found that human herpesvirus 8
(HHV-8), also known as Kaposi’s sarcoma-associated herpesvirus, can activate NIX-mediated mitophagy via viral IFN regulatory factor 1 (vIRF-1) [17]. vIRF-1 appears to play important roles
in blocking IFN and other stress responses, such as mitochondria-mediated apoptosis via BH3-only-proteins (BIM and BID) and MAVS (also known as IPS-1, VISA, and Cardif), to virus infection
and replication through inhibitory interactions with cellular signaling proteins [18,19,20,21]. In studies to further understand the role of vIRF-1 in mitochondria, we identified the
mitochondrial translation elongation factor Tu, encoded by the _TUFM_ gene, as a vIRF-1 binding protein. TUFM is known to activate mitophagy upon virus infection [15, 22,23,24]. However, the
exact localization of TUFM in mitochondria and role of TUFM in mitophagy have not been fully elucidated. Here, we characterize the role of TUFM in the inhibition of caspase-8-mediated
apoptosis amplified by altered mitochondria and identify the existence of and structural requirement for autophagy-competent TUFM on the OMM. RESULTS VIRF-1 BINDS DIRECTLY TO TUFM vIRF-1
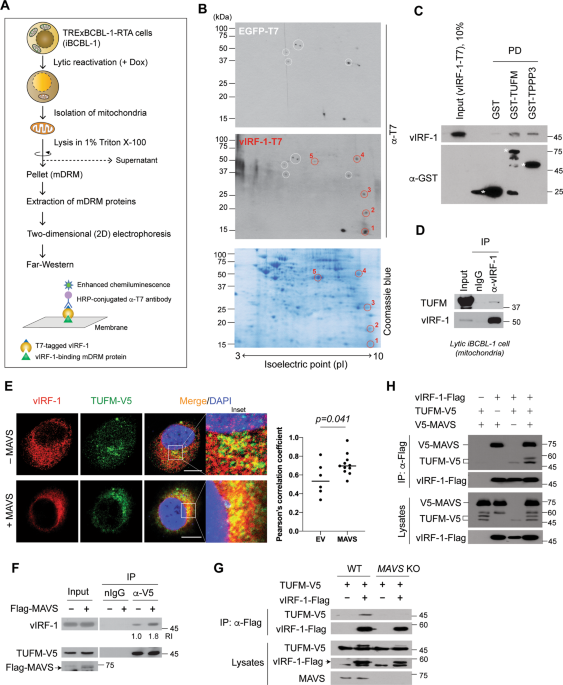
localizes in part to mitochondria by targeting to the detergent-resistant microdomains (DRM) [21]. To examine the role of mitochondrial DRM (mDRM)-localized vIRF-1, we sought to identify
vIRF-1-interacting mDRM proteins from the TRExBCBL-1-RTA (hereafter simply termed iBCBL-1) cell line, which is an HHV-8-infected primary effusion lymphoma (PEL) cell line with doxycycline
(Dox)-inducible expression of RTA, a lytic switch protein [25]. Due to poor solubility of mDRM proteins in standard lysis buffer, we performed Far-western blotting instead of a
co-precipitation method (Fig. 1A). Five distinct individual dots (Fig. 1B) highly reactive with vIRF-1-T7 compared to control EGFP-T7 were found, and the corresponding spots were excised
from a replica gel for mass spectrometry analysis. One of the spots was identified as TUFM (Fig. S1). An in vitro pull-down assay confirmed the direct interactions of vIRF-1 with TUFM and
TPPP3, another hit protein (Fig. 1C). Furthermore, a co-immunoprecipitation (co-IP) assay showed that vIRF-1 interacted with endogenous TUFM on mitochondria isolated from lytic iBCBL-1 cells
(Fig. 1D). In addition, an immunofluorescence assay (IFA) showed that vIRF-1 co-localized with TUFM in transfected HeLa.Kyoto cells, and their co-localization was promoted by overexpression
of MAVS (Fig. 1E), which was reported previously to promote the mitochondrial targeting of vIRF-1 [21]. Consistent with this, co-IP assays showed that vIRF-1 interaction with TUFM was
promoted by MAVS overexpression and greatly diminished by MAVS deficiency (Fig. 1F, G). However, vIRF-1 interaction with MAVS was not affected by TUFM overexpression (Fig. 1H), indicating
that TUFM and MAVS interactions with vIRF-1 are not mutually exclusive. TUFM DEPLETION POTENTIATES APOPTOSIS IN LYTIC VIRF-1-DEFICIENT CELLS Mitochondria-localized vIRF-1 plays a crucial
role for survival of lytically infected cells [17, 21]. So, we examined whether TUFM is involved in vIRF-1 regulation of cell survival using four different iBCBL-1 cell lines expressing
Dox-inducible vIRF-1 and/or TUFM shRNAs (Fig. 2A). Luciferase shRNA (shLuc) was used as a control. Note that both lytic reactivation and shRNA expression are induced simultaneously by Dox
treatment in the cell lines. Dox-inducible knockdown (KD) of TUFM and vIRF-1 proteins was verified using immunoblotting (Fig. 2A). As expected, vIRF-1-KD resulted in reduced numbers of
iBCBL-1 cells after 4 days of Dox treatment (Fig. 2B). TUFM-KD also led to a significant decrease in numbers of shLuc and vIRF-1-KD lytic cells (Fig. 2B). To determine if the reduced numbers
of TUFM-depleted lytic cells were associated with increased cell death, we counted the number of dead cells using a trypan blue exclusion assay. TUFM depletion indeed increased the
proportion of dead cells within cultures of both cell lines, shLuc and shvIRF-1, during lytic replication; however, cell death was induced earlier in the vIRF-1-KD cells compared to in the
shLuc cells (Fig. 2C). Overall, these results suggest that both vIRF-1 and TUFM are important for the survival of lytically infected cells. We next determined the type of cell death promoted
by TUFM depletion. Pan-caspase inhibitors zVAD-FMK and Emricasan, but neither DMSO nor the necroptosis inhibitor Necrostatin-1, inhibited cell death induced by TUFM-KD in lytic vIRF-1-KD
cells (Fig. 2D). Specific inhibitors of caspase-8 (CASP8) and caspase-3 (CASP3) also inhibited the TUFM-KD-induced cell death (Fig. 2D), indicating that TUFM depletion is associated with
caspase-dependent apoptosis. Indeed, TUFM-KD significantly promoted apoptosis in lytic vIRF-1-KD cells, as evidenced by Annexin V, TUNEL, and PARP cleavage assays (Fig. 2E, F). Moreover,
TUFM-KD potentiated cytochrome c release to the cytosol in the vIRF-1-KD cells (Fig. 2G), suggesting that TUFM could inhibit mitochondria-mediated apoptosis. TUFM deficiency could lead to
reduced mitochondrial respiratory activity and promote the production of ROS, which are probably involved in causing apoptosis. Indeed, mitochondrial superoxide was elevated significantly by
TUFM-KD in not only lytic vIRF-1-KD, but also lytic control cells (Fig. 2H). However, TUFM depletion-induced oxidative stress is unlikely to be involved directly in apoptosis initiation
