- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT C1GALT1 controls the crucial step of GalNAc-type O-glycosylation and is associated with both physiologic and pathologic conditions, including cancers. EPH receptors comprise the
largest family of receptor tyrosine kinases (RTKs) and modulate a diverse range of developmental processes and human diseases. However, the role of C1GALT1 in the signaling of EPH receptors
remains largely overlooked. Here, we showed that C1GALT1 high expression in gastric adenocarcinomas correlated with adverse clinicopathologic features and is an independent prognostic factor
for poor overall survival. Silencing or loss of C1GALT1 inhibited cell viability, migration, invasion, tumor growth and metastasis, as well as increased apoptosis and cytotoxicity of
5-fluorouracil in AGS and MKN45 cells. Phospho-RTK array and western blot analysis showed that C1GALT1 depletion suppressed tyrosine phosphorylation of EPHA2 induced by soluble Ephrin A1-Fc.
O-glycans on EPHA2 were modified by C1GALT1 and both S277A and T429A mutants, which are O-glycosites on EPHA2, dramatically enhanced phosphorylation of Y588, suggesting that not only
overall O-glycan structures but also site-specific O-glycosylation can regulate EPHA2 activity. Furthermore, depletion of C1GALT1 decreased Ephrin A1-Fc induced migration and reduced Ephrin
A1 binding to cell surfaces. The effects of C1GALT1 knockdown or knockout on cell invasiveness in vitro and in vivo were phenocopied by EPHA2 knockdown in gastric cancer cells. These results
suggest that C1GALT1 promotes phosphorylation of EPHA2 and enhances soluble Ephrin A1-mediated migration primarily by modifying EPHA2 O-glycosylation. Our study highlights the importance of
GalNAc-type O-glycosylation in EPH receptor-regulated diseases and identifies C1GALT1 as a potential therapeutic target for gastric cancer. SIMILAR CONTENT BEING VIEWED BY OTHERS ST6GAL1
TARGETS THE ECTODOMAIN OF ERBB2 IN A SITE-SPECIFIC MANNER AND REGULATES GASTRIC CANCER CELL SENSITIVITY TO TRASTUZUMAB Article Open access 04 May 2021 THE EMERGING ROLES OF GΑ12/13 PROTEINS
ON THE HALLMARKS OF CANCER IN SOLID TUMORS Article Open access 23 October 2021 NEU4-MEDIATED DESIALYLATION ENHANCES THE ACTIVATION OF THE ONCOGENIC RECEPTORS FOR THE DISSEMINATION OF OVARIAN
CARCINOMA Article 14 October 2024 INTRODUCTION Gastric cancer is the third leading cause of cancer-related deaths in the world [1]. Surgery remains the main treatment for operable gastric
cancer. For advanced gastric cancer, new therapeutic approaches are currently being researched, such as monoclonal antibodies that target HER2 and immune checkpoint inhibitors, or a
combination of these with chemotherapy [2]. Glycosylation is the most common posttranslational modification of proteins, and aberrant glycosylation is often observed in cancers [3].
Mucin-type O-glycosylation modifies proteins in the gastrointestinal tract, and this modification is initiated through the transfer of N-acetylgalactosamine (GalNAc) to serine or threonine
residues forming Tn antigen [4]. This reaction is catalyzed by a large group of enzymes named UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases (GALNTs) [5]. Core 1
β1,3-galatosyltransferase (C1GALT1), with the help of its endoplasmic reticulum chaperone Cosmc, transfers galactose (Gal) to Tn antigen to form Galβ1, 3GalNAc (T-antigen) [6]. T-antigen is
then modified by other glycosyltransferases to form complex O-glycans [7]. Mucin-type O-glycosylation regulates multiple biological functions such as apoptosis and angiogenesis of cancer
cells [8, 9]. In our previous studies, C1GALT1 promoted the invasive behavior of colon cancer cells [10] and enhanced the proliferation of hepatocellular carcinoma cells [11]. Altered
O-glycosylation was reported to influence gastric cancer carcinogenesis cascade and the ensuing cancer progression [12]; however, the underlying mechanisms remain unclear. Receptor tyrosine
kinases (RTKs), such as EGFR, MET, and FGFR2, have been reported to carry O-glycans, and the O-glycosylation can modulate RTK activities [10, 11, 13,14,15]. Alterations in RTK activities,
