- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Recent advances in the detection and treatment of prostate cancer (PCa) have reduced morbidity and mortality from this common cancer. Despite these improvements, PCa remains the second
leading cause of cancer death in men in the United States. Further understanding of the genetic underpinnings of lethal PCa is required to drive risk detection and prevention and ultimately
reduce mortality. We therefore set out to identify germline variants associated with cases of lethal prostate cancer (LPCa).
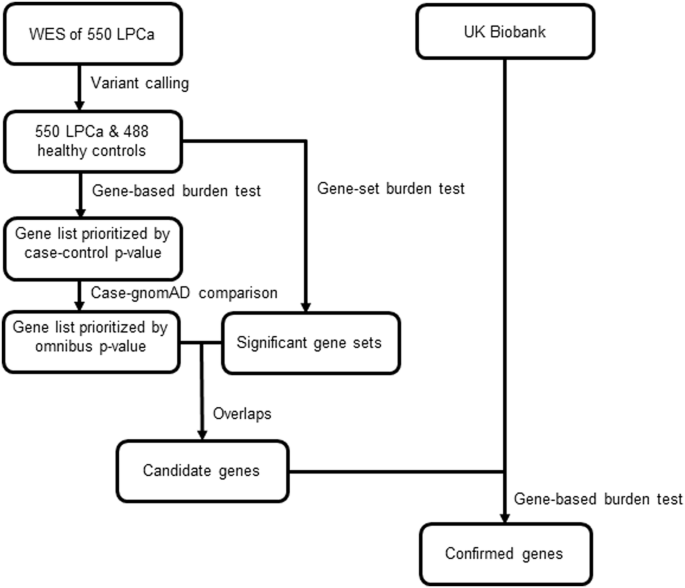
Using a two-stage study design, we compared whole-exome sequencing data of 550 LPCa patients to 488 healthy male controls. Men were classified as having LPCa based on medical record review.
Candidate genes were identified using gene- and gene-set-based rare truncating variant association tests. Case-control burden testing through Firth’s penalized logistic regression and
case-gnomAD allelic burden testing through a one-sided mid-p Fisher’s exact test were conducted. Each gene’s p-values from these tests were combined into an omnibus p-value for candidate
gene selection. In the subsequent validation stage, genes were assessed using the UK Biobank and Firth’s penalized logistic regression for each ancestry, combined through meta-analysis.
Gene-based rare variant association tests identified 12 genes nominally associated with LPCa. Rare-variant association tests identified a gene set with a significantly higher burden of
truncating germline mutations in LPCa patients than controls. Combining gene- and gene-set test results, four nominally significant genes (PPP1R3A, TG, PPFIBP2, and BTN3A3) were selected as
candidates. Subsequent validation using the UK Biobank found that PPP1R3A was significantly associated with LPCa risk (odds ratio 2.34, CI 1.20–4.59). Specifically, pGln662ArgfsTer7 was
identified as the predominant variant in PPP1R3A among LPCa patients in our dataset.
Both individual gene and gene-set analyses identified candidates associated with LPCa. The novel association of PPP1R3A and LPCa risk merits further investigation.
The data used in this study will be made available on reasonable request.
The program used in this study, VICTOR, is freely accessible at http://bjfenglab.org/.
The computational resources at the University of Utah Center for High-Performance Computing were partially funded by the NIH Shared Instrumentation Grant 1S10OD021644-01A1. We acknowledge
the Utah Genetic Reference Project, University of Utah’s Center for Genomic Medicine, for providing healthy controls for this study. The authors gratefully acknowledge the Chan Soon-Shiong
Family Foundation for providing the funds for sequencing the Heritage 1000 Projects (H1K) samples. The authors gratefully acknowledge the generous support from donors to The Patrick C. Walsh
Hereditary Prostate Cancer Research Program at The Brady Urological Institute.
This study was funded by the Department of Defense Impact Award grant PC150797. The computational resources used were partially funded by the NIH Shared Instrumentation Grant
1S10OD021644-01A1.
Department of Dermatology, University of Utah, Salt Lake City, UT, USA
Department of Family and Preventative Medicine, University of Utah, Salt Lake City, UT, USA
Program for Personalized Cancer Care, NorthShore University HealthSystem, Evanston, Illinois, USA
Department of Medicine and the Duke Cancer Institute, Duke University School of Medicine, Durham, NC, USA
Department of Urology and the James Buchanan Brady Urologic Institute, Johns Hopkins University School of Medicine, Baltimore, MD, USA
BF – conception and design, data analysis and interpretation, drafting manuscript, critical revision of manuscript, statistical analysis; JLB – conception and design, data analysis and
interpretation, drafting manuscript, critical revision of manuscript; JW – data analysis and interpretation, statistical analysis; CC – data analysis and interpretation, drafting manuscript,
critical revision of manuscript, statistical analysis; NAS – drafting manuscript, critical revision of manuscript; ZS – data analysis and interpretation, statistical analysis; SLZ – DNA
analysis and data interpretation; JX – critical revision of manuscript, data analysis and interpretation, supervision; WBI – critical revision of manuscript, supervision; KAC – conception
and design, critical revision of manuscript, data analysis and interpretation, supervision.
The PERCH software, for which BJF is the inventor, has been non-exclusively licensed to Ambry Genetics Corporation for their clinical genetic testing services and research. BJF also reports
funding and sponsorship to his institution on his behalf from Pfizer Inc. and Regeneron Genetics Center LLC.
This study was performed using consented human germline exome sequencing and clinical characteristic data under approval from the respective institutional review boards of the authors in
accordance with the Declaration of Helsinki.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author
self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Anyone you share the following link with will be able to read this content:






