- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Reinforced cellular responses to endoplasmic reticulum (ER) stress are caused by a variety of pathological conditions including cancers. Human rhomboid family-1 protein (RHBDF1), a multiple
transmembrane protein located mainly on the ER, has been shown to promote cancer development, while the binding immunoglobulin protein (BiP) is a key regulator of cellular unfolded protein
response (UPR) for the maintenance of ER protein homeostasis. In this study, we investigated the role of RHBDF1 in maintaining ER protein homeostasis in breast cancer cells. We showed that
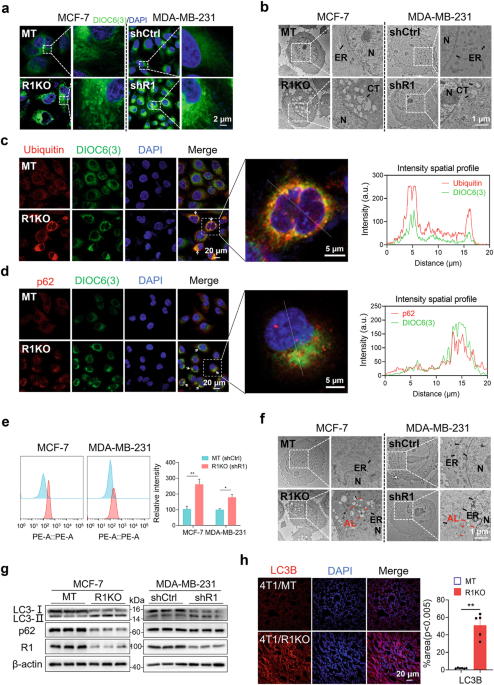
deleting or silencing RHBDF1 in breast cancer cell lines MCF-7 and MDA-MB-231 caused marked aggregation of unfolded proteins in proximity to the ER. We demonstrated that RHBDF1 directly
interacted with BiP, and this interaction had a stabilizing effect on the BiP protein. Based on the primary structural motifs of RHBDF1 involved in BiP binding, we found a pentapeptide (PE5)
targeted BiP and inhibited BiP ATPase activity. SPR assay revealed a binding affinity of PE5 toward BiP (Kd = 57.7 μM). PE5 (50, 100, 200 μM) dose-dependently promoted ER protein
aggregation and ER stress-mediated cell apoptosis in MCF-7 and MDA-MB-231 cells. In mouse 4T1 breast cancer xenograft model, injection of PE5 (10 mg/kg, s.c., every 2 days for 2 weeks)
significantly inhibited the tumor growth with markedly increased ER stress and apoptosis-related proteins in tumor tissues. Our results suggest that the ability of RHBDF1 to maintain BiP
protein stability is critical to ER protein homeostasis in breast cancer cells, and that the pentapeptide PE5 may serve as a scaffold for the development of a new class of anti-BiP
inhibitors.
It has been well documented that cancer cells are capable of sustaining active protein synthesis under various conditions of intracellular or extracellular stress, especially endoplasmic
reticulum (ER) stress [1,2,3]. Cancer cells under ER stress can activate the unfolded protein response (UPR), which involves a number of protein machineries, including the binding
immunoglobulin protein (BiP; also known as HSPA5 or GRP78) and signaling pathways related to inositol-requiring enzyme 1 (IRE1), PRKR-like ER kinase (PERK), and activating transcription
factor 6 (ATF6) [2, 4,5,6,7,8,9]. BiP, a major molecular chaperone of the UPR, functions in assisting proteins in folding correctly and is important in reducing unfolded protein accumulation
and restoring ER homeostasis [10,11,12,13]. BiP expression in cancer cells is increased in response to UPR sensor signaling under ER stress [12,13,14]. An elevated level of BiP has been
attributed to the ability of cancer cells to resist stress-induced cell death [15,16,17,18]. BiP has also been attributed to poor prognosis and drug resistance in cancer [19,20,21].
Therefore, BiP has become an important target for anticancer drug development [22]. It is therefore highly desirable to pursue new strategies to develop BiP inhibitors.
The human rhomboid family-1 protein (RHBDF1; also known as inactive rhomboid-1, iRhom1) [23, 24], the expression of which is significantly increased in cancer tissues compared to
tumor-adjacent tissues [23, 25, 26], is an ER-resident membrane protein [27]. It has been shown to be critically involved in breast cancer progression, poor prognosis, and resistance to
chemotherapy [28,29,30,31,32,33]. Additionally, RHBDF1 can facilitate the epithelial-to-mesenchymal transition (EMT) and cell proliferation in colorectal cancer [31, 34], thereby acting as
an oncogene in cervical cancer [32], mediate G-protein coupled receptor agitator-driven transactivation of EGFR signaling pathways [35], and maintain the stability of hypoxia-inducible
factor-1α (HIF-1α) in cancer cells under hypoxic and inflammatory conditions [29]. An increasing amount of evidence indicates that the RHBDF1 protein can directly interact with a variety of
proteins, such as RACK (the receptor of activated protein-C kinase-1) [29], JNK (c-Jun N-terminal kinase) [36], Par6a (a component of a protein complex critical to the establishment of
epithelial cell apicobasal polarity) [37], and iTAP (iRhom Tail-associated protein) [38], often with a tangible impact on the activities of these proteins. It is thus plausible that RHBDF1,
as an ER-resident membrane protein, may function as a molecular chaperone in the modulation of critical cellular processes, including ER stress and the UPR.
We demonstrate in this study that RHBDF1 plays a crucial role in maintaining ER protein homeostasis in breast cancer cells. This effect is achieved at least partly through RHBDF1 binding to
the BiP protein, enhancing the protein stability of the latter. In addition, we identified segments of the primary structure of the RHBDF1 protein involved in BiP interaction. We show that a
pentapeptide, PE5, which resembles a BiP-binding motif in RHBDF1, can effectively inhibit the ATPase activity of BiP, resulting in greatly increased aggregation of ER proteins and a higher
ER stress-mediated apoptosis rate. Our findings not only provide new insights into the molecular mechanism underlying cancer cell responses to ER stress but also highlight the utility of
targeting RHBDF1-BiP interactions as a plausible strategy in anticancer drug development.
The following reagents were used: DMSO (Sigma, D2438, Stamford, USA), DIOC6(3) (MKBio, MX4009, Shanghai, China), MG132 (Selleckchem, S2619, Houston, USA), Cycloheximide (Sigma, C4859,
Stamford, USA), Thapsigargin (GLPBIO, GC11482, Montclair, CA, USA), and Leupeptin hemisulfate (MCE, HY-18234A, Nashville, TN 37212, USA). The following antibodies were used: RHBDF1 (Abcam,
ab81342, Cambridge, MA, USA), BiP (Proteintech, 11587-1-AP, Rosemont, IL 60018, USA), ER marker PDI (Proteintech, 11245-1-AP and 66422-1-Ig, Rosemont, IL 60018, USA), ATF4 (Beyotime, AF2560,
Shanghai, China), CHOP (CST, 2895, Boston, USA), cleaved Caspase-3 (CST, 9661, Boston, USA), cleaved PARP (CST, 5625, Boston, USA), Ubiquitin (Abcam, 134953, Cambridge, MA, USA), p62
(Proteintech, 66184-1-Ig, Rosemont, IL 60018, USA), LC3A/B (Affinity, AF5402, Cincinnati, OH, USA), LC3B (Affinity, AF4650, Cincinnati, OH, USA), HA (Invitrogen, 26183, California, USA), MYC
(Proteintech, 16286-1-AP, Rosemont, IL 60018, USA), and FLAG (Sigma, F7425, Stamford, USA). The anti-MYC, anti-HA, and anti-FLAG magnetic beads were purchased from Bimake (Houston, TX77014,
USA). The Streptavidin magnetic beads were purchased from MedchemExpress (MCE, HY-K0208, Nashville, TN 37212, USA).
