- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Chronic obstructive pulmonary disease (COPD) is characterised by persistent airway inflammation even after cigarette smoking cessation. Neutrophil extracellular traps (NETs) have
been implicated in COPD severity and acute airway inflammation induced by short-term cigarette smoke (CS). However, whether and how NETs contribute to sustained airway inflammation in COPD
remain unclear. This study aimed to elucidate the immunoregulatory mechanism of NETs in COPD, employing human neutrophils, airway epithelial cells (AECs), dendritic cells (DCs), and a
long-term CS-induced COPD mouse model, alongside cyclic guanosine monophosphate-adenosine monophosphate synthase and toll-like receptor 9 knockout mice (_cGAS__-−/−_, _TLR9__−/−_);
Additionally, bronchoalveolar lavage fluid (BALF) of COPD patients was examined. Neutrophils from COPD patients released greater cigarette smoke extract (CSE)-induced NETs (CSE-NETs) due to
mitochondrial respiratory chain dysfunction. These CSE-NETs, containing oxidatively-damaged DNA (NETs-DNA), promoted AECs proliferation, nuclear factor kappa B (NF-κB) activation,
NF-κB-dependent cytokines and type-I interferons production, and DC maturation, which were ameliorated/reversed by silencing/inhibition of cGAS/TLR9. In the COPD mouse model, blocking
NETs-DNA-sensing via _cGAS_−_/−_ and _TLR9_−_/−_ mice, inhibiting NETosis using mitoTEMPO, and degrading NETs-DNA with DNase-I, respectively, reduced NETs infiltrations, airway inflammation,
NF-κB activation and NF-κB-dependent cytokines, but not type-I interferons due to IFN-α/β receptor degradation. Elevated NETs components (myeloperoxidase and neutrophil elastase activity)
in BALF of COPD smokers correlated with disease severity and NF-κB-dependent cytokine levels, but not type-I interferon levels. In conclusion, NETs-DNA promotes NF-κB-dependent autoimmunity
via cGAS/TLR9 in long-term CS exposure-induced COPD. Therefore, targeting NETs-DNA and cGAS/TLR9 emerges as a potential strategy to alleviate persistent airway inflammation in COPD. SIMILAR
CONTENT BEING VIEWED BY OTHERS N6-METHYLADENOSINE-MODIFIED CIRCSAV1 TRIGGERS FERROPTOSIS IN COPD THROUGH RECRUITING YTHDF1 TO FACILITATE THE TRANSLATION OF IREB2 Article 24 February 2023 A
DISINTEGRIN AND METALLOPROTEINASE DOMAIN-15 DEFICIENCY LEADS TO EXAGGERATED CIGARETTE SMOKE-INDUCED CHRONIC OBSTRUCTIVE PULMONARY DISEASE (COPD)-LIKE DISEASE IN MICE Article 20 July 2020
NEUTROPHIL EXTRACELLULAR TRAPS PROMOTE IMMUNOPATHOGENESIS OF VIRUS-INDUCED COPD EXACERBATIONS Article Open access 09 July 2024 INTRODUCTION Chronic obstructive pulmonary disease (COPD) is a
leading cause of global morbidity and mortality,1 and is characterised by persistent airflow limitation owing to progressive airway remodelling and alveolar destruction. Cigarette smoke (CS)
is the primary risk factor for COPD. CS induces an abnormal inflammatory response, characterised by increased neutrophils, macrophages and T and B lymphocytes recruited from the
circulation. These inflammatory cells, together with lung structural cells, including airway epithelial cells (AECs), secrete numerous proinflammatory cytokines to induce chronic airway
inflammation, which essentially drives tissue injury in COPD.2 Anti-inflammation is considered the mainstream strategy for COPD therapy. Bronchodilators, including long-acting muscarinic
antagonists (LAMAs) and long-acting beta-2 agonists (LABAs), either as standalone agents or in combination with anti-inflammation agents (inhaled corticosteroids, ICS), constitute the
cornerstone of pharmacological treatment for patients with COPD. Nevertheless, ICS administration alone does not decelerate the rate of lung function decline in patients with COPD.3
Moreover, in contrast to the transient acute airway inflammation induced by short-term CS exposure, the airway inflammation observed in COPD persists even after smoking cessation,4,5
suggesting that the efficacy of the current therapy is limited by persistent airway inflammation. However, researchers have not completely elucidated the underlying mechanism. A key aspect
of persistent airway inflammation lies in the delayed immunological reactions caused by a complex interplay between innate and adaptive immune systems, particularly in response to
pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). DAMPs are notably augmented in COPD due to hazardous substances from CS exposure, oxidative
stress-induced apoptosis, and impaired phagocytosis by alveolar macrophages.6 Pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) and cyclic guanosine
monophosphate-adenosine monophosphate synthase (cGAS), recognise these DAMPs and initiate signalling cascades, often culminating in the nuclear factor kappa B (NF-κB) activation in COPD.7,8
Once activated, NF-κB translocates to the nucleus within AECs and alveolar macrophages,9,10 stimulating transcription of inflammatory cytokines genes, including C-X-C motif chemokine ligands
(CXCL5), interleukins (such as IL-8, IL-1β), granulocyte-macrophage colony-stimulating factor (GM-CSF), tumour necrosis factor-alpha (TNF-α), and type-I interferons (IFNs, such as IFN-β).
These cytokines and IFNs further perpetuate and exacerbate airway inflammation by recruiting and activating neutrophils, macrophages, and lymphocytes (including B cells, T cells and Natural
Killer Cells).2,11 Additionally, viral infections, such as those caused by adenovirus, can amplify inflammation through their persistent DNA recognised by TLRs and cGAS, to further stimulate
cytokines production through NF-κB pathway.12,13 On the other hand, there is evidence of autoimmune elements within the COPD pathology, where self-DNA released from apoptosis and necrosis
of lung tissue may trigger an inappropriate immune response via TLRs and cGAS activation, thereby contributing to sustained airway inflammation.14,15 Recently, research has increasingly
highlighted neutrophil extracellular traps (NETs), a web-like structure composed of self-DNA released by the neutrophils, as an active contributor to COPD pathogenesis16: (1) Clinically, the
induced sputum of patients with both stable and exacerbated COPD shows increased level of NETs,17,18 which have been associated with the severity of airflow limitation and microbiota
diversity;19,20,21 (2) In vitro, the CS extract (CSE) is capable of inducing NETosis, a releasing process of NETs, on both human22,23 and mouse24 neutrophils; (3) NETs stimulated by
phorbol-12-myristate-13-acetate (PMA)25,26 and IL-827 cause cytotoxicity in the human primary AECs and AECs cell lines; (4) PMA-,25,28 uric acid (UA)-, and monosodium urate crystal (MSU)-29
induced NETs promote, whereas IL-8-induced NETs prohibit,27 the expressions of IL-8 and IL-6 on AECs; (5) CSE-induced NETs (CSE-NETs) activate the maturation of myeloid dendritic cells (DCs)
to initiate a T-cell-mediated immune response;22,24 and (6) In vivo, NETs or self-DNA have been implicated in the development of acute airway inflammation in short-term CS-treated
mice.23,30 Therefore, NETs participate in short-term CS-induced acute airway inflammation by affecting the function of AECs and DCs. However, researchers have not yet investigated the
contribution of DNA components in NETs (NETs-DNA) to persistent airway inflammation in COPD by initiating an autoimmune response. In this study, we investigated whether and how NETs
contribute to long-term CS-induced COPD, with a focus on the immunoregulatory effects of NETs-DNA on AECs, and the role of NETs and targeted pathways as therapeutic targets to alleviate
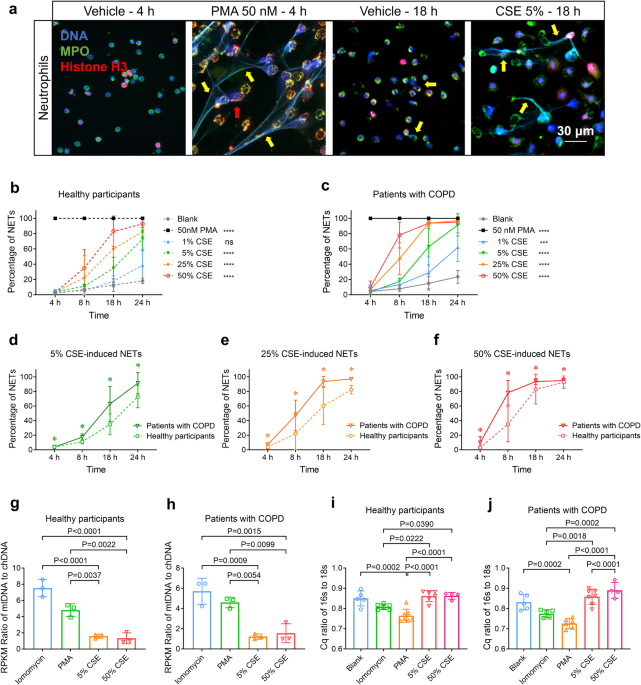
persistent airway inflammation in COPD. RESULTS CSE INDUCES GREATER NETOSIS OF NEUTROPHILS DERIVED FROM PATIENTS WITH COPD; CSE-NETS CONTAIN CHDNA AND MTDNA CSE induces NETosis in human
peripheral blood neutrophils (hPBNs);22,23 CS exposure also triggers chDNA and mtDNA release from lung tissue. We asked whether hPBNs from healthy participants and patients with COPD
(supplementary Tables S1, S2) exhibit the different capacities of NETosis upon CSE stimulation, and whether the mtDNA:chDNA ratio differs between CSE-induced NETs and those produced by PMA
or lomomycin, other known NETosis inducers. We quantified NETosis percentage in hPBNs treated with escalating doses of CSE and varying incubation time, then measured the mtDNA:chDNA ratio
within NETs generated by CSE, PMA and iomomycin, respectively, by using next-generation sequencing, and assessing the 16s (mtDNA marker) to 18s (chDNA marker) ratio. We observed that hPBNs
derived from patients with COPD (COPD-neutrophils) display a higher potency to release NETs upon stimulation with increasing CSE (Fig. 1a–f, Supplementary Figs. S2d–j, S3a and Supplementary
Movie S1). CSE-induced NETs contained both mtDNA and chDNA at a ratio of approximately 1.3:1, whereas those stimulated by lomomycin and PMA contained a higher ratio of mtDNA to chDNA (Fig.