including EGFR, HER2 (ErbB2, EGFR2), and MET (HGFR), are associated with gastric cancer progression [16,17,18]. Several lines of evidence have indicated that RTKs actively contribute to
gastric oncogenesis and disease progression and are recognized to be the target of cancer therapy [19, 20]. Although C1GALT1 is the critical O-glycosylating enzyme, its expression and role
in RTK activities in gastric cancer remain unclear. EPH receptors are the largest family of RTKs. More surprisingly, the functional significance of O-glycosylation for EPH receptors has
never been reported. Human EPH receptors are composed of nigh EPHAs and five EPHBs which preferentially bind to their Ephrin A and Ephrin B ligands, respectively. The Ephrin-EPH system
controls a diverse range of developmental processes and pathogenesis of diseases ranging from neuronal disorders to cancer [21]. In human cancers, EPH receptors are frequently overexpressed
and contribute to tumor progression [22,23,24,25]. Therefore, EPH receptors receive much attention and are attractive drug targets [22, 26, 27]. EPHA2 and its ligand Ephrin A1 are
overexpressed in gastric adenocarcinoma and elevated EPHA2 expression is associated with poor survival [28, 29]. EPHA2 has been reported to promote epithelial–mesenchymal transition [30, 31]
and silencing of EPHA2 inhibits gastric cancer cell growth and invasion [32]. Although EPHA2 plays a critical role in cancers, how EPHA2 activity is regulated remains unclear. RESULTS
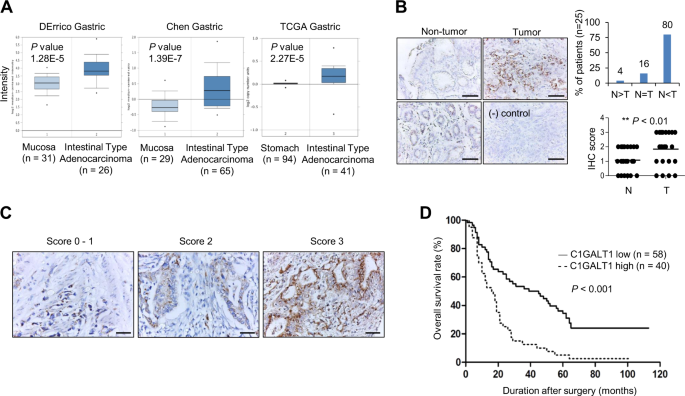
C1GALT1 IS OVEREXPRESSED IN GASTRIC CANCER From the Oncomine database, DErrico Gastric, Chen Gastric, and TCGA gastric showed that _C1GALT1_ mRNA expression was overexpressed in gastric
adenocarcinoma compared with normal gastric mucosa tissue (Fig. 1a). Our immunohistochemical staining indicated that 80% (_n_ = 25) of gastric adenocarcinoma tissues showed higher C1GALT1
protein levels than paired nontumor gastric tissues (Fig. 1b). Only 4% of cases exhibited lower C1GALT1 expression in gastric adenocarcinomas. In addition, we observed that glandular
epithelial cells in the lower part of nontumor gastric mucosa expressed higher C1GALT1 than surface epithelial cells (Fig. 1b, left panel). These results suggest that C1GALT1 is
significantly overexpressed in gastric adenocarcinomas compared with their adjacent nontumor tissues. C1GALT1 EXPRESSION CORRELATES WITH POOR SURVIVAL IN PATIENTS WITH GASTRIC ADENOCARCINOMA
We analyzed the expression of C1GALT1 in gastric cancers from tissue microarray and correlated these with clinicopathologic features and prognoses of gastric cancer. Of the tissue
microarray of 111 patients, 13 were excluded because of unqualified slides. The expression of C1GALT1 was scored from 0 to 3 on the basis of the staining intensity of immunohistochemistry
(Fig. 1c). Scores 0–2 and 3 were grouped as low and high C1GALT1 expression, respectively. The TNM stage was based on the classification system depending on the invasion of gastric wall (T),
the involvement of lymph node (N), and the presence of distant metastasis (M) [33]. Histological grading was based on the Goseki grading system [34]. The results indicated that high C1GALT1
expression correlated with advanced cancer (stages III and IV), higher histological grade, advanced tumor invasion, and nodal metastasis (Table 1). Kaplan–Meier survival curves indicated
that high C1GALT1 expression correlated with poor overall survival (_p_ < 0.001, Fig. 1d). Univariate analysis indicated that high C1GALT1 expression, advanced cancer stage, higher
histological grade, and nodal metastasis significantly correlated with mortality (Table 2). Furthermore, Cox regression analysis revealed that C1GALT1 and advanced cancer stage were
independent predictors of mortality. These findings suggest that C1GALT1 is an independent prognostic factor for poor survival in patients with gastric adenocarcinoma. C1GALT1 PROMOTES
MALIGNANT BEHAVIORS OF GASTRIC CANCER CELLS To assess the effect of C1GALT1 on gastric cancer cells, we analyzed cell viability, migration, invasion, and chemoresistance using MTT, transwell
migration, Matrigel invasion, and flow cytometry assays, respectively. Q-RT-PCR (Fig. 2a) and western blotting (Fig. 2b) showed variable C1GALT1 expression in five gastric cancer cell
lines. C1GALT1 knockdown, knockout, and overexpression in gastric cancer cells were confirmed by western blotting (Fig. 2c). Flow cytometry showed that C1GALT1 knockdown or knockout indeed
affected O-glycan expression on the surfaces of AGS and MKN45 cells, as revealed through VVA and PNA staining (Supplementary Fig. S1). Phenotypic assays indicated that C1GALT1 knockdown or
knockout significantly suppressed the viability (Fig. 2d), migration (Fig. 2e), and invasion (Fig. 2f) in AGS cells and MKN45 cells, respectively. By contrast, C1GALT1 overexpression
enhanced these phenotypes in AGS and SNU-1 cells. Moreover, we observed that si-C1GALT1-2 siRNA with lower C1GALT1 knockdown efficiency exerted a weaker effect on these phenotypes compared
with the other two siRNAs. Because si-C1GALT1-1 and si-C1GALT1-3 exhibited excellent knockdown efficiency, we used these two siRNAs for other experiments. Because altered glycosylation has
been reported to modulate chemoresistance [35], we examined whether C1GALT1 could regulate 5-FU cytotoxicity in gastric cancer cells. Flow cytometry with FITC-annexin V and PI showed that
C1GALT1 knockdown significantly increased apoptosis in both AGS and MKN45 cells compared with control siRNA knockdown cells (Fig. 2g). Taken together, these results suggest that C1GALT1
promotes malignant behaviors of gastric cancer cells. EFFECTS OF C1GALT1 ON TUMOR GROWTH AND METASTASIS IN NOD/SCID MICE To further investigate the effect of C1GALT1 on gastric tumor growth,
C1GALT1 knockdown AGS and C1GALT1 knockout MKN45 cells were xenografted in NOD/SCID mice. We established stable C1GALT1 knockdown AGS cells using short hairpin (sh) RNA and the C1GALT1
knockdown was confirmed by western blotting (Fig. 3a). We found that silencing of C1GALT1 significantly decreased tumor sizes and tumor weights in NOD/SCID mice subcutaneously injected with
AGS cells. (Fig. 3b). Next, we analyzed the effect of C1GALT1 on lung metastasis of AGS cells using a mouse tail vein injection model. H&E staining of the lung sections revealed that
C1GALT1 knockdown decreased the number of metastatic tumor nodules in the lungs (Fig. 3c). To further confirmed the effect of C1GALT1 on gastric tumor growth, C1GALT1 knockout MKN45 cells
were subcutaneously injected into NOD/SCID mice. The results also showed that the loss of C1GALT1 inhibited tumor sizes and tumor growth (Fig. 3d). Moreover, we tested the effect of C1GALT1
knockout on peritoneal tumor growth because peritoneal dissemination is the most frequent metastatic pattern of gastric cancer. Results showed that C1GALT1 knockout significantly decreased
the number of MKN45 tumor nodules and total tumor weights in the peritoneal cavity of NOD/SCID mice (Fig. 3e, f). These results suggest that silencing or loss of C1GALT1 inhibits
subcutaneous and peritoneal tumor growth of gastric cancer cells in vivo. EFFECTS OF C1GALT1 KNOCKDOWN ON MULTIPLE PHOSPHO(P)-RTKS IN GASTRIC CANCER CELLS We have demonstrated that
phosphorylation of multiple RTKs, such as EGFR, FGFR2, IGF1R, and MET, can be regulated by O-glycosylation in various cancers [10, 11, 36, 37]. Consistently, we found that C1GALT1 knockdown
decreased the level of several p-RTKs using p-RTK array analysis, including EGFR, MET (HGFR), HER2 (ErbB2), Flt-3, Insulin R, IGF-1R, and ROR2 in AGS cells treated with 10% FBS
(Supplementary Fig. S2). Western blotting confirmed that C1GALT1 knockdown decreased the phosphorylation of EGFR, HER2, and AKT in both AGS and MKN45 cells (Supplementary Fig. S3A). Because
the presence of O-glycans on HER2 and the effect of C1GALT1 on its O-glycans are unclear, we further analyzed HER2 O-glycosylation using VVA and PNA pull-down assays. Our data indicated that
VVA lectin could pull down HER2 in parental AGS and MKN45 cells, although the extent was low (Supplementary Fig. S3B), which could be because of low HER2 expression in these cells.