The female BALB/c mice (20–22 g, 6–8 weeks old) were purchased from Taconic Bioscience (Shanghai, China). They were housed in the Laboratory Animal Center, Nankai University, Tianjin, China.
All animal experimental procedures were approved by the Animal Ethics Committee of Nankai University and strictly followed by the Guidelines for the Care and Use of Laboratory Animals
(National Institutes of Health, USA). All animals were placed in pathogen-free conditions with a temperature of 24–25 °C and a relative humidity of 50%–55%, a light/dark cycle for 12 h, and
free access to standard rodent feed and tap water.
Human breast cancer cell lines MCF-7 (RRID: CVCL_0031) and MDA-MB-231 (RRID: CVCL_0062), mouse breast cancer 4T1 cells (RRID: CVCL_0125), and Human embryonic kidney AD293 cells (RRID:
CVCL_9804) and 293 T cells (RRID: CVCL_RU09) were purchased from the American Type Culture Collection (ATCC). The MCF-7 cells with the stable knockout of RHBDF1 used in this paper are the
cells constructed by our group before [36]. MCF-7 and AD293 cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM), MDA-MB-231 cells were grown in Roswell Park Memorial Institute 1640
Medium (RPMI-1640), and 4T1 cells were grown in DMEM/F12 with 10% fetal bovine serum. Cells were cultured in cell culture dishes or multi-well plates with the indicated medium supplemented
with 10% FBS and penicillin/streptomycin (100 U/mL, 50 mg/mL) at 37 °C in mixed gas consisting of 20% O2, 5% CO2, and 75% N2. Cells were transfected with indicated plasmids using
Lipofectamine2000 (Thermo Fisher Scientific, Massachusetts, USA) with reduced serum medium Opti-MEM (Life Technologies, California, USA) following the manufacturer’s instructions.
Full-length human RHBDF1 gene (NM_022450) and Full-length human HSPA5 gene (NM_005347) were amplified from complementary DNA of MCF-7 cells. Then full-length human RHBDF1 tagged with
C-terminal HA was cloned into the pLVX-EF1α-IRES-Puro (LMAI Bio, LM-2015, Shanghai, China) and full-length human BiP was cloned into pcDNA3.1 MYC (RRID: Addgene_176045, Cambridge, MA, USA).
The different coding sequences (for 1–411 aa, 411–655 aa, 655–855 aa, 655–828 aa, and 800–855 aa) of RHBDF1 were further amplified from full-length human RHBDF1 and cloned as described
above. The 70–125 aa of human ubiquitin was amplified from the complementary DNA of MCF-7 cells and cloned into pLVX-EF1α-IRES-Puro/6 × His.
To construct cell lines with stable knockdown of the indicated gene, the pLKO.1 puro vector (RRID: Addgene_8453, Cambridge, MA, USA) containing the shRNA for indicated mRNA was constructed
and then transfected into 293 T cells with psPAX2 (RRID: Addgene_12260, Cambridge, MA, USA) and pMD2.G (RRID: Addgene_12259, Cambridge, MA, USA). After 48 h, the cell culture medium was
collected and filtered by a 0.45 μm filter to use as a virus crude solution. The obtained lentivirus was infected into MCF-7 or MDA-MB-231 cells with 10 μg/mL Polybrene, followed by
screening infected cells with puromycin. The sequences of shRNAs for RHBDF1 are: forward 5′-CCGGCAGTGACAGCACCCAGAAATGCTCGAGCATTTCTGGGTGCTGTCACTGTTTTTTG-3′ and reverse
5′-AATTCAAAAACCGGCAGTGACAGCACCCAGAAATGCTCGAGCATTTCTGGGTGCTGTCACTG-3′.