1g, h). These results were further corroborated by the quantification cycle (Cq) value ratio of _16s_ to _18s_ (Fig. 1i, j). Together, COPD-neutrophils show increased NETosis upon CSE
stimulation, and CSE-NETs contain lower ratios of mtDNA:chDNA compared to those induced by PMA and lomomycin. CSE-INDUCED NETOSIS REQUIRES MITOCHONDRIAL ROS, BUT NOT NOX NETosis triggered by
PMA is nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX)-dependent, but the mechanism of NETosis induced by CSE remains elusive. Given that CSE-NETs exhibit heightened mtDNA
content compared to PMA-NETs, we asked whether mitochondrial reactive oxygen species (mtROS) or NOX played a pivotal role in CSE-induced NETosis. We thus evaluated the inhibitory effects on
NETosis induced by 5% CSE or PMA using respective inhibitors for mtROS (mitoTEMPO), mitochondrial respiration (thenoyltrifluoroacetone, TTFA), and NOX (diphenyleneiodonium chloride [DPI]
and VAS2870 [VAS]). Inhibiting mtROS and mitochondrial respiration using mitoTEMPO and TTFA selectively suppressed 5% CSE-induced, but not PMA-induced, NETosis (Fig. 2a, b) in hPBNs derived
from healthy participants, whereas NOX inhibitors DPI and VAS specifically attenuated PMA-induced, but not 5% CSE-induced NETosis (Fig. 2c, d); Deoxyribonuclease-I (DNase-I, endonuclease for
single/double-stranded DNA) and GW311616A (GW, inhibitor of neutrophil elastase [NE]) treatments were effective against both 5% CSE- and PMA-induced NETosis (Fig. 2e, f and Supplementary
Fig. S3b), with comparable results observed for COPD patient-derived hPBNs (Supplementary Fig. S4a–f). Treatment with 5% CSE resulted in elevated mtROS levels, particularly in hPBNs from
COPD patients (Fig. 2g–i), with co-staining suggesting that mitochondria are the primary source of cellular ROS (supplementary Fig. S4g). Additionally, 5% CSE exposure triggered
co-localisation of oxidative DNA damage marker 8-OHdZG (8-hydroxy-2’-deoxyguanosine) with mitochondrial membrane protein TOMM20 (outer mitochondrial membrane complex subunit 20) on the hPBNs
(Fig. 2j) and enhanced 8-OHdZG staining in CSE-NETs derived from both healthy participants and patients with COPD (Fig. 2k and supplementary Fig. S4h). Together, CSE-induced NETosis relies
on mtROS and mitochondrial respiration; CSE also provokes DNA damage within the mitochondrial membrane of hPBNs and in the formed CSE-NETs. CSE-NETS INCREASE THE PROLIFERATION AND PRODUCTION
OF BOTH NF-ΚB-DEPENDENT CYTOKINES AND TYPE-I IFNS IN HUMAN AECS (HAECS), AND THE MATURATION OF HUMAN DCS (HDCS) Previous studies have shown that PMA, IL-8, UA, and MSU-induced NETs can
exert cytotoxic effects or modulate the expression of IL-8 and IL-6 on AECs;25,26,27,28,29 however, the impact of CSE-induced NETs on primary human AECs remains uncertain. To address this,
we prepared 5% CSE-induced NETs from healthy participants (Supplementary Method 9 and Supplementary Fig. S5a), and treated hAECs with escalating doses of NETs-DNA. We found that both 6 μg/mL
and 12 μg/mL, but not 24 μg/mL, CSE-NETs promoted hAECs proliferation (Fig. 3a–c). In hAEC, the NETs treatment led to a dose-dependent increase in gene expression and soluble protein levels
of NF-κB-dependent cytokines (CXCL5, IL-8, TNFα and IL-1β), as well as type-I IFNs (IFN-β1 and IL-12, Fig. 3d–l). Additionally, NETs treatment facilitated NF-κB (P65 and P50) activation in
hAECs (Supplementary Figs. S5b, S6a–e), and maturation of hDCs (Fig. 3m, n). Together, CSE-NETs promote the proliferation and production of NF-κB-dependent cytokines and type-I IFNs in
hAECs, and enhance the maturation of hDCs. CGAS/TLR9 IS REQUIRED FOR CSE-NETS-INDUCED PROLIFERATION AND PRODUCTION OF BOTH NF-ΚB-DEPENDENT CYTOKINES AND TYPE-I IFNS IN HAECS, AND MATURATION
OF HDCS cGAS/TLR9 senses mtDNA/chDNA to activate downstream signalling cascades. Given the presence of mtDNA and chDNA in CSE-NETs, we asked whether these receptors contribute to the
proliferation and production of cytokines in hAECs treated with CSE-NETs. We found that silencing both _cGAS_ and _TLR9_ reduced NETs-mediated hAECs proliferation (Supplementary Fig. S5c–f
and Fig. 4a–c, m–o). _cGAS_ silencing ameliorated the NETs-induced gene expression and/or soluble protein levels of CXCL8, IL-1β, IFN-β1, and IL-12 (Fig. 4d–l), whereas _TLR9_ silencing
ameliorated NETs-induced gene expression and/or soluble protein levels of CXCL5, TNFα, IL-1β and IL-12 (Fig. 4p–x). Furthermore, NETs-induced NF-κB (P65 and P50) activation in hAECs was
inhibited upon _cGAS_ and _TLR9_ silencing (Supplementary Fig. S6a–e). The inhibitors RU.521 and ODN 2088, targeting cGAS and TLR9, respectively, also decreased NETs-mediated DC maturation
(Fig. 4y, z). Together, DNA sensor cGAS/TLR9 is essential for CSE-NETs-induced proliferation, production of NF-κB-dependent cytokines and type-I IFNs in hAECs, and hDCs maturation. KNOCKOUT
OF CGAS AND TLR9, RESPECTIVELY ALLEVIATES AIRWAY INFLAMMATION, NETS INFILTRATION AND PRODUCTION OF NF-ΚB-DEPENDENT CYTOKINES, BUT NOT TYPE-I IFNS, IN A COPD MOUSE MODEL We then investigated
whether the knockout of NETs-DNA sensor cGAS/TLR9 could mitigate long-term CS exposure-induced airway inflammation in an established COPD mouse model (Supplementary Method 19). We exposed
_cGAS_ and _TLR9_ knockout mice (_cGAS__−/−_ and _TLR9__−/−_) and corresponding littermate to CS, and evaluated their severity of airway inflammation and changes in lung functions (Fig. 5a
and Supplementary Fig. S7). Compared to CS-littermate (CS-littermate) mice, CS-treated _cGAS__−/−_ mice (CS-_cGAS__−/−_) displayed reduced total cells, neutrophils and lymphocytes in
bronchoalveolar lavage fluid (BALF, Fig. 5b–d), decreased NF-κB-dependent cytokines, including CXCL5, TNFα, IL-1β in BALF (Fig. 5e–h), CXCL5, GM-CSF, IL-1β in serum (Supplementary Fig.