because TUFM depletion alone was not sufficient to induce apoptosis (Fig. 2E–G). Together, these results suggest that TUFM may play a key and direct role in the inhibition of apoptosis in
vIRF-1-deficient cells after lytic reactivation. TUFM INHIBITS CASP8 ACTIVATION INDUCED BY DYSREGULATED MITOCHONDRIA To define the mechanism of apoptosis promoted by TUFM depletion, we first
examined caspase activation. Immunoblotting revealed that TUFM-KD promoted CASP8 activation, as evidenced by increased levels of the p41/43 fragments of CASP8, in lytic vIRF-1-KD cells, but
not in lytic control cells (Fig. 3A). CASP3 was also activated but to a lesser extent (Fig. 3A). However, the levels of MCL-1, a key regulator of PEL cell survival [26], were not affected
by depletion of either vIRF-1 or TUFM. Importantly, rescue with an shRNA-resistant version of TUFM substantially reduced CASP8 activation in lytic vIRF-1/TUFM-double-KD (dKD) cells (lane 8
in Fig. 3B). These results suggest that TUFM plays a key role in the inhibition of CASP8-mediated apoptosis induced by vIRF-1 deficiency. As TUFM is implicated in mitophagy activation upon
infection by certain RNA viruses [15, 27], we postulated that TUFM-KD leads to excessive accumulation of dysregulated mitochondria and thereby potentiates apoptosis in vIRF-1-deficient
cells. To test this, we examined mitochondria using TOM20-IFA. The number of cells with both highly clustered mitochondria and apoptotic nuclei (fragmented or condensed chromatin) greatly
increased in the vIRF-1/TUFM-dKD cells compared to vIRF-1-KD cells after lytic reactivation (Fig. S3). Emricasan inhibited nuclear fragmentation but not mitochondrial aggregation (Fig. S4),
suggesting that mitochondrial aggregation precedes caspase-mediated apoptosis. Together, the results suggest that TUFM plays a key role in the inhibition of apoptosis via regulation of
mitochondrial dynamics rather than direct inhibition of apoptosis-related caspases. We examined the role of TUFM in the regulation of mitochondrial dynamics and apoptotic caspase activation
using a combined approach of TOM20-IFA and CASP8 Fluorochrome Inhibitor of Caspases (C8-FLICA). C8-FLICA assay detects active CASP8 in cells. The number of vIRF-1/TUFM-dKD cells with both
aggregated mitochondria and CASP8 activation was around 1.9-fold higher compared to vIRF-1-KD cells upon lytic reactivation (Fig. 3C, D). Intriguingly, the number of C8-FLICA-negative but
still abnormal nuclear cells with aggregated mitochondria was equivalent between vIRF-1-KD and vIRF-1/TUFM-dKD cells (Fig. 3D), suggesting a selective role of TUFM in the inhibition of CASP8
induced by altered mitochondria. There was a modest but positive association between aggregated mitochondria and CASP8 activation in individual vIRF-1-KD and vIRF-1/TUFM-dKD cells (Fig.
3E). However, the dKD cells showed an increase in the number of cells with highly aggregated mitochondria together with intensive CASP8 activation compared to vIRF-1-KD cells (above the
arbitrary threshold in Fig. 3E). Moreover, C8-FLICA co-localized in part with aggregated mitochondria in apoptotic cells (Fig. 3F), and the p41/43 fragments of CASP8 were detected at a
higher level in the mitochondrial fraction of lytic vIRF-1/TUFM-dKD cells (Fig. 3G). Together, our results suggest that TUFM depletion-induced CASP8 activation is largely dependent on
altered mitochondria. To confirm that the proapoptotic effect of TUFM-KD is attributable to an accumulation of altered mitochondria, we transduced the control and TUFM-KD cells with
DRP1K38A, a dominant negative form of DRP1 that inhibits mitochondrial fission [28]. Indeed, C8-FLICA assays showed that DRP1K38A expression rendered the both cell lines highly sensitive to
lytic reactivation-induced CASP8 activation but to a higher extent in TUFM-KD cells compared with control cells (Figs. 3H and S5). Similar results were obtained with mdivi-1 (Fig. S6), a
mitochondrial division inhibitor [29]. Taken together, these results suggest that the proapoptotic effect of TUFM-KD in the context of vIRF-1 deficiency is associated with dysregulation of
mitochondria quality control. TUFM CAN INHIBIT TRAIL-INDUCED CASP8 ACTIVATION IN THE OMM Mdivi-1 is known to promote death receptor-mediated apoptosis through enhanced CASP8 activity in
cancer cells [30, 31]. This synergistic effect appears to be associated with dysregulation of mitochondrial dynamics. Thus, we investigated whether TUFM depletion potentiates apoptosis
induced by combined treatment with TNF-related apoptosis-inducing ligand (TRAIL) and mdivi-1. For this experiment, we generated HeLa.Kyoto cell lines expressing Dox-inducible shTUFM or
shLuc. After Dox incubation for 3 days, the cells were treated with TRAIL and/or mdivi-1 for 6 h. Consistent with published findings, mdivi-1 promoted TRAIL-induced CASP8 activation in
control cells (compare lane 5 with 7 in Fig. 3I). Interestingly, we found that mdivi-1 could not further enhance CASP8 activation in the TUFM-KD cells (compare lane 6 with 8 in Fig. 3I),
while TRAIL greatly enhanced CASP8 activation in the TUFM-KD cells compared to the control cells treated with DMSO or mdivi-1 (compare lane 6 with lanes 5 and 7 in Fig. 3I). The negation of
mdivi-1 activation of CASP8 by TUFM depletion and the efficacy of TUFM depletion alone on CASP8 activation in the context of these cervical cancer cells provides further evidence that TUFM