Cells were cultured in 6 or 12-well tissue culture plates and lysed in RIPA buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, and 1% protease inhibitors
(Sigma-Aldrich, P8215, Stamford, USA). Cell lysates were separated by SDS-PAGE and transferred onto PVDF membranes (Millipore, IPFL00010, Massachusetts, USA). Membranes were blocked with 5%
nonfat dry milk powder in Tris-buffered saline-Tween 20 buffer containing 20 mM Tris-HCl (pH 7.4), 137 mM NaCl, and 0.1% Tween-20 for 1 h at room temperature, incubated overnight at 4 °C
with primary antibodies against the target proteins, further incubated with appropriate horseradish peroxidase (HRP)–conjugated secondary antibody for 1 h, and the protein expression is
detected by enhanced ECL luminescence solution. The expression is analyzed by Image J software.
To obtain cDNA, total RNA was extracted using TRIzol (Invitrogen, California, USA) from indicated cells and reverse transcribed using SuperScript III First-Strand Synthesis SuperMix
(Invitrogen, 11752-50, California, USA). A 20 µL volume reaction consisted of 1 µL reverse transcription product and 250 nM of each primer, and the thermal cycle program was as follows:
denaturing for 15 s at 95 °C, annealing, and extension at 60 °C for 30 s. The relative quantity of samples was calculated according to the comparative −ΔΔCt method and normalized to ActB.
The primers for BiP are: forward 5′-GAAAGAAGGTTACCCATGCAGT-3′ and reverse 5′-CAGGCCATAAGCAATAGCAGC-3′.
For immunoprecipitation assays, AD293 cells overexpressing the indicated plasmids were lysed for 15 min on ice with NP40 lysis buffer containing 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP40,
0.5% deoxycholate, and 1% protease-inhibitor cocktail (Sigma-Aldrich, P8215, Stamford, USA). The lysates were centrifuged at 13,000 r/min at 4 °C for 10 min, and the supernatant was rotated
lightly with anti-HA, anti-MYC, or anti-FLAG magnetic beads for 5 h at 4 °C. The beads were washed two times for 20 s at 4 °C with rotation using the Triton X-100 lysis buffer containing
300 mM NaCl. Samples were boiled at 100 °C for 10 min with 1 × SDS-PAGE sample buffer and then loaded.
For endogenous IP analysis, cells were lysed according to the described above and the supernatant was incubated with the indicated primary antibodies overnight at 4 °C and mixed with protein
A/G agarose beads (Santa Cruz, sc-2003, Santa Cruz, USA) for 3 h at 4 °C. The beads were washed two times at 4 °C using the same lysis buffer by centrifugation at 2500 × g. The following
processes were the same as described above.
For pull-down assay, MCF-7 cells were transiently transfected with pLVX-RHBDF1-HA or control pLVX-HA, and pull-down analysis was performed with anti-HA magnetic beads as described above. The
immunoprecipitants were electrophoresed by 10% SDS-PAGE and stained using the Fast Silver Stain Kit (Beyotime Biotechnology, P0017S, Shanghai, China) according to the manufacturer’s
instructions. The bands of interest were cut into small pieces, decolorized in 50% acetonitrile with 25 mM ammonium bicarbonate, reduced with dithiothreitol, alkylated with iodoacetamide,
and digested with trypsin (working concentration 10 ng/μL) at 37 °C overnight. The digestion solution after trypsin peptide extraction and lyophilization was analyzed by LC-MS/MS. The
LC/MS/MS analysis of Pinpoint and Proteome Discoverer1.4 was carried out using Thermo Scientific triple quadruplex ion trap and Orbitrap fusion technology.
For biotin-pull-down analysis, the MCF-7 cells were lysed and centrifuged at 13,000 r/min at 4 °C for 10 min, and the supernatants were rotated lightly with streptavidin beads and biotin-PE5
(100 µM) or biotin-mPE5 for 5 h at 4 °C and then the beads were washed, boiled, and loaded as the described above.
For determination of the effect of PE5 on the RHBDF1-BiP interaction, MCF-7 cells were co-transfected with R1-HA and BiP-MYC, and the lysates were centrifuged at 13,000 r/min at 4 °C for 10
min, and then the supernatants were rotated lightly with anti-HA beads and PE5 (100 µM) at 4 °C for 5 h and the following processes were same as the described above.