S9n–p), and CXCL5, IL-1β in lung tissue slices (Supplementary Fig. S9c, d, h–k), suppressed lung tissue NF-κB P65 activation (Supplementary Fig. S9q), improved lung function indicated by
reduced FRC/BW (functional residual capacity/body weight) and increased FEV100/FVC (forced expiratory volume at 100 ms/forced vital capacity, Fig. 5k, l), and reduced histological score,
mucin stain score, and mean linear intercept (MLI) of the alveoli (Fig. 5m, n and Supplementary Fig. S9a, b, f, g). The severity of NETs infiltration, as assessed by immunofluorescence
co-staining of main components of NETs (Supplementary Method 24 and Supplementary Fig. S8), decreased in the CS-_cGAS__−/−_ mice (Fig. 5o, p), and was correlated with neutrophil count,
histological score, and FEV100/FVC in CS-treated mice (Supplementary Fig. S9t–v). However, no change in type-I IFNs was observed in either CS-littermates or CS-_cGAS__−/−_ mice (Fig. 5i, j
and Supplementary Fig. S9e, l, m, r, s), potentially due to impaired expression of IFN-α/β receptor subunit-1 (IFNAR1) in hAECs upon CS exposure (Supplementary Fig. S6f). A similar
amelioration pattern was seen in CS-_TLR9__−/−_ mice (Supplementary Figs. S10, S11). Collectively, cGAS and TLR9 Knockout independently mitigated long-term CS-induced NF-κB (but not typer-I
IFNs)-related airway inflammation. INHIBITION OF NETOSIS BY MITOTEMPO ALLEVIATES AIRWAY INFLAMMATION, NETS INFILTRATION AND PRODUCTION OF NF-ΚB-DEPENDENT CYTOKINES, BUT NOT TYPE-I IFN, IN
THE COPD MOUSE MODEL To further investigate the contribution of NETs to long-term CS exposure-induced airway inflammation, we treated the COPD mouse model with mitoTEMPO, the specific mtROS
inhibitor that inhibits CSE-induced NETosis as shown above. Compared to CS-treated controls, CS-treated mice receiving intraperitoneal injection of mitoTEMPO (CS-MT i.p mice, Fig. 6a)
displayed reduced total cells, neutrophils, and lymphocytes in BALF (Fig. 6b–d), decreased NF-κB-dependent cytokines (CXCL5, GM-CSF and IL-1β) in both BALF (Fig. 6e–h), serum (Supplementary
Fig. S12n–p) and lung tissue slices (Supplementary Fig. S12c, d, h–k), suppressed lung tissue NF-κB P65 activation (Supplementary Fig. S12q), improved lung function indicated by reduced
FRC/BW and increased FEV100/FVC (Fig. 6k, l), and reduced histological score, mucin stain score, and MLI of alveoli (Fig. 6m, n and Supplementary Fig. S12a, b, f, g). The severity of NETs
infiltration decreased in the CS-MT i.p mice (Fig. 6o, p), and was correlated with IL-1β and CXCL5 levels, but not IFN-β1 level, in the BALF of CS-treated mice (Supplementary Fig. S12t–v).
No changes in type-I IFNs were observed (Fig. 6i, j and Supplementary Fig. S12e, l, m, r, s). Overall, mitoTEMPO alleviated long-term CS-induced NETs infiltration and NF-κB (but not typer-I
IFNs)-related airway inflammation. DEGRADATION OF NETS-DNA BY DNASE-I ALLEVIATES NETS INFILTRATION AND EMPHYSEMA-PHENOTYPE IN THE COPD MOUSE MODEL DNase-I has been shown to reduce airway
inflammation induced by acute CS exposure.23 To explore the therapeutic potential of targeting NETs-DNA to mitigate airway inflammation induced by long-term CS exposure, we employed DNase-I
to degrade NETs-DNA in the COPD mouse model. Compared with CS-treated control, CS-treated mice receiving nebulised DNase-I (DNase-I _Neb_. mice, Fig. 7a) showed alleviated
emphysema-phenotype, as indicated by reduced FRC/BW (Fig. 7k) and MLI of alveoli (Fig. S13a, f), although there were only downward trends for total and different cell counts in BALF (Fig.
7b–d), histological scores (Fig. 7m, n), and mucin staining scores (Supplementary Fig. S13b, g) were observed in DNase-I _Neb_. mice. Nevertheless, NF-κB-dependent cytokines CXCL5 and IL-1β
levels in BALF (Fig. 7e–h) and lung tissue slices (Supplementary Fig. S13c, d, h–k), IL-1β level in serum (Supplementary Fig. S13n–p), and NF-κB P65 activation in lung tissues (Supplementary
Fig. S13q) significantly decreased in DNase-I _Neb_. mice. The severity of NETs infiltration also decreased in CS-DNase-I _Neb_. mice (Fig. 7o, p), and was correlated with MLI of alveoli
and FRC/BW in CS-treated mice (Supplementary Fig. S13t, u). No changes in type-I IFNs were observed (Fig. 7i, j and Supplementary Fig. S13e, l, m, r, s). Together, DNase-I treatment
alleviates NETs infiltration and emphysema features in the COPD mouse model. Supplementary Table S3 summarises the improved indicators seen in CS-_cGAS__−/−_, _TLR9_−_/−_, MT i.p, and
DNase-I _Neb_. mice. DISORDERED NF-ΚB-DEPENDENT CYTOKINES, BUT NOT TYPE-I IFNS, ARE CORRELATED WITH MPO AND NE ACTIVITY IN THE BALF OF PATIENTS WITH COPD Patients with COPD, both stable and
exacerbated, exhibit increased NETs in induced sputum17,18 that correlate with airflow limitation severity and microbiota diversity.19,20,21 However, the relationship between NETs
components, NF-κB-dependent cytokines, and type-I IFNs in BALF of COPD patients remains unexplored. We collected BALF from 34 healthy participants (13 without and 21 with smoking history)
and 32 COPD patients with smoking history (Supplementary Methods 1–3 and Supplementary Tables S1, S2). In comparison to healthy non-smokers and smokers, patients with COPD showed increased
levels of NF-κB-dependent cytokines (IL-1β, CXCL8 in BALF, Fig. 8a, d, and CXCL5, IL-1β in lung tissue slices, Supplementary Fig. S16e–j) and NETs components (MPO, NE activity in BALF, Fig.
8j, Supplementary Fig. S15a), but decreased the level of type-I IFN (IFN-β in BLAF, Fig. 8g, and in lung tissue slices, Supplementary Fig. S16k–m). The levels of IL-1β, CXCL8, and MPO
correlated negatively with FEV1/FVC and FEV1%Pred (the ratio of tested FEV1 to predicted FEV1), respectively, after adjusting for age, sex, body mass index (BMI), and smoking history (Fig.
8b, c, e, f, k, l). NE activity showed a negative correlation with FEV1/FVC (supplementary Fig. S15b, c). Conversely, IFN-β level did not correlate with either FEV1/FVC or FEV1%Pred (Fig.