functions to promote mitophagy and prevent the accumulation of damaged mitochondria. TUFM IS LOCALIZED ON THE OMM IN MODIFIED AND/OR AGGREGATE FORMS We next examined the effect of TUFM
overexpression on TRAIL-induced CASP8 activation. The TUFM variant (ΔMTS), which lacks the mitochondrial targeting sequences (MTS), was used as a control. Native TUFM, but not the ΔMTS form,
inhibited CASP8 activation induced by TRAIL treatment (Fig. 4A). However, the inhibitory effect of TUFM was modest (~30–40% inhibition). As TUFM is mostly imported into the mitochondrial
matrix, we hypothesized that a minority of TUFM protein on the OMM, particularly in mDRM, has an ability to inhibit CASP8. To test this, we enforced TUFM and TUFM.ΔMTS localization to mDRM
by fusing them to the mDRM-targeting N-terminal sequence (V11–150) of vIRF-1 (Fig. 4B). Proteinase K (PK) sensitivity and mitochondrial fractionation assays confirmed that the V11–150 tag
could drive TUFM to the cytosolic side and mDRM of the OMM (Fig. S7). Indeed, V11–150-TUFM, but not V11–150-TUFM.ΔMTS, could inhibit TRAIL-induced CASP8 activation by more than 50% compared
to the empty vector control (Fig. 4B). Moreover, we found that the OMM-targeting sequence (amino acids 1–33) of TOM20 could drive TUFM to mDRM of the OMM (Fig. S7), and the fusion protein,
TOM201–33-TUFM, could significantly inhibit TRAIL-induced CASP8 activation (Fig. 4B). These results suggest that TUFM can inhibit CASP8 activation via the MTS and localization in mDRM of the
OMM. Since TUFM interacts with the cytosolic autophagy machinery, such as the ATG12-ATG5 conjugate, TUFM involved in autophagy activation and/or regulation of CASP8 is likely to locate on
the cytosolic side of the OMM. As TUFM exists mostly in the cleaved (mature) form (ΔMTS), an OMM-localized TUFM is expected to comprise a preprotein that is larger than the cleaved form
(Fig. 4C). Transfection experiments showed, as expected, that the major species produced from a wild-type TUFM vector was equivalent to the size of the ΔMTS protein (arrowhead in Fig. 4D),
but additional bands, including a form migrating above the ΔMTS band (arrow in Fig. 4D), were detected upon prolonged exposure of the immunoblot. Mitochondrial processing peptidase (MPP)
recognizes basic sequences of mitochondrial preproteins and cleaves a single site, often including arginine (R), at the −2 position[32]. Indeed, TUFM contains a putative MPP recognition
arginine residue at position 41. To assess whether the slower-migrating band is the preprotein of TUFM, we generated the TUFM variant R41A by replacing the arginine with alanine (Fig. 4C).
However, the size of TUFM.R41A was indistinguishable from that of TUFM.ΔMTS (Fig. 4D), indicating that R41 is not involved in MPP recognition and cleavage. It is unclear whether the upper
band is the preprotein or a modified form of ΔMTS. Protease sensitivity assays showed that the slow migrating TUFM band disappeared with PK treatment (Fig. 4E), indicating that it is located
on the cytosolic side of the OMM. Intriguingly, a new TUFM fragment of about 40 kDa was detected after PK treatment (red asterisk in Fig. 4E), indicating that a Flag tag-containing
C-terminal segment of the larger protein might be protected from PK digestion by the OMM with only a small region of the N-terminus exposed to the cytosol. To investigate the possibility
that there exists a form of TUFM orientated inversely with cytoplasmic exposure of containing the functional domains for regulation of autophagy or CASP8 activity, we used pharmacological
inhibitors of protein degradation to potentially stabilize such a species. Amongst the tested inhibitors, only the proteasome inhibitors bortezomib and MG132 could stabilize the
high-molecular-weight TUFM species observed previously (Fig. 4D, E) and other, slower-migrating forms (Fig. S8), which may represent aggregated and/or post-translationally modified forms of
TUFM. Bortezomib-stabilized high-molecular-weight species present on isolated mitochondria were susceptible to PK digestion (Fig. 4F), providing evidence that they are located mainly on the
cytosolic side of the OMM, but rapidly targeted for degradation mainly by the Ub-proteasome system. Taken together, we hypothesize that TUFM in part localizes to, and may aggregate within
(Fig. S8), the OMM, with the C-terminus exposed to the cytoplasm, and thus is susceptible to cytosolic Ub modification and proteasomal degradation (Fig. 4G). TUFM IS STABILIZED ON MDRM UPON
MITOPHAGY OR AUTOPHAGY ACTIVATION We examined whether endogenous TUFM can be detected in the modified/aggregated forms in virus-infected cells. Indeed, higher-molecular-weight TUFM proteins
were readily detected in mDRM of lytic cells along with vIRF-1 and stabilized by bortezomib (Fig. 5A). Likewise, the ATG12-ATG5 conjugate and NIX, but not NLRX1, MAVS, and VDAC, also were
more abundant on mDRM of lytic cells (Fig. 5A). Furthermore, ATG12-IFA showed that ATG12 could be localized to mitochondria (Fig. 5B). The modified TUFM and ATG12-ATG5 proteins on the
mitochondria isolated from lytic iBCBL-1 cells were susceptible to PK digestion (Fig. 5C). Together, these results suggest that mDRM on the OMM may act as an assembly platform for
TUFM-mediated autophagy. We next examined the role of vIRF-1 in the modifications of TUFM using transfection. Indeed, vIRF-1 could promote TUFM modifications and/or aggregates in mDRM (Fig.