Cells were attached to the circular glass sheet and the frozen tumor tissue was sectioned into 6–8 mm sections, then were fixed with 4% paraformaldehyde, followed by permeabilization with
0.3% Triton X-100. Then samples were blocked with 5% bovine serum albumin and probed with the primary antibodies overnight at 4 °C. The following samples were washed and incubated with
secondary antibodies for 1 h at room temperature, then stained with DAPI (Invitrogen, 33342, California, USA). Samples were sealed by an antifading mounting medium and imaged using LSM 800.
To stain ER using DIOC6(3), the cells were attached to the circular glass sheet. After overnight, the cells were probed with DIOC6(3) (5 μM) for 5 min at room temperature, and then washed
and stained with DAPI, followed by imaging using LSM 800.
For the immunofluorescence assay of PE5, MCF-7 cells were attached to a 24-well cell culture dish with a glass slide and cultured at 37 °C in 5% CO2 for 6–12 h. Then 100 μM Mca-PE5 was added
into the medium and cultured for 1 h and then the following processes were the same as described above.
For the immunofluorescence assay of mouse breast cancer tissue, the frozen tumor tissue was sectioned into 6–8 mm sections and then the following processes were the same as described above.
The cell pellets were collected and fixed with 2.5% glutaraldehyde (Sigma-Aldrich, G5882, Stamford, USA). The samples were washed three times using 100 mM sodium phosphate buffer and were
post-fixed in 1% osmium tetroxide for 1 h. After blocking with 1% uranyl acetate, the samples were dyed for 1 h and dehydrated through a graded series of alcohol to 100% and embedded in
epoxy resin. The embedded samples were cut using Leica ultramicrotome EM UC6 (Leica). Later, the samples were stained with uranyl acetate and lead citrate in a Leica EM Stainer and imaged
under a Spirit Transmission Electron Microscope (FEI Company) operating at 120 kV.
To detect intracellular aggresome, the cells were collected and then stained using PROTEOSTAT® Aggresome detection kit (ENZO, ENZ-51035-K100, New York, NY 10022, USA) according to the
manufacturer’s instructions and quantified via flow cytometry using a BD FACSCanto II cytometer (BD Biosciences, San Jose, CA, USA).
To create the functional peptide segment engaging with the ATP-binding domain of BiP, we performed the molecular docking analysis using Schrodinger 2015 suite (Schrodinger, version 3.1, LLC,
New York, NY, 2015). Five peptide candidates surrounding Y713/R714 (PV6, YV5, PE5, PA4, YE4) (Supplementary Fig. S2-1) were selected for molecular docking with BiP’s N-terminal ATP-binding
domain (PDB: 3IUC). Since peptide binding sites in the N-terminal ATP-binding domain of BiP have not yet been published, we found five potential peptide binding sites in the N-terminal
ATP-binding domain of BiP using the Schrodinger 2015 suite (Supplementary Fig. S2-2), and then used one of them as a ligand binding pocket of five peptides for molecular docking
(Supplementary Fig. S2-2, Site 1). The structure of the human BiP protein was retrieved from the Uniprot database (https://www.uniprot.org) and downloaded from the RCSB database
(http://www1.rcsb.org) in PDB format. The 2D to 3D structural transformation and conformational optimization of BiP protein were performed using Schrodinger’s LigPrep module, and the Protein
Preparation Wizard module was used for protein hydrogenation, hydrogen bonding, removal of water molecules, and energy minimization under OPLS 2005 field. Then the Receptor Grid Generation
module is used to generate the docking grid. Finally, the molecular docking process of peptides and proteins is completed with the Ligand Docking panel of the Glide module. The peptide with
the highest Glide GScore was selected for further SPR experiments.
The human BiP (full-length) DNA was inserted in a pET28a vector (Yeasen, 11905ES03, Shanghai, China) with N-terminal 6 × His-tag and expressed in Escherichia coli BL21 (DE3) cells (Agilent).