8h, i). Notably, MPO level positively correlated with CXCL8 level, and NE activity correlated with IL-1β and CXCL8 levels, respectively (Fig. 8m, n and Supplementary Fig. S15d, e); however,
neither MPO nor NE activity correlated with IFN-β level (Fig. 8o and Supplementary Fig. S15f). Additionally, we observed increased nuclear expression of NF-κB P65 protein in airway
epithelial cells of COPD patient lung slices, compared with that of non-smokers and smokers in the healthy group (Supplementary Table S4 and Supplementary Fig. S16a–d). Together, NETs
components are linked to dysregulated NF-κB-dependent cytokines, but not type-I IFNs, in BALF of COPD patients. DISCUSSION NETs are initially recognised to entrap and kill pathogens via a
DNA framework comprising abundant antimicrobial granular proteins, such as MPO and NE.31 However, they are immunogenic and have been implicated in several autoimmune and non-infectious
diseases.25,32,33 In COPD, augmented self-DNA from increased lung cell apoptosis acts as DAMPs, impeding inflammation resolution. NETs, rich in DNA content, have been implicated in the loss
of microbiota diversity among patients with COPD20 and CS-induced acute airway inflammation in murine models.23,30 Nevertheless, the role of NETs in mediating innate and adaptive immune
responses to sustain airway inflammation in COPD remains unknown. Herein, we demonstrated the immunomodulatory role of NETs in long-term CS-induced COPD as follows: (1) hPBNs derived from
patients with COPD exhibited a stronger ability to release CSE-NETs, which contained high levels of oxidatively-damaged mtDNA and chDNA; (2) CSE-NETs not only stimulated the proliferation
and production of both NF-κB-dependent cytokines and type-I IFNs on hAECs, but also promoted the maturation of hDCs; (3) these effects were ameliorated by the silencing/inhibition of cGAS
and TLR9 in vitro; (4) in vivo, the knockout of DNA sensors (_cGAS__−/−_ and _TLR9__−/−_ mice), the inhibition of NETs (by mitoTEMPO i.p), and the degradation of NETs (by DNase-I _Neb_.)
respectively resulted in alleviated airway inflammation, reduced production of NF-κB-dependent cytokines (but not type-I IFNs), suppressed NF-κB P65 activation, and improved lung function in
the long-term CS-induced COPD mouse model; (5) the severity of NETs infiltration was correlated with that of airway inflammation in the CS-treated mice; and (6) In COPD patients, the levels
or activity of NETs components (MPO and NE activity) in BALF correlate with disease severity and the abundance of NF-κB-dependent cytokines (IL-1β and CXCL8), but not those of type-I IFN
(IFN-β). Taken together, the DNA components in CSE-NETs promoted NF-κB-, but not type-I IFNs-, dependent autoimmunity via cGAS/TLR9 in long-term CS-induced COPD. The cause of excessive
NETosis in COPD is largely unknown;34 despite spontaneous NETosis regulated by CXCR2 in patients with COPD,35 NETosis induced by CS exposure might be a more likely contributor. Notably, we
observed a stronger capacity for NETosis in COPD-neutrophils upon CSE stimulation. Consistently, COPD-neutrophils demonstrated increased NETosis following exposure to diesel exhaust,36 thus
suggesting greater susceptibility to undergo NETosis following exposure, potentially linked to augmented mtROS release in COPD-neutrophils. Unlike PMA-induced NETosis that relies on NOX,
CSE-triggered NETosis responds primarily to mtROS. Consistently, ribonucleoprotein (RNP)-induced NETosis required mtROS instead of NOX in patients with systemic lupus erythematosus,37
thereby indicating that NETosis induced by different inducers may rely on dissimilar mechanisms.38 Moreover, CSE-NETs consist of abundant oxidatively-damaged DNA, which is more capable of
promoting autoimmune reactions33 and the formation of autologous NETs.39 Furthermore, CSE-NETs comprised a lower ratio of mtDNA to chDNA compared to PMA- and iomomycin-NETs, since mtDNA
exhibits different immunoregulatory features from chDNA,40 NETs induced by different stimuli may exert diverse cellular effects. This variability could account for the controversial effects
of PMA- and IL-8-induced NETs on IL-8/IL-6 expression in hAECs.25,27,28 Accordingly, we investigated the effects of CSE-NETs on the function of hAECs, which are essential for maintaining
immune homoeostasis in the lungs.41 CSE-NETs stimulated proliferation, production of both NF-κB-dependent cytokines and type-I IFNs, and NF-κB (P65 and P50) activation in hAECs. Moreover,
both cGAS and TLR9 were required to facilitate these effects, thus emphasising the importance of NETs-DNA sensing on hAECs to mediate an autoimmune response. Previously, CSE-NETs were shown
to promote DC maturation to prime T-cell-mediated immune response,22,24 and we further showed that inhibition of cGAS and TLR9 suppressed the CSE-NETs-induced maturation of hDCs. Taken
together, CSE-NETs triggered hAEC and hDC dysfunction, and the sensing of NETs-DNA by both airway structural cells (such as hAECs) and immune cells (such as hDCs) promoted autoimmune
reactions. TLR4 and TLR9 are critical PPRs involved in COPD pathogenesis.42,43,44 TLR4 recognises exogenous ligands (such as lipopolysaccharide), respiratory syncytial virus fusion protein,
and endogenous ligands like heat shock proteins (HSP60, HSP70) and high mobility group box 1 (HMGB1), whereas TLR9 specialises in the detection of unmethylated CpG motifs prevalent in
bacterial and viral DNA, as well as in human mtDNA.45 cGAS is a crucial innate immune sensor that detects cytoplasmic DNA from pathogens or cellular injury. Both cGAS and TLR9 are reported
DNA sensors capable of sensing cytosolic/extracellular mtDNA/chDNA.46,47 The recognition of self-DNA by cGAS and TLR9 not only activates the NF-κB signalling pathway to release
NF-κB-dependent cytokines but also induces the robust production of type-I IFNs.15 These cytokines and IFNs serve as prominent immunomodulators in autoimmune contexts.48,49 Excessive
activation of cGAS/TLR9 has been linked to allergic and autoimmune diseases.50,51 For example, sensing of cytosolic DNA by cGAS in hAECs52 and TLR9 in innate lymphoid cells53 plays a role in
asthma pathogenesis. However, the role of NETs-DNA recognition by cGAS/TLR9 in eliciting NF-κB-dependent cytokines and type-I IFNs response in COPD is unclear. Interestingly, our in vitro
data showed that CSE-NETs stimulated type-I IFNs production in hAECs, while in vivo data suggested that CS exposure resulted in decreased type-I IFNs levels in both CS-treated mice and
patients with COPD; moreover, only levels of NF-κB-dependent cytokines, but not type-I IFNs, were correlated with disease severity in COPD mouse models and patients with COPD. This
controversy between in vitro and in vivo results might stem from the impairment of IFNAR1 expression on hAECs, a crucial receptor of type-I IFNs to facilitate the downstream signalling
cascade,54 by long-term CS exposure (supplementary Fig. S6f),55 thus hindering the overall amplification and production of type-I IFNs. Notably, the decreased expression of type-I IFNs has
been previously reported in COPD contexts, the suppressed type-I IFNs in COPD might compromise the antiviral defence ability, thus contributing to persistent airway inflammation and even
acute exacerbations.56,57 These findings emphasised the key role of NETs-DNA and the associated NF-κB signalling, but not type-I IFNs response, in sustained airway inflammation in COPD. The
interplay and differences between cGAS and TLR9 in their downstream signalling upon DNA recognition remain poorly understood. In hDCs, the pathways regulated by cGAS and TLR9 may exist in
parallel and display interdependencies to suppress each other.58 Notably, mitochondrial DNA itself has been shown to activate NETosis via the cGAS-stimulator of interferon genes (STING)
axis, with enhancing NE production and extracellular DNA release within NETs during sterile inflammation,39 suggesting a functional involvement of cGAS-STING in NETosis. Therefore, it is
possible that NETs-DNA could stimulate a feedback loop in neutrophils by further promoting NETosis under CS exposure conditions. Consistently, we observed significantly less NETs
infiltration in CS-treated cGAS−/− (but not TLR9−/−) mice, indicating that cGAS, but not TLR9, participates in CS-induced NETosis. Consequently, _cGAS_ deficiency might not only reduce
NETs-DNA sensing but also suppress CS-induced NETosis, thus providing potentially superior protection against CS exposure than _TLR9_ deficiency. Regarding NETs-DNA sensing in hAECs,
although silencing _cGAS_ or _TLR9_ individually led to varied reductions in CSE-NETs-induced cytokines/IFNs expressions/productions, no significant differences emerged from two-way ANOVA
analysis (supplementary Fig. S14). Nevertheless, further investigation into the intricate regulatory roles that cGAS and TLR9 play in DNA sensing and response is warranted, to better clarify
their differences in downstream signalling upon DNA recognition. MitoTEMPO is a superoxide inhibitor that specifically targets the mitochondria.59 We observed a desirable control of
CS-induced COPD following mitoTEMPO treatment probably owing to the following reasons: (1) mitoTEMPO efficiently inhibits CSE-induced NETosis; (2) mitoTEMPO helps in restoring the
equilibrium of oxidant and antioxidant molecules,60 which are characterised in CS-induced COPD;61 (3) oxidatively-damaged mtDNA/chDNA is more capable of inducing the disordered release of
cytokines, which is likely controlled by mitoTEMPO. Previously, DNase-I was shown to protect mice from type-2 immunopathology33 and CS-induced acute airway inflammation,23 we further showed
that the degradation of NETs-DNA by DNase-I treatment alleviated NF-κB-dependent cytokines and emphysema-phenotype in the long-term CS-treated mice, suggesting that DNase-I could offer
modest protective effects against the sustained airway inflammation and subsequent alveolar destruction. It should be noted that the therapeutic potential of DNase-I might be limited by the
suboptimal nebulised administration methods used in this study. Nevertheless, we further showed that the severity of NETs infiltration was correlated with that of airway inflammation and
emphysema, as well as the levels of NF-κB-dependent cytokines in the CS-treated mice, thus supporting the importance of NETs-DNA in CS-induced airway inflammation and alveolar destruction.