5D). Moreover, treatment with carbonyl cyanide _m_-chlorophenyl hydrazone (CCCP), a proton ionophore that triggers mitophagy [21], led to increased levels of the observed
high-molecular-weight TUFM (>100-kDa) in both the absence and presence of vIRF-1, and to a more pronounced effect in the latter (Fig. 5E). Kaempferide, which is an inducer of
TUFM-mediated autophagy [33], also enhanced the expression of mDRM-associated high-molecular-weight TUFM by 3.4-fold compared to the level in control cells (Fig. 5F). Together, our data
correlate TUFM modification with autophagy activation and suggest that vIRF-1-effected TUFM modification may contribute to vIRF-1-promoted mitophagy in infected cells. TUFM ACTIVATES
AUTOPHAGY VIA THE CANONICAL PATHWAY To assess if vIRF-1 promotes TUFM-mediated autophagy flux, a tandem TUFM-mCherry-EGFP (TUFM-mCE) fusion protein was generated and transfected into
HeLa.Kyoto cells with or without vIRF-1 (Fig. 6A). Once TUFM-mCE is delivered to lysosomes upon autophagy, the signal of EGFP, but not mCherry, is quenched. About 2% of TUFM-mCE-transfected
cells showed punctate structures with only red fluoresce (basal autophagy), but vIRF-1 increased up to 15% the population of cells showing this pattern (Fig. 6A). To assess mitophagy flux,
we generated a mitophagy reporter, mito-mCE, in which the tandem mCherry-EGFP tag was attached to the TOM201–33 sequence for its mitochondrial targeting (Fig. 6B). The reporter worked for
assessment of AO (antimycin A and oligomycin)-induced mitophagy in Parkin-transfected cells (Fig. 6B). The mitophagy assays using mito-mCE showed that TOM201–33-TUFM, and to a lesser extent
native TUFM, promoted mitophagy flux upon AO or TRAIL treatment (Fig. 6C). However, TOM201–33-TUFM-medated mitophagy induced by TRAIL was abolished in _ATG7_ or _ATG12_ KO cells (Fig. 6D,
E), indicating that TUFM-mediated mitophagy is activated via the canonical autophagy pathway. TUFM DIMERIZATION VIA THE GXXXG MOTIF WITHIN THE MTS IS REQUIRED FOR AUTOPHAGY We examined
whether TUFM homodimerizes, using NanoBiT protein fragment complementation assay[34] (Fig. 7A). The NanoBiT subunits, Large BiT (LgB) and Small BiT (SmB), were fused to the C-terminus of
TUFM-V5 (Fig. 7B). To verify its true homodimeric interaction on mitochondria, TUFM.ΔMTS and the unrelated HaloTag protein were included as negative controls; V11–150-TUFM.ΔMTS and
N-terminal point variants (see below) of full-length TUFM fused to LgB were also included (Fig. 7B). Co-transfected with TUFM-V5-LgB, TUFM-V5-SmB exhibited stronger luminescence intensity
compared to HaloTag-SmB (Fig. 7C). The luminescence intensity was also reduced to background levels by deletion of the MTS. V11–150-TUFM.ΔMTS also could not bind to TUFM (Fig. 7C). These
results suggest that the MTS is involved in TUFM dimerization. The dimerization of membrane proteins is often mediated by their transmembrane helix and highly stabilized by GxxxG motif
within the helix[35]. TUFM contains GxxxG motif, 15GLAAG19, within the MTS. To examine if the introduced GxxxG motif is involved in TUFM dimerization, we replaced the glycine residues with
alanine (A), leucine (L), or isoleucine (I). The NanoBiT assays showed that the self-interaction of TUFM was diminished by the introduced mutations, and most substantially by LxxxL and IxxxI
that present bulky hydrophobic side chains (Fig. 7C). It is noteworthy that the TUFMLxxxL and TUFMIxxxI variants showed reduced levels of the predominant ΔMTS species, probably due to an
interference with import to the mitochondrial matrix. TUFM.R41A also showed reduced dimerization (Fig. 7C). These results suggest that the MTS contains primary-structure determinants of TUFM
dimerization. TUFM DIMERIZATION IS REQUIRED FOR AUTOPHAGY REGULATION OF CASP8 ACTIVATION We examined the involvement of the GxxxG motif in TUFM aggregation. Immunoblotting revealed
TUFMAxxxA, and to a lesser extent TUFMIxxxI, failed to form high-molecular-weight species (Fig. 7D). Moreover, TUFMAxxxA failed to inhibit TRAIL-induced CASP8 activation (Fig. 7E),
suggesting that TUFM dimerization may be required for the inhibition of CASP8 activation. Furthermore, co-IP assays showed that ATG12-ATG5 was co-immunoprecipitated with TUFM and
V11–150-TUFM, but to a lesser extent with TUFMAxxxA (Fig. 7F). Importantly, TUFMAxxxA-mCE failed to mediate mitophagy upon TRAIL treatment (Fig. 7G). Taken together, we propose a model for
autophagy-competent TUFM (Fig. 7H), in which newly synthesized TUFM preprotein is inserted into the OMM, through the action of a putative mammalian mitochondrial import (Mim) complex [36],
where it is able to activate autophagy by interacting with the autophagy machinery. vIRF-1 or a cellular mitophagic factor could stabilize autophagy-competent TUFM by protecting it from
Ub-proteasome-mediated degradation. DISCUSSION Our findings suggest that TUFM on the cytosolic side of the OMM may play a crucial role in the inhibition of CASP8 via autophagy activation.
TUFM has been reported to interact with several mitophagy-related proteins. For example, TUFM is known to mediate autophagy by interacting with the mitochondrial protein NLRX1 upon virus
infection [22]. However, NLRX1 is located mainly in the mitochondrial matrix [37], and its expression in mitochondria is unchanged during HHV-8 lytic replication (Fig. 5A). TUFM is also
known to mediate mitophagy by interacting with PINK1 upon mitochondrial damage [38, 39]. Intriguingly, PINK1 phosphorylation of TUFM at serine 222 was proposed to restrict TUFM to the
cytosol and inhibit mitophagy in a negative feedback manner [38]. However, it is unlikely that PINK1 regulation of TUFM-mediated mitophagy occurs because cytosolic TUFM was not detected in
lytically infected cells even in the presence of proteasome inhibitor (Fig. 5A). Consistent with our previous finding [17], NIX was abundant in mDRM of lytic cells (Fig. 5A). It still
remains unclear how vIRF-1 activates NIX-mediated mitophagy [17]. In this regard, it would be interesting to examine in future how vIRF-1 recruitment of the autophagy machinery via TUFM
facilitates NIX-mediated mitophagy during lytic replication. The mechanism by which TUFM is localized to the OMM remains elusive. In yeast, the precursors of proteins with an N-terminal
signal anchor sequence are typically inserted into the OMM by the mitochondrial import (Mim) complex (Mim1/Mim2)[40]. However, the Mim complex has been found only in yeast so far; it has not
been possible to identify a homolog in mammals or other species by simple sequence comparisons. Interestingly, recent functional studies with trypanosomes revealed that the OMM protein
pATOM36 can replace the Mim complex in yeast, although it does not show any sequence similarity to the Mim proteins [41]. Thus, the functional analog may have arisen by convergent evolution.