The strain was cultured in Luria-Bertani (LB) medium at 37 °C for 3 h. When OD600 nm was 0.6, 1 mM gratuitous inducer IPTG (MCE, HY-15921, Nashville, TN 37212, USA) was mixed with medium
and cultured continuously at 16 °C for 20 h. The bacterial cells were centrifuged at 9000 r/min for 15 min and the supernatant was lysed by sonication. The proteins were purified in the
buffer containing 50 mM HEPES, pH 8.0, and 400 mM NaCl using Ni-NTA agarose (Qiagen, 30230, Shanghai, China). The agarose was washed with 20 mM imidazole buffer to remove the miscellaneous
proteins. The BiP protein was eluted in 250 mM imidazole buffer and incubated with PreScission protease overnight at 4 °C to remove 6 × His-tag. To further purify the protein, anion exchange
was performed using a HiTrap Q HP column (GE Healthcare) and size exclusion chromatography was performed using a HiLoad 16/60 Superdex 200 column. The protein buffer was exchanged into
buffer B (50 mM HEPES, pH 8.0, 50 mM NaCl, 10 mM MgCl2, 10 mM KCl) for the experiments.
To evaluate the interaction between PE5 and BiP, surface plasmon resonance (SPR) was performed using a BIAcore T200 (Uppsala, Sweden). The recombinant BiP protein (1 nM) as the ligand was
immobilized onto the surface of carboxylate glucans in the HTG sensor chip and PBS was used as the running buffer for PE5 or mPE5 and PBS-P with 5% DMSO was used as the running buffer for
HA15. Each affix sample was prepared and injected onto the chip surface at seven gradient concentrations ranging from 156 nM to 5000 nM to record the sample binding to the surface. The
experimental temperature was 25 °C and the flow rate was 20 μL/min. According to the 1:1 Langmuir binding model, the affinity constants (Kd) were calculated by BIAcore T200 software 3.0.2.
ATP-to-ADP conversion was measured by the ADP-GloTM Kinase Assay Kit (Promega, V6930, Madison, WI 53711-5399, USA). A reaction mixture containing 40 μM recombinant full-length BiP protein
and a standard ATPase assay buffer with increased concentrations of PE5, mPE5, or HA15 was prepared in 384 Wells of white OptiPlate (Perkin Elmer, Massachusetts, USA) and then was
preincubated for 30 min at 37 °C. Then 1 mM ATP was added to the reaction and incubated for 2 h. Luminescence was read on a Multifunctional microporous plate detector (TECAN, 03030923,
Austria).
The cells (1 × 105~ 2 × 105/well) were attached in 6-well plates and cultured for 6~12 h at 37 °C in 5% CO2. PE5 or mPE5 was added to the medium at the indicated concentrations and
continuously cultured for 48 h. After washing with cold PBS and resuspending, the cells were stained with Annexin V-FITC and propidium iodide (PI) using Annexin V–FITC Apoptosis Detection
Kit (Elabscience, E-CK-A211, Wuhan, China) according to the manufacturer’s protocol. The fluorescence was measured by flow cytometry using a BD FACSCanto II cytometer (BD Biosciences, San
Jose, CA, USA).
1.0 × 106 4T1 cells were suspended in 100 mL PBS and then were injected subcutaneously into the right-side chest of 8-week-old female BALB/c mice (Taconic Bioscience). When the average tumor
size reached 60 mm3, the mice were randomly divided into 5 mice in each group. The control group or the treatment group was injected subcutaneously with PBS or PE5 in the pericancerous
epithelium, once every 2 days. The tumor size and volume were measured by calipers once every two days and calculated by the formula: 0.5 × D × d2, where D and d were the longest and
shortest vertical diameters, respectively. When the tumor size in the control group reached 1000 mm3, the experiment was concluded and the mice were euthanized and tumors were collected and
frozen for histology.
All data were determined from at least three independent experiments and presented as mean ± SD. The unpaired two-tailed Student t-test was used to analyze the statistical difference between
the two sets of data. One-way ANOVA was used for statistical analysis of the difference between multiple groups. *P-values