In summary, our in vivo data demonstrate that sensing-blockage/inhibition/degradation of NETs-DNA confers therapeutic benefits in the long-term CS-induced COPD mouse model. However, when
translating these observations to clinical practice, it is essential to acknowledge the complexity of COPD pathophysiology, where targeting NETs or cGAS/TLR9 alone might not be sufficient to
fully address the intricate interactions between innate and adaptive immunity, DAMPs, cytokines, and concurrent pathogen challenges. Potential synergistic benefits in reducing inflammation
and improving lung function could be achieved through combination therapies targeting multiple aspects of COPD, including but not limited to NETosis and cGAS/TLR9 pathways. Moreover, given
the critical role of cGAS/TLR9 signalling in pathogen recognition and antiviral responses, as well as NETs’ function in pathogen clearance and lung microbiota maintenance, targeted
interventions must be judiciously designed to minimise disruption of host defence mechanisms. Our study has limitations: while we establish correlations between NETs components, disease
severity, and NF-kB-dependent cytokines levels in BALF of patients with COPD adhering to standard LAMA or ICS + LABA therapies, we lack information on individual pharmacological regimens
that might impact MPO and NE levels. This is particularly pertinent given previous studies suggesting an association between NETs abundance and ICS resistance or ICS regular use in patients
with asthma,62,63 implying potential interactions between NETosis and ICS therapy. Future research could benefit from incorporating detailed clinical records to elucidate how current
therapeutic strategies influence CS-induced NETosis. Herein, we present evidence that CS-induced NETs-DNA sensed by cGAS and TLR9 in AECs and DCs promotes NF-κB-, but not type-I IFNs,
dependent autoimmunity in long-term CS-induced COPD. Despite their roles in eliminating pathogens and maintaining microbiota diversity in the lungs, NETs contribute self-DNA as a significant
DAMP, promoting disordered production of inflammatory cytokines and thus sustaining airway inflammation in an autoimmune fashion in COPD. Consequently, NETs-DNA and its sensing receptors
(cGAS/TLR9) represent potential therapeutic targets to reduce persistent airway inflammation in COPD. Supplementary Fig. S17 illustrates the proposed mechanism. MATERIALS AND METHODS STUDY
APPROVAL AND ETHICS STATEMENTS The protocol of human study was reviewed and approved by the Chinese Ethics Committee for Registering Clinical Trials (approval number: ChiCTR900022271). All
animal experiments were approved and conducted in accordance with the guidelines of the Animal Ethics Committee of the West China Hospital (approval number: 2018049A). PATIENTS AND SAMPLES
COPD diagnosis adhered to the Global Strategy for the Diagnosis, Management, and Prevention of COPD (GOLD) criteria. From January 2016 to March 2020, the study recruited 45 healthy
individuals and 42 COPD patients at West China Hospital, all providing informed consent. Among them, 11 healthy volunteers and 10 COPD patients contributed peripheral blood samples for NETs
and DCs experiments. Additionally, 34 healthy participants (divided into 13 non-smokers and 21 smokers) and 32 COPD patients with a history of smoking underwent BALF testing (Supplementary
Tables S1, S2). The collections of BALF, circulating neutrophils, and monocytes were described in the supplementary materials. NEUTROPHILS ISOLATION AND NETS STIMULATION Neutrophils from
COPD patients and healthy participants were isolated using the MACSxpress Neutrophil Isolation Kit (Miltenyi Biotec, Cat No. 130-104-434) according to the manufacturer’s instructions, with
purity confirmed by flow cytometry for CD15 and CD16. Freshly isolated cells (1.0 × 106) were seeded on fibrinogen-coated coverslips and incubated in serum-free RPMI 1640 for adhesion.
Subsequently, NETs were induced by varying concentrations of CSE at 37 °C for 4 to 24 h to assess CSE-induced NETosis; PMA and ionomycin stimulation were used as positive control. We
administered mitoTEMPO, TTFA, DPI, VAS, DNase-I, and GW as described in the supplementary materials. QUANTIFICATION OF NETS-RELEASING NEUTROPHILS The evaluation of NETs-releasing neutrophils
percentage was conducted by immunofluorescence co-staining of the following NETs components: myeloperoxidase (MPO), histone H3, and DNA. Briefly, NETs on coverslips were washed twice with
phosphate buffer saline (PBS), permeabilized with ice-cold acetone:methanol (−20 °C), fixed in neutral formalin, then blocked and incubated overnight at 4 °C with primary antibodies for MPO
and histone H3. After PBS washes, secondary antibodies were applied for 2 h. Following mounting with DAPI medium, samples were imaged on whole-filed fluorescence microscopy and evaluated as
described in the supplementary materials. COLLECTION OF NETS-CONTAINING SUPERNATANTS CSE-induced NETs were prepared under 5% CSE for 18 h, which induces approximately 50% NETosis without
neutrophil necrosis. After CSE exposure, NETs were thoroughly washed and detached with MNase (Thermo Fisher Scientific, Cat No. 88216, 20 units/mL, 37 °C, 30 min). DNA concentration was
quantified using the QuantiFluor dsDNA system (Promega, Cat No. E2670). NETs were enriched and sequenced by next-generation sequencing as described in the supplementary materials. CULTURE
AND SIMULATION OF HAECS Primary hAECs were obtained from Lifeline Cell Technology (Cat No. FC-0016) and maintained in BrochiaLife Medium Complete Kit (Cat No. LL-0023) on collagen-coated
plates. Cells at passages 3–5 were subjected to 6, 12 or 24 μg/mL NETs for 48 or 72 h. RNA and protein extraction ensued using E.Z.N.A. HP Total RNA Isolation Kit (Omega Bio-Tek, Cat No.