Identification of a mammalian functional counterpart of Mim involved in OMM targeting of TUFM would be of considerable significance. TUFM is highly expressed in several cancers including
glioblastoma, cholangiocarcinoma, and colorectal cancer (CRC) [42,43,44], and increased expression of TUFM is associated with shorter patient survival in CRC[45]. CRC is one of the most
common cancers with TRAIL resistance. Thus, therapeutic targeting of TUFM-mediated autophagy might be clinically beneficial in CRC. In summary, TUFM-mediated mitophagy appears to play an
important role in host antiviral responses and carcinogenesis by downregulation of CASP8-mediated apoptosis. We found new, complexed and modified, forms of OMM-localized TUFM, which
potentially are involved in promotion of mitophagy and consequent inhibition of CASP8 activation. In particular, TUFM dimerization seems to be required for CASP8 regulation. Thus,
pharmacological or genetic targeting of TUFM dimerization, without disrupting its mitochondrial translation activity, would facilitate experimental dissection of the multiple functions of
TUFM and could provide a basis for the development of selective antiviral and cancer therapies specifically targeting OMM-localized TUFM interactions and activity. Overall, our findings
further understanding of the role of TUFM in connecting autophagy and apoptosis and have broader therapeutic implications with respect to virus infections and cancer. MATERIALS AND METHODS
CELL CULTURE TRExBCBL-1-RTA (a gift from Dr. Jae U. Jung, herein termed iBCBL-1) and its derivative lines were cultured in RPMI 1640 medium (Quality Biological) supplemented with 15%
heat-inactivated fetal bovine serum (FBS), stable L-alanyl-glutamine (Glutamine XL), and streptomycin and penicillin, at 37 °C and 5% CO2. 293T, HeLa.Kyoto (a gift from Dr. Ron R. Kopito),
and their derivative cell lines were cultured in DMEM supplemented with 10% FBS and antibiotics. The cell lines were tested for mycoplasma contamination (R&D systems) and if necessary
cultured in plasmocin™ treatment or prophylactic (InvivoGen). Transient transfection with plasmids was performed using GenJet version II (SignaGen Laboratories). For stable and doxycycline
(Dox)-inducible expression of short hairpin RNAs (shRNAs), culture cells were lentivirally transduced with shRNA-specifying sequences in the presence of 10 µg/ml polybrene overnight, and
stably transduced cells were selected by growing in the presence of 1 µg/ml puromycin or 400 µg/ml geneticin for more than 1 month, and pooled clones were collected. For detection of
mitochondrial superoxide, cells were incubated with 5 µM MitoSOX red indicator (Invitrogen) in Hank’s buffered salt solution containing calcium and magnesium for 10 min at 37°C just before
cell fixation. LENTIVIRUS PRODUCTION To produce infectious lentiviruses, 293T cells were co-transfected with the lentiviral vector together with the packaging plasmid psPAX2 and the
vesicular stomatitis virus G protein expression plasmid pVSV-G at a ratio of 5:4:1. Two days later, virions were collected from the culture medium by ultracentrifugation in an SW28 rotor at
25,000 rpm for 2 h at 4 °C. The virion pellets were resuspended in an appropriate volume of phosphate-buffered saline (PBS) to achieve 100× concentration. Transduction titers of lentiviruses
were determined in 293T cells in the presence of appropriate antibiotic to select transduced cells. ISOLATION OF MITOCHONDRIA Pure mitochondria were isolated using Axis-Shield OptiPrep
iodixanol (Sigma-Aldrich). In brief, latent and lytic iBCBL-1 cells were homogenized in buffer B (0.25 M sucrose, 1 mM EDTA, 20 mM HEPES-NaOH [pH 7.4]) with 50 strokes of a Dounce glass
homogenizer and centrifuged at 1000 × _g_ for 10 min. An aliquot of homogenate was used as total-cell extract. The supernatant was further centrifuged at 13,000 × _g_ for 10 min. The pellet
was collected as a crude mitochondrial fraction. For further enrichment, the pellet was resuspended in 36% iodixanol, bottom-loaded under 10% and 30% iodixanol gradients, and centrifuged at
50,000 × _g_ for 4 h. The mitochondria were collected at the 10%/30% iodixanol interface. For isolation of mitochondrial detergent-resistant membrane microdomains (mDRM), enriched
mitochondria were incubated in TNE buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, and 1 mM EDTA) containing 1% Triton X-100 on ice for 30 min, and centrifuged at 21,000 × _g_ for 10 min. The
supernatant was used as a soluble mitochondrial fraction, and the pellet was used as the mDRM fraction. The pellet was boiled in 1× sodium dodecyl sulfate (SDS) sample buffer for SDS-PAGE.
DNA MANIPULATION All polymerase chain reaction amplification and site-directed mutagenesis including point and deletion mutations were performed using SuperFi™ DNA polymerase (Thermo Fisher
Scientific). Subcloning of open reading frames and their derivatives into expression plasmids including pICE (a gift from Steve Jackson; Addgene plasmid #46960), pcDNA3.1(+) (Invitrogen),
plenti.puro (a gift from Melina Fan; Addgene plasmid #74218), pGEX-4T-1 (GE Healthcare Life Sciences), and pTYB4 (New England Biolab). Plasmids were purified using ZymoPURE II Plasmid
Midiprep kit for transfection (Zymo Research). The plasmids and oligonucleotides used in this study are listed in Tables S1 and S2, respectively. IMMUNOLOGICAL ASSAYS Antibodies used in
immunological assays including immunoblotting, immunoprecipitation, and immunostaining are listed in Table S3. For the preparation of total extracts, cells were resuspended in RIPA buffer
(50 mM Tris [pH 7.4], 150 mM NaCl, 1% Igepal CA-630, 0.1% SDS, and 0.25% deoxycholate) containing a protease inhibitor cocktail and protein phosphatase inhibitors including 10 mM NaF and 5
mM Na3VO4 and sonicated using Bioruptor (Diagenode) for 5 min in ice water at a high-power setting (320 W). For immunoblotting, cell lysates were separated by SDS-PAGE, transferred to
nitrocellulose or polyvinylidene difluoride (PVDF) membranes, and immunoblotted with appropriate primary antibodies diluted in SuperBlock™-PBS blocking buffer (Thermo Fisher Scientific).