R6812) and Minut Total Protein Extraction Kit (Invent Biotechnologies, Cat No. SD-001/SN-002), respectively. Proliferation was assessed via Click-iT EdU Assay (Invitrogen, C10499). cGAS and
TLR9 expression in hAECs was silenced using small interfering RNA (siRNA) (Life Technologies) transfected with TransIT-TKO Reagent (Mirus Bio, _Cat No_. MIR 2150), with transfection
efficiency and cytotoxicity monitored using BLOCK-iT Alexa Fluor Red Control (Invitrogen, Cat No. 14750100). Optimal siRNA sequences (Cat No. S28872 for cGAS, Cat No_._ S41746 for TLR9) were
applied prior to NETs stimulation. The supplementary materials described the detailed methods of cell culture, stimulation, siRNA knockdown of _cGAS_ and _TLR9_, quantitation of
proliferation, reverse-transcription quantitative PCR (RT-qPCR), western blotting on primary hAECs. DIFFERENTIATION AND STIMULATION OF PERIPHERAL MONOCYTES-DERIVED DCS Monocytes were
isolated from fresh peripheral blood of healthy donors or COPD patients via lymphoprep density gradient medium (StemCell Technologies, _Cat No_. 07801). Briefly, blood diluted with PBS
containing 2% FBS (StemCell Technologies, Cat No. 07905) was centrifuged at 800×_g_ for 30 min. Monocytes were harvested and differentiated into DCs using the ImmunoCult Dendritic Cell
Culture Kit (StemCell Technologies, Cat No. 10985) at 37 °C for 3 days, and further differentiated for 2 days with fresh medium to yield immature DCs. These cells were then stimulated with
12 μg/mL NETs for 24 h at 37 °C for subsequent flow cytometry analysis. ImmunoCult Dendritic Cell Maturation Supplement (StemCell Technologies, Cat No. 10985) was employed as a positive
control for DC maturation. To explore cGAS and TLR9’s role in NETs-induced DC maturation, RU.521 (Aobious Inc., _Cat No_. AOB37877, a selective cGAS inhibitor), and ODN 2088 (InvivoGen,
tlr1-2088, a recognised TLR9 antagonist disrupting CpG ODNs and TLR9 interaction) were employed. The detailed methods of differentiation, stimulation, and flow cytometry assessment of
monocyte-derived DCs were described in the supplementary materials. ANIMAL EXPERIMENTS We obtained cGAS knockout mice (_cGAS__−/−_, _Cat No_. 026554) and TLR9 knockout mice (_TLR9__−/−_, Cat
No. 034329) from Jackson Laboratories. The COPD mouse model was established using an established nose-only CS exposure method.64 Briefly, 8-week-old, sex-matched mice were placed in custom
nose-only exposure tubes (China pattern No. ZL201821367875.5) for smoke inhalation. Smoke from Marlboro cigarettes (1.0 mg nicotine, 11 mg tar) was generated using a smoking machine (CH
Technologies) and diluted with fresh air. Mice were exposed to smoke from ~30 cigarettes in two 75-min sessions per day, 5 days a week, for 12 weeks, interspersed with recovery periods.
MitoTEMPO (Sigma-Aldrich, Cat No. SML0737) was prepared at 0.5 mg/mL in sterile saline, filtered and administered intraperitoneally (50 μg in 100 μL) to 25 g mice on smoking days, with
saline as control. DNase-I (Sigma-Aldrich, Cat No. 11284932001) was dissolved to 2 mg/mL; 4 mL dissolved DNase-I containing 16,000 units was aerosolized over 15 min on smoking days, and
saline was used for controls. We then assessed lung function, NETs infiltration, histology, cell counts in BALF, and levels of proinflammatory cytokines in serum and BALF as described in the
supplementary materials. QUANTIFICATION OF NETS INFILTRATION IN LUNG SLICE OF MOUSE The NETs in lung sections of mice were immunofluorescence co-stained with MPO, histone H3 and DNA. Lung
sections underwent dewaxing, hydration, and antigen retrieval in citrate buffer at 95 °C. Autofluorescence reduction was achieved with sodium borohydride incubation, followed by PBS washing.
Post-blocking, primary antibodies for MPO and histone H3 were applied overnight at 4 °C, followed by secondary antibodies (Alexa Fluor 555 and 647) for 2 h. Slides were washed, mounted with
DAPI medium, and stored at 4 °C. Negative controls were included. Imaging was conducted on a Zeiss Imager Z2 microscope. ImageJ was used to analyse fluorescence images. Briefly, the lung
areas in images were measured, followed by demarcation of NETs areas and background areas from negative controls. Normalised NETs areas were calculated as the average percentage of NETs
areas minus background areas across three random views per slice, with detailed methods described in the supplementary materials. ASSESSMENT OF NF-ΚB-DEPENDENT-CYTOKINES, TYPE-I IFNS LEVELS
Cytokine and interferon levels in mouse serum and BALF were determined using the Bio-Plex Pro Mouse Chemokine Luminex Assay Kit (BioRad, Lot No. 17005875). Human BALF analysis was executed
with the Magnetic Luminex Screening Assay Kit (R&D systems, Cat No. LXSAHM) on a Bio-Plex 200 system (BioRad) following standard procedures as detailed in the supplementary materials. In
vitro, hAECs were stimulated with TLR9/cGAS siRNA and NETs, and culture supernatants post-centrifugation were stored at −80 °C. ELISA (R&D Systems, Cat No. VAL101, VAL103 and VAL137)
quantified IL-1β, CXCL8 and IFN-β levels per manufacturer instructions. DNA-BINDING ELISAS FOR ACTIVATED NF-ΚB P65 AND P50 Nuclear extracts of hAECs stimulated with TLR9/cGAS siRNA and/or
NETs were prepared with a nuclear extract kit (Active Motif, Cat No. 40410). Whole-cell extracts of frozen lung tissues from _TLR9__−/−_, _cGAS__−/_− and control mice post-CS exposure were
also obtained. Protein content was quantified via Bradford assay (BioRad, Cat No. 5000201). NF-κB P65 and P50 activities were assessed using NF-κB P65 (or P50) Activity Assay (Active Motif,
Cat No. 40096 and 41096) according to the manufacturer’s instructions as described in the supplementary materials. STATISTICAL ANALYSIS Data are expressed as mean ± SD. Statistical details,
including analysis methods, significance thresholds (_P_ < 0.05), _n_ values, scatter plots, and _p_ values, are presented within figure legends and supplementary materials. Briefly, for
two-group comparisons, data normality was assessed via Shapiro–Wilk or Kolmogorov–Smirnov tests; normally distributed datasets were analysed by unpaired _t_-tests, while non-normal datasets
underwent Mann–Whitney _U_-tests. For multiple-group comparisons, one-way or two-way ANOVA, complemented by Tukey’s honest significant test, were employed. For partial correlation analysis,
Pearson’s partial correlation tests controlled for covariates (age, sex, BMI, smoking history of COPD patients), succeeded by multiple linear regression analyses. DATA AVAILABILITY The raw
sequence data have been deposited in the Genome Sequence Archive in National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of
Sciences, under accession number HRA001059 that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human. Other data were available from the supplemental material, or from the
corresponding author upon request. REFERENCES * Global Initiative for Chronic Obstructive Lung Disease_._ Global strategy for the diagnosis, management, and prevention of COPD.
http://goldcopd.org/ (2024). * Brightling, C. & Greening, N. Airway inflammation in COPD: progress to precision medicine. _Eur. Respir. J._ 54, 1900651 (2019). Article CAS PubMed
Google Scholar * Yang, I. A., Clarke, M. S., Sim, E. H. A. & Fong, K. M. Inhaled corticosteroids for stable chronic obstructive pulmonary disease. _Cochrane Database Syst. Rev_. 2012,
CD002991 (2012). * Hogg, J., Paré, P. & Hackett, T. The contribution of small airway obstruction to the pathogenesis of chronic obstructive pulmonary disease. _Physiol. Rev._ 97, 529–552
(2017). Article PubMed PubMed Central Google Scholar * Oelsner, E. et al. Lung function decline in former smokers and low-intensity current smokers: a secondary data analysis of the
NHLBI Pooled Cohorts Study. _Lancet Respir. Med._ 8, 34–44 (2020). Article PubMed Google Scholar * Brusselle, G. G., Joos, G. F. & Bracke, K. R. New insights into the immunology of
chronic obstructive pulmonary disease. _Lancet_ 378, 1015–1026 (2011). Article CAS PubMed Google Scholar * Barnes, P. J. Inflammatory mechanisms in patients with chronic obstructive
pulmonary disease. _J. Allergy Clin. Immunol._ 138, 16–27 (2016). Article CAS PubMed Google Scholar * Alharbi, K. S. et al. Nuclear factor-kappa B and its role in inflammatory lung
disease. _Chem. Biol. Interact._ 345, 109568 (2021). Article CAS PubMed Google Scholar * Di Stefano, A. et al. Increased expression of nuclear factor- B in bronchial biopsies from
smokers and patients with COPD. _Eur. Respir. J._ 20, 556–563 (2002). Article PubMed Google Scholar * Caramori, G. et al. Nuclear localisation of p65 in sputum macrophages but not in
sputum neutrophils during COPD exacerbations. _Thorax_ 58, 348–351 (2003). Article CAS PubMed PubMed Central Google Scholar * Gao, W. et al. Bronchial epithelial cells: the key effector
cells in the pathogenesis of chronic obstructive pulmonary disease? _Respirology_ 20, 722–729 (2015). Article PubMed Google Scholar * Haeberle, H. A., Nesti, F., Dieterich, H. J.,
Gatalica, Z. & Garofalo, R. P. Perflubron reduces lung inflammation in respiratory syncytial virus infection by inhibiting chemokine expression and nuclear factor-kappa B activation.