Following incubation with HRP-labeled Ig-specific secondary antibody, immunoreactive bands were visualized using enhanced chemiluminescent (ECL) substrates, such as Clarity (Bio-Rad) or
SuperSignal™ West Femto (Thermo Fisher Scientific), on an ECL film. ImageJ software (NIH) was used to quantify the signal from immunoblots. For immunoprecipitation (IP), total-cell or
mitochondrial extracts were prepared in RIPA-B buffer lacking SDS and incubated with specific primary antibody at 4 °C overnight and incubated with protein G-agarose beads (Cell Signaling
Technology) for an additional 3 h. For the IP of V5- or Flag-tagged proteins, anti-V5-agarose (Bethyl Laboratories) and anti-DYKDDDDK tag(L5) affinity gel (BioLegend) were used.
Immunoprecipitated complexes were washed with RIPA-B buffer, followed by elution of bound proteins with 1× SDS sample buffer. If necessary, Clean-Blot™ IP Detection reagent (Thermo Fisher
Scientific) was used to avoid detection of IgG used in IP assays. For detection of ubiquitinated proteins, an extra wash was performed using RIPA buffer supplemented with 1 M urea after IP,
and nitrocellulose membrane was heat-activated by autoclaving at 121 °C for 30 min prior to blocking with 5% nonfat dry milk in PBS containing 0.25% Tween 20 (PBS-T). For indirect
immunofluorescent assay (IFA), cells grown on a coverslip (and transfected) were fixed in Image-iT™ fixative solution (Thermo Fisher Scientific) and permeabilized in 0.5% Triton X-100 in
PBS. iBCBL-1 cells were attached on poly-L-lysine-coated coverslips. For certain experiments, cells grown and transfected on poly-L-lysine-coated coverslips were permeabilized with 25 µg/ml
of saponin in PBS containing 100 mM potassium chloride for 5 min at room temperature before fixation. Following incubation with SuperBlock™-PBS blocking buffer for 1 h at room temperature,
coverslips were incubated with primary antibodies, washed with PBS, and then incubated with appropriate fluorescent dye-conjugated secondary antibodies. Coverslips were mounted in ProLong™
Gold Antifade Mountant containing DAPI (Thermo Fisher Scientific) on glass slides, and cells were imaged by Zen software on a Zeiss confocal laser scanning microscope 700 with a ×20 or
oil-immersion ×40 and ×63 objective. For co-localization analysis, images were randomly acquired under the microscope, and co-localization was scored using the ‘Coloc2’ tool of ImageJ. The
integrated intensity (IntDen) of stained proteins were determined using ImageJ. FAR-WESTERN BLOT AND MASS SPECTROMETRY mDRM proteins were extracted in sample lysis buffer (9 M urea, 2 M
thiourea, 100 mM DTT, 2% CHAPS (w/v), 60 mM n-octyl β-D-glucopyranoside, 0.5% Zoom carrier ampholyte (pH 3 to 10), and protease inhibitor cocktail), separated by isoelectric focusing over
immobilized pH gradient strips (Invitrogen), and further separated by SDS-PAGE in the second dimension (2D). The 2D gel electrophoresis was conducted in triplicate: two gels were used for
Far-western blotting, and the third for Coomassie blue staining. For Far-western blotting, mDRM proteins in the 2D gels were transferred to PVDF membranes, which were then blocked with 5%
nonfat dry milk in Tris buffered saline containing 0.1% Tween-20 (TBS-T) and incubated with 1 µg/ml of purified recombinant protein vIRF-1-T7 or EGFP-T7 for 3 h at room temperature. After
washes in TBS-T (3 × 10 min), the blots were incubated for 1 h with HRP-conjugated anti-T7 antibody. Reactive spots were visualized by ECL. The corresponding spots were excised from the
Coomassie blue stained gel, digested by trypsin, and analyzed by matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometry (Voyager, Applied Biosystems).
vIRF-1-T7 and EGFP-T7 proteins were expressed as chitin-binding domain-fusion proteins in BL21 derivative Rosetta cells (Novagen). The T7-tagged proteins were purified using chitin beads as
previously performed [21]. GST-PRECIPITATION ASSAYS Recombinant glutathione-S-transferase (GST) and GST-fusion proteins were expressed in Rosetta cells and purified by standard methods. 1 µg
GST or GST-fusion protein was incubated with 20 µl bed volume of washed glutathione sepharose-4B beads for 1 h at room temperature. After washing in binding buffer (PBS plus 1% Triton
X-100), the protein-bead complexes were incubated with 1 µg recombinant vIRF-1 or 293T cell lysates containing vIRF-1-Flag proteins at 4 °C overnight, washed in binding buffer four times,
separated on SDS-PAGE, and subjected to immunoblotting. CELL VIABILITY AND APOPTOSIS ASSAYS Dead cells existing prior to the experiments were removed using Histopaque-1077 (Sigma) or Dead
Cell Removal kit (Miltenyl Biotec). Freshly isolated intact iBCBL-1 cell lines were treated with 1 µg/ml Dox for the indicated times. Cell viability was then measured using trypan blue
exclusion assays. For apoptosis assays, cells were analyzed by FITC-annexin V staining (BioLegend) or terminal deoxynucleotide transferase (TdT)-mediated dUTP nick labeling (TUNEL) assay kit
(AAT Bioquest). All imaging and quantitation of apoptotic cells were performed using Cellometer Vision CBA Image Cytometry (Nexcelom), which employs four independent images for cell
counting. FCS Express 6 Flow software (De Novo Software) was used for data analysis. MITOPHAGY FLUX ASSAYS HeLa.Kyoto cells expressing mitophagy fluorescence reporters, TUFM-mCE and
mito-mCE, were imaged using the ZOE fluorescent cell imager (Bio-Rad) for quantitative analysis. The number of total and mitophagy-positive cells from three randomly selected images were
counted. Representative cell images were taken using Zeiss confocal laser scanning microscope 700 with oil-immersion ×63 objective. REAGENTS Chemical reagents were purchased from the
following companies: MG132 and bortezomib from Cell Signaling Technology; proteinase K, necrostatin-1, doxycycline, bafilomycin A1, leupeptin, antimycin A, oligomycin, and CCCP from
Millipore Sigma; Liensinine from AK Scientific; mdivi-1 from Cayman Chemical; TRAIL from BioLegend; z-IETD-FMK (iCASP8) and zVAD-FMK from Enzo Life Sciences; AZ 10417808 (iCASP3) from
ApexBio; emricasan from SelleckChem; furimazine from Promega. QUANTIFICATION AND STATISTICAL ANALYSIS Statistical analyses were performed using Prism 8 software (Graphpad Software, Inc.).
Statistical significance is stated in the Figure legends and Supplemental Figure legends. Differences between controls and samples were determined by paired _t_-tests (two-sided) and were
considered significant when the _p_ value was less than 0.05 (_p_ < 0.05). DATA AVAILABILITY All relevant data are available from the authors upon reasonable request. REFERENCES * Kong Y,
Trabucco SE, Zhang H. Oxidative stress, mitochondrial dysfunction and the mitochondria theory of aging. Interdiscip Top Gerontol. 2014;39:86–107. Article PubMed Google Scholar * Malik
AN, Czajka A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion. 2013;13:481–92. Article CAS PubMed Google Scholar * Hoffmann RF, Jonker MR,
Brandenburg SM, de Bruin HG, Ten Hacken NHT, van Oosterhout AJM, et al. Mitochondrial dysfunction increases pro-inflammatory cytokine production and impairs repair and corticosteroid
responsiveness in lung epithelium. Sci Rep. 2019;9:15047. Article CAS PubMed PubMed Central Google Scholar * Wanderoy S, Hees JT, Klesse R, Edlich F, Harbauer AB. Kill one or kill the
many: interplay between mitophagy and apoptosis. Biol Chem. 2020;402:73–88. Article PubMed Google Scholar * Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular
homeostasis, physiology and pathology. Nat Cell Biol. 2018;20:1013–22. Article CAS PubMed Google Scholar * Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The
ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015;524:309–14. Article CAS PubMed PubMed Central Google Scholar * Novak I, Kirkin V, McEwan DG, Zhang
J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. Article CAS PubMed Google Scholar * Band M, Joel A, Hernandez
A, Avivi A. Hypoxia-induced BNIP3 expression and mitophagy: in vivo comparison of the rat and the hypoxia-tolerant mole rat, Spalax ehrenbergi. FASEB J. 2009;23:2327–35. Article CAS PubMed
Google Scholar * Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol.
2012;14:177–85. Article PubMed Google Scholar * Wei Y, Chiang WC, Sumpter R Jr., Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell.
2017;168:224–38. Article CAS PubMed Google Scholar * Zhang L, Qin Y, Chen M. Viral strategies for triggering and manipulating mitophagy. Autophagy 2018;14:1665–73. Article CAS PubMed
PubMed Central Google Scholar * Choi YB, Harhaj EW. Functional implications of mitochondrial reactive oxygen species generated by oncogenic viruses. Front Biol. 2014;9:423–36. Article CAS
Google Scholar * Kim SJ, Khan M, Quan J, Till A, Subramani S, Siddiqui A. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS
Pathog. 2013;9:e1003722. Article PubMed PubMed Central Google Scholar * Kim SJ, Syed GH, Siddiqui A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent
mitophagy. PLoS Pathog. 2013;9:e1003285. Article CAS PubMed PubMed Central Google Scholar * Ding B, Zhang L, Li Z, Zhong Y, Tang Q, Qin Y, et al. The matrix protein of human
parainfluenza virus type 3 induces mitophagy that suppresses interferon responses. Cell Host Microbe. 2017;21:538–47. Article CAS PubMed Google Scholar * Vilmen G, Glon D, Siracusano G,
Lussignol M, Shao Z, Hernandez E, et al. BHRF1, a BCL2 viral homolog, disturbs mitochondrial dynamics and stimulates mitophagy to dampen type I IFN induction. Autophagy. 2021;17:1296–1315.
Article CAS PubMed Google Scholar * Vo MT, Smith BJ, Nicholas J, Choi YB. Activation of NIX-mediated mitophagy by an interferon regulatory factor homologue of human herpesvirus. Nat
Commun. 2019;10:3203. Article PubMed PubMed Central Google Scholar * Choi YB, Cousins E, Nicholas J. Novel functions and virus-host interactions implicated in pathogenesis and
replication of human herpesvirus 8. Recent Results Cancer Res. 2021;217:245–301. Article CAS PubMed Google Scholar * Choi YB, Nicholas J. Bim nuclear translocation and inactivation by
viral interferon regulatory factor. PLoS Pathog. 2010;6:e1001031. Article PubMed PubMed Central Google Scholar * Choi YB, Sandford G, Nicholas J. Human herpesvirus 8 interferon
regulatory factor-mediated BH3-only protein inhibition via Bid BH3-B mimicry. PLoS Pathog. 2012;8:e1002748. Article CAS PubMed PubMed Central Google Scholar * Hwang KY, Choi YB.
Modulation of mitochondrial antiviral signaling by human herpesvirus 8 interferon regulatory factor 1. J Virol. 2016;90:506–20. Article CAS PubMed Google Scholar * Lei Y, Wen H, Yu Y,
Taxman DJ, Zhang L, Widman DG, et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 2012;36:933–46. Article CAS PubMed
PubMed Central Google Scholar * Kuo SM, Chen CJ, Chang SC, Liu TJ, Chen YH, Huang SY, et al. Inhibition of avian influenza A virus replication in human cells by host restriction factor
TUFM is correlated with autophagy. MBio. 2017;8:e00481–17. Article CAS PubMed PubMed Central Google Scholar * Wang R, Zhu Y, Ren C, Yang S, Tian S, Chen H, et al. Influenza A virus
protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy. 2021;17:496–511. Article CAS PubMed Google Scholar * Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU.