_Am. J. Respir. Crit. Care Med._ 165, 1433–1438 (2002). Article PubMed Google Scholar * Hogg, J. C. Role of latent viral infections in chronic obstructive pulmonary disease and asthma.
_Am. J. Respir. Crit. Care Med._ 164, S71–S75 (2001). Article CAS PubMed Google Scholar * Caramori, G. et al. Autoimmunity and COPD: clinical Implications. _Chest_ 153, 1424–1431 (2018).
Article PubMed Google Scholar * Benmerzoug, S., Ryffel, B., Togbe, D. & Quesniaux, V. Self-DNA sensing in lung inflammatory diseases. _Trends Immunol._ 40, 719–734 (2019). Article
CAS PubMed Google Scholar * Twaddell, S., Baines, K., Grainge, C. & Gibson, P. The emerging role of neutrophil extracellular traps in respiratory disease. _Chest_ 156, 774–782 (2019).
Article PubMed Google Scholar * Obermayer, A. et al. New aspects on the structure of neutrophil extracellular traps from chronic obstructive pulmonary disease and in vitro generation.
_PLoS ONE_ 9, e97784 (2014). Article PubMed PubMed Central Google Scholar * Pedersen, F. et al. Neutrophil extracellular trap formation and extracellular DNA in sputum of stable COPD
patients. _Respir. Med._ 109, 1360–1362 (2015). Article PubMed Google Scholar * Grabcanovic-Musija, F. et al. Neutrophil extracellular trap (NET) formation characterises stable and
exacerbated COPD and correlates with airflow limitation. _Respir. Res._ 16, 59 (2015). Article PubMed PubMed Central Google Scholar * Dicker, A. et al. Neutrophil extracellular traps are
associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. _J. Allergy Clin. Immunol._ 141, 117–127 (2018). Article PubMed PubMed
Central Google Scholar * Wright, T. et al. Neutrophil extracellular traps are associated with inflammation in chronic airway disease. _Respirology_ 21, 467–475 (2016). Article PubMed
Google Scholar * Zhang, H. et al. Erythromycin suppresses neutrophil extracellular traps in smoking-related chronic pulmonary inflammation. _Cell Death Dis._ 10, 678 (2019). Article PubMed
PubMed Central Google Scholar * Zou, Y. et al. Neutrophil extracellular traps induced by cigarette smoke contribute to airway inflammation in mice. _Exp. Cell Res._ 389, 111888 (2020).
Article CAS PubMed Google Scholar * Qiu, S. et al. Neutrophil extracellular traps induced by cigarette smoke activate plasmacytoid dendritic cells. _Thorax_ 72, 1084–1093 (2017). Article
PubMed Google Scholar * Lachowicz-Scroggins, M. et al. Extracellular DNA, neutrophil extracellular traps, and inflammasome activation in severe asthma. _Am. J. Respir. Crit. Care Med._
199, 1076–1085 (2019). Article CAS PubMed PubMed Central Google Scholar * Saffarzadeh, M. et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a
predominant role of histones. _PLoS ONE_ 7, e32366 (2012). Article CAS PubMed PubMed Central Google Scholar * Pham, D. et al. Neutrophil autophagy and extracellular DNA traps contribute
to airway inflammation in severe asthma. _Clin. Exp. Allergy_ 47, 57–70 (2017). Article CAS PubMed Google Scholar * Wan, R. et al. Neutrophil extracellular traps amplify neutrophil
recruitment and inflammation in neutrophilic asthma by stimulating the airway epithelial cells to activate the TLR4/ NF-κB pathway and secrete chemokines. _Aging_ 12, 16820–16836 (2020).
Article CAS PubMed PubMed Central Google Scholar * Sabbione, F. et al. Neutrophil extracellular traps stimulate proinflammatory responses in human airway epithelial cells. _J. Innate
Immun._ 9, 387–402 (2017). Article CAS PubMed PubMed Central Google Scholar * Nascimento, M. et al. Self-DNA release and STING-dependent sensing drives inflammation to cigarette smoke
in mice. _Sci. Rep._ 9, 14848 (2019). Article PubMed PubMed Central Google Scholar * Brinkmann, V. et al. Neutrophil extracellular traps kill bacteria. _Science_ 303, 1532–1535 (2004).
Article CAS PubMed Google Scholar * Jorch, S. & Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. _Nat. Med._ 23, 279–287 (2017). Article CAS
PubMed Google Scholar * Toussaint, M. et al. Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. _Nat. Med._ 23, 681–691 (2017). Article CAS
PubMed PubMed Central Google Scholar * Trivedi, A., Khan, M., Bade, G. & Talwar, A. Orchestration of neutrophil extracellular traps (Nets), a unique innate immune function during
chronic obstructive pulmonary disease (COPD) development. _Biomedicines_ 9, 53 (2021). Article CAS PubMed PubMed Central Google Scholar * Pedersen, F. et al. Neutrophil extracellular
trap formation is regulated by CXCR2 in COPD neutrophils. _Eur. Respir. J._ 51, 1700970 (2018). * Wooding, D. et al. Acute air pollution exposure alters neutrophils in never-smokers and
at-risk humans. _Eur. Respir. J._ 55, 1901495 (2020). * Lood, C. et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like
disease. _Nat. Med._ 22, 146–153 (2016). Article CAS PubMed PubMed Central Google Scholar * Kenny, E. et al. Diverse stimuli engage different neutrophil extracellular trap pathways.
_eLife_ 6, e24437 (2017). * Liu, L. et al. Induction of neutrophil extracellular traps during tissue injury: Involvement of STING and Toll-like receptor 9 pathways. _Cell Prolif._ 52, e12579
(2019). Article PubMed PubMed Central Google Scholar * Zhong, F., Liang, S. & Zhong, Z. Emerging role of mitochondrial DNA as a major driver of inflammation and disease progression.
_Trends Immunol._ 40, 1120–1133 (2019). Article CAS PubMed Google Scholar * Hewitt, R. & Lloyd, C. Regulation of immune responses by the airway epithelial cell landscape. _Nat. Rev.