Global changes in Kaposi’s sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol. 2003;77:4205–20. Article CAS
PubMed PubMed Central Google Scholar * Kojima Y, Hayakawa F, Morishita T, Sugimoto K, Minamikawa Y, Iwase M, et al. YM155 induces apoptosis through proteasome-dependent degradation of
MCL-1 in primary effusion lymphoma. Pharm Res. 2017;120:242–51. Article CAS Google Scholar * Wang K, Ma H, Liu H, Ye W, Li Z, Cheng L, et al. The glycoprotein and nucleocapsid protein of
hantaviruses manipulate autophagy flux to restrain host innate immune responses. Cell Rep. 2019;27:2075–91. Article CAS PubMed Google Scholar * Smirnova E, Shurland DL, Ryazantsev SN,
van der Bliek AM. A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol. 1998;143:351–8. Article CAS PubMed PubMed Central Google Scholar *
Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer
membrane permeabilization. Dev Cell. 2008;14:193–204. Article CAS PubMed PubMed Central Google Scholar * Wang J, Hansen K, Edwards R, Van Houten B, Qian W. Mitochondrial division
inhibitor 1 (mdivi-1) enhances death receptor-mediated apoptosis in human ovarian cancer cells. Biochem Biophys Res Commun. 2015;456:7–12. Article CAS PubMed Google Scholar * Akita M,
Suzuki-Karasaki M, Fujiwara K, Nakagawa C, Soma M, Yoshida Y, et al. Mitochondrial division inhibitor-1 induces mitochondrial hyperfusion and sensitizes human cancer cells to TRAIL-induced
apoptosis. Int J Oncol. 2014;45:1901–12. Article CAS PubMed Google Scholar * Kitada S, Yamasaki E, Kojima K, Ito A. Determination of the cleavage site of the presequence by mitochondrial
processing peptidase on the substrate binding scaffold and the multiple subsites inside a molecular cavity. J Biol Chem. 2003;278:1879–85. Article CAS PubMed Google Scholar * Kim D,
Hwang HY, Ji ES, Kim JY, Yoo JS, Kwon HJ. Activation of mitochondrial TUFM ameliorates metabolic dysregulation through coordinating autophagy induction. Commun Biol. 2021;4:1. Article CAS
PubMed PubMed Central Google Scholar * Dixon AS, Schwinn MK, Hall MP, Zimmerman K, Otto P, Lubben TH, et al. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein
Interactions in Cells. ACS Chem Biol. 2016;11:400–8. Article CAS PubMed Google Scholar * Liu Y, Engelman DM, Gerstein M. Genomic analysis of membrane protein families: abundance and
conserved motifs. Genome Biol. 2002;3:research0054. PubMed PubMed Central Google Scholar * Becker T, Wenz LS, Kruger V, Lehmann W, Muller JM, Goroncy L, et al. The mitochondrial import
protein Mim1 promotes biogenesis of multispanning outer membrane proteins. J Cell Biol. 2011;194:387–95. Article CAS PubMed PubMed Central Google Scholar * Singh K, Sripada L, Lipatova
A, Roy M, Prajapati P, Gohel D, et al. NLRX1 resides in mitochondrial RNA granules and regulates mitochondrial RNA processing and bioenergetic adaptation. Biochim Biophys Acta Mol Cell Res.
2018;1865:1260–76. Article CAS PubMed Google Scholar * Lin J, Chen K, Chen W, Yao Y, Ni S, Ye M, et al. Paradoxical mitophagy regulation by PINK1 and TUFm. Mol Cell. 2020;80:607–20.
Article CAS PubMed Google Scholar * Rakovic A, Grunewald A, Voges L, Hofmann S, Orolicki S, Lohmann K, et al. PINK1-interacting proteins: proteomic analysis of overexpressed PINK1.
Parkinsons Dis. 2011;2011:153979. PubMed PubMed Central Google Scholar * Wiedemann N, Pfanner N. Mitochondrial machineries for protein import and assembly. Annu Rev Biochem.
2017;86:685–714. Article CAS PubMed Google Scholar * Vitali DG, Kaser S, Kolb A, Dimmer KS, Schneider A, Rapaport D. Independent evolution of functionally exchangeable mitochondrial
outer membrane import complexes. Elife. 2018;7:e34488. Article PubMed PubMed Central Google Scholar * Xi HQ, Zhang KC, Li JY, Cui JX, Zhao P, Chen L. Expression and clinicopathologic
significance of TUFM and p53 for the normal-adenoma-carcinoma sequence in colorectal epithelia. World J Surg Oncol. 2017;15:90. Article PubMed PubMed Central Google Scholar * Tamai K,
Nakamura-Shima M, Shibuya-Takahashi R, Kanno SI, Yasui A, Mochizuki M, et al. BEX2 suppresses mitochondrial activity and is required for dormant cancer stem cell maintenance in intrahepatic
cholangiocarcinoma. Sci Rep. 2020;10:21592. Article CAS PubMed PubMed Central Google Scholar * Weng X, Zheng S, Shui H, Lin G, Zhou Y. TUFM-knockdown inhibits the migration and
proliferation of gastrointestinal stromal tumor cells. Oncol Lett. 2020;20:250. Article CAS PubMed PubMed Central Google Scholar * Shi H, Hayes M, Kirana C, Miller R, Keating J,
Macartney-Coxson D, et al. TUFM is a potential new prognostic indicator for colorectal carcinoma. Pathology. 2012;44:506–12. Article CAS PubMed Google Scholar Download references
ACKNOWLEDGEMENTS We thank Dr. Edward W. Harhaj for a critical review of the manuscript. We thank Dr. Nikolaus Pfanner for helpful discussion on mammalian analogs of Mim proteins. FUNDING
This work was supported by National Cancer Institute (NCI) grant R01CA214131 to YBC; the work was initiated under grant R21CA136356 to JN. This publication was made possible by the Johns
Hopkins Institute for Clinical and Translational Research (ICTR) which is funded in part by Grant Number UL1 TR003098 from the National Center for Advancing Translational Sciences (NCATS)
and NIH Roadmap for Medical Research. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Oncology, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of
Medicine, Baltimore, MD, 21287, USA Chang-Yong Choi, Mai Tram Vo, John Nicholas & Young Bong Choi Authors * Chang-Yong Choi View author publications You can also search for this author
inPubMed Google Scholar * Mai Tram Vo View author publications You can also search for this author inPubMed Google Scholar * John Nicholas View author publications You can also search for
this author inPubMed Google Scholar * Young Bong Choi View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS CYC designed the study, performed the
experiments, and analyzed the results. MTV performed the experiments. JN provided valuable suggestions, discussed the results, and proofread the manuscript. YBC designed the study,
performed the experiments, analyzed the results, and wrote the manuscript. CORRESPONDING AUTHOR Correspondence to Young Bong Choi. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare
no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Edited
by: E. Baehrecke SUPPLEMENTARY INFORMATION SUPPLEMENTAL MATERIALS RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License,
which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a
link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license,
unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory
regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Choi, CY., Vo, M.T., Nicholas, J. _et al._ Autophagy-competent mitochondrial
translation elongation factor TUFM inhibits caspase-8-mediated apoptosis. _Cell Death Differ_ 29, 451–464 (2022). https://doi.org/10.1038/s41418-021-00868-y Download citation * Received: 24
March 2021 * Revised: 31 August 2021 * Accepted: 01 September 2021 * Published: 12 September 2021 * Issue Date: February 2022 * DOI: https://doi.org/10.1038/s41418-021-00868-y SHARE THIS
ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard
Provided by the Springer Nature SharedIt content-sharing initiative