Immunol._ 21, 347–362 (2021). Article CAS PubMed PubMed Central Google Scholar * Foronjy, R. F. et al. TLR9 expression is required for the development of cigarette smoke-induced
emphysema in mice. _Am. J. Physiol. Lung Cell Mol. Physiol._ 311, L154–L166 (2016). Article PubMed PubMed Central Google Scholar * Doz, E. et al. Cigarette smoke-induced pulmonary
inflammation is TLR4/MyD88 and IL-1R1/MyD88 signaling dependent. _J. Immunol._ 180, 1169–1178 (2008). Article CAS PubMed Google Scholar * Di Stefano, A. et al. Bronchial inflammation and
bacterial load in stable COPD is associated with TLR4 overexpression. _Eur. Respir. J_. 49, 1602006 (2017). * Fitzgerald, K. A. & Kagan, J. C. Toll-like receptors and the control of
immunity. _Cell_ 180, 1044–1066 (2020). Article CAS PubMed PubMed Central Google Scholar * Kumar, V. The trinity of cGAS, TLR9, and ALRs guardians of the cellular galaxy against
host-derived self-DNA. _Front. Immunol._ 11, 624597 (2020). Article CAS PubMed Google Scholar * Huang, L. et al. mtDNA activates cGAS signaling and suppresses the YAP-mediated
endothelial cell proliferation program to promote inflammatory injury. _Immunity_ 52, 475–486.e475 (2020). Article CAS PubMed PubMed Central Google Scholar * Barnabei, L., Laplantine,
E., Mbongo, W., Rieux-Laucat, F. & Weil, R. NF-kappaB: at the borders of autoimmunity and inflammation. _Front. Immunol._ 12, 716469 (2021). Article CAS PubMed PubMed Central Google
Scholar * Turnier, J. & Kahlenberg, J. The role of cutaneous type I IFNs in autoimmune and autoinflammatory diseases. _J. Immunol._ 205, 2941–2950 (2020). Article CAS PubMed Google
Scholar * Ma, R., Ortiz Serrano, T., Davis, J., Prigge, A. & Ridge, K. The cGAS-STING pathway: the role of self-DNA sensing in inflammatory lung disease. _FASEB J_. 34, 13156–13170
(2020). Article CAS PubMed Google Scholar * Riley, J. & Tait, S. Mitochondrial DNA in inflammation and immunity. _EMBO Rep._ 21, e49799 (2020). Article CAS PubMed PubMed Central
Google Scholar * Han, Y. et al. Airway epithelial cGAS is critical for induction of experimental allergic airway inflammation. _J. Immunol._ 204, 1437–1447 (2020). Article CAS PubMed
Google Scholar * Thio, C., Lai, A., Chi, P., Webster, G. & Chang, Y. Toll-like receptor 9-dependent interferon production prevents group 2 innate lymphoid cell-driven airway
hyperreactivity. _J. Allergy Clin. Immunol._ 144, 682–697.e689 (2019). Article CAS PubMed Google Scholar * McNab, F., Mayer-Barber, K., Sher, A., Wack, A. & O’Garra, A. Type I
interferons in infectious disease. _Nat. Rev. Immunol._ 15, 87–103 (2015). Article CAS PubMed PubMed Central Google Scholar * HuangFu, W. C., Liu, J., Harty, R. N. & Fuchs, S. Y.
Cigarette smoking products suppress anti-viral effects of type I interferon via phosphorylation-dependent downregulation of its receptor. _FEBS Lett._ 582, 3206–3210 (2008). Article CAS
PubMed PubMed Central Google Scholar * García-Valero, J. et al. Deficient pulmonary IFN-β expression in COPD patients. _PLoS ONE_ 14, e0217803 (2019). Article PubMed PubMed Central
Google Scholar * Mehta, M. et al. Interferon therapy for preventing COPD exacerbations. _EXCLI J._ 19, 1477–1480 (2020). PubMed PubMed Central Google Scholar * Deb, P., Dai, J., Singh,
S., Kalyoussef, E. & Fitzgerald-Bocarsly, P. Triggering of the cGAS-STING pathway in human plasmacytoid dendritic cells inhibits TLR9-mediated IFN production. _J. Immunol._ 205, 223–236
(2020). Article CAS PubMed Google Scholar * Liang, H., Sedlic, F., Bosnjak, Z. & Nilakantan, V. SOD1 and MitoTEMPO partially prevent mitochondrial permeability transition pore
opening, necrosis, and mitochondrial apoptosis after ATP depletion recovery. _Free Radical Biol. Med._ 49, 1550–1560 (2010). Article CAS Google Scholar * Barzegari, A. et al.
Mitochondria-targeted antioxidant mito-TEMPO alleviate oxidative stress induced by antimycin A in human mesenchymal stem cells. _J. Cell Physiol._ 235, 5628–5636 (2020). Article CAS PubMed
Google Scholar * Barnes, P. Oxidative stress-based therapeutics in COPD. _Redox Biol._ 33, 101544 (2020). Article CAS PubMed PubMed Central Google Scholar * Tsai, C. H. et al.
Neutrophil extracellular trap production and CCL4L2 expression influence corticosteroid response in asthma. _Sci. Transl. Med._ 15, eadf3843 (2023). Article CAS PubMed Google Scholar *
Gál, Z. et al. Plasma neutrophil extracellular trap level is modified by disease severity and inhaled corticosteroids in chronic inflammatory lung diseases. _Sci. Rep._ 10, 4320 (2020).
Article PubMed PubMed Central Google Scholar * Chen, J. et al. The elevated CXCL5 levels in circulation are associated with lung function decline in COPD patients and cigarette
smoking-induced mouse model of COPD. _Ann. Med._ 51, 314–329 (2019). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by the
1•3•5 project for disciplines of excellence, West China Hospital, Sichuan University [grant numbers ZYGD23009]; the National Natural Science Foundation of China (NSFC) [grant numbers
81830001, 81670038, 81800015 and 31871157]; the Natural Science Foundation of Sichuan Province [Grant number 2022NSFSC0603]; the China Postdoctoral Science Foundation [Grant numbers
2019T120851 and 2018M643501]; the Fundamental Research Funds for the Central Universities (the Postdoctoral Foundation of Sichuan University) [Grant number 2018SCU12028]. AUTHOR INFORMATION
AUTHORS AND AFFILIATIONS * Department of Respiratory and Critical Care Medicine, West China Hospital, West China School of Medicine, and Division of Pulmonary Diseases, State Key Laboratory
of Biotherapy, Sichuan University, Chengdu, Sichuan, 610041, China Jun Chen, Tao Wang, Xiaoou Li, Lijuan Gao, Ke Wang, Mengxin Cheng, Zijian Zeng, Lei Chen, Yongchun Shen & Fuqiang Wen
Authors * Jun Chen View author publications You can also search for this author inPubMed Google Scholar * Tao Wang View author publications You can also search for this author inPubMed
Google Scholar * Xiaoou Li View author publications You can also search for this author inPubMed Google Scholar * Lijuan Gao View author publications You can also search for this author
inPubMed Google Scholar * Ke Wang View author publications You can also search for this author inPubMed Google Scholar * Mengxin Cheng View author publications You can also search for this
author inPubMed Google Scholar * Zijian Zeng View author publications You can also search for this author inPubMed Google Scholar * Lei Chen View author publications You can also search for
this author inPubMed Google Scholar * Yongchun Shen View author publications You can also search for this author inPubMed Google Scholar * Fuqiang Wen View author publications You can also
search for this author inPubMed Google Scholar CONTRIBUTIONS Conceptualization: J.C., X.L. and F.W.; Methodology: J.C., T.W. and X.L.; Acquisition of data: J.C., L.G., K.W., M.C. and Z.Z.;
Analysis and interpretation of data: J.C., Y.S. and L.C.; Project administration: T.W.; Drafting the article and illustrator: J.C.; Revising the article critically for important intellectual
content: J.C., T.W. and F.W. All authors have read and approved the article. CORRESPONDING AUTHOR Correspondence to Fuqiang Wen. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare
no competing interests. SUPPLEMENTARY INFORMATION SUPPLEMENTARY MATERIALS SUPPLEMENTARY MOVIE S1 RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original
author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the
article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use
is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Chen, J., Wang, T., Li, X. _et al._ DNA of neutrophil extracellular traps promote
NF-κB-dependent autoimmunity via cGAS/TLR9 in chronic obstructive pulmonary disease. _Sig Transduct Target Ther_ 9, 163 (2024). https://doi.org/10.1038/s41392-024-01881-6 Download citation *
Received: 04 July 2023 * Revised: 08 May 2024 * Accepted: 20 May 2024 * Published: 17 June 2024 * DOI: https://doi.org/10.1038/s41392-024-01881-6 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative



:max_bytes(150000):strip_icc():focal(999x0:1001x2)/princess-anne-timothy-lawrence-garter-service-061724-d320e7616f8d48fb846f24b47c0a0da2.jpg)


:max_bytes(150000):strip_icc():focal(319x0:321x2)/people_social_image-60e0c8af9eb14624a5b55f2c29dbe25b.png)