Subsequently, we overexpressed HER2 in HEK293FT cells and found that VVA and PNA could also pull down HER2 after benzyl-α-GalNAc treatment (Supplementary Fig. S3B). We used another gastric
cancer cell line, N87, which expresses high levels of HER2, and found that C1GALT1 knockdown drastically increased HER2 pulled down through VVA (Supplementary Fig. S3C). C1GALT1 knockdown
also decreased the phosphorylation of HER2 in N87 cells. Furthermore, C1GALT1-mediated cell growth was suppressed by lapatinib, a dual inhibitor for EGFR and HER2 (Supplementary Fig. S4).
These results suggest that C1GALT1 knockdown modifies O-glycosylation and decreases 10% FBS-mediated phosphorylation of multiple RTKs including EGFR and HER2 in gastric cancer cells. IMPACTS
OF C1GALT1 ON EPHRIN A1-TRIGGERED PHOSPHORYLATION OF EPHA2 Surprisingly, the effect of O-glycosylation on EPH receptors, the largest family of RTKs, has never been reported. Ephrin A1 is
the ligand for EPHA receptors and is overexpressed in gastric cancer. In addition, soluble Ephrin A1 has been reported to be secreted in conditioned media of several cancer cell lines and be
detected in serum of cancer patients [38,39,40]. We found that soluble Ephrin A1 was present in conditioned media of AGS and MKN cells (Supplementary Fig. S5). To systematically examine
whether C1GALT1-mediated O-glycosylation can regulate the activity of the EPH receptor family, soluble Ephrin A1-Fc was used to treat AGS cells transfected with control siRNA or C1GALT1
siRNA and then p-RTK array analysis was performed. Interestingly, C1GALT1 knockdown decreased levels of p-EPHA1 and p-EPHA2 as well as p-EGFR and p-MET (Fig. 4a). Using short exposure, EPHA2
was the first activated by Ephrin A1-Fc. To know whether decreased p-EGFR and p-MET were mediated by Ephrin A1-Fc treatment, AGS cells transfected with control or C1GALT1-specific siRNA
were serum starved and then treated with or without Ephrin A1-Fc. The results showed that p-EGFR and p-MET were decreased by C1GALT1 siRNA even without Ephrin A1-Fc treatment (Supplementary
Fig. S6), suggesting that the decrease in p-EGFR and p-MET in the p-RTK array does not depend on Ephrin A1-mediated signaling. However, we noticed that Ephrin A1-Fc could slightly increase
phosphorylation of EGFR but not MET. To examine whether EPHA2 was the major EPHA receptor for Ephrin A1, we knocked down EPHA2 with two independent siRNAs and analyzed the Ephrin A1-Fc
binding to AGS and MKN45 cells. Flow cytometry showed that EPHA2 knockdown decreased EPHA2 levels by 76.6%–78.2% and 53.8%–55.2% in AGS and MKN45 cells, respectively, and inhibited Ephrin A1
binding by 79.5%–82.9% and 72.2%–79.7%, respectively (Fig. 4b), indicating that EPHA2 is the predominant EPH receptor for Ephrin A1-Fc in gastric cancer cells. Western blot analysis
validated that C1GALT1 knockdown and knockout decreased Ephrin A1-Fc induced phosphorylation of EPHA2 at Y588 in AGS and MKN45 cells, respectively (Fig. 4c). Next, we performed VVA lectin
pull-down assays to examine whether EPHA2 was decorated with O-glycans and whether C1GALT1 could modify the O-glycans. We found that EPHA2 could be pulled down with VVA and the levels were
enhanced by C1GALT1 knockdown and knockout in AGS and MKN45 cells, respectively (Fig. 4d). By contrast, C1GALT1 overexpression in AGS cells decreased the amounts of EPHA2 pulled down by VVA.
To rule out the possibility that the VVA binding to EPHA2 is dependent on N-glycans, we further used PNGaseF to remove N-glycans in lysates and then performed VVA pull-down assays. Results
showed that C1GALT1 knockdown still increased the amounts of EPHA2 pulled down by VVA after removal of N-glycans (Supplementary Fig. S7). These findings suggest that C1GALT1 can modify
O-glycans on EPHA2 and regulate soluble Ephrin A1-induced tyrosine phosphorylation of EPHA2. Henrik Clausen’s team identified two O-glycosites, S277 and T429, on EPHA2 in gastric cancer
cells using proteomics approaches [41]. To further determine whether site-specific O-glycosylation can modulate EPHA2 activity, we constructed S277A and T429A mutants using site-directed
mutagenesis. Interestingly, our results showed that both S277A and T429A mutants dramatically enhanced phosphorylation of Y588 (Fig. 4e). These results suggest that not only overall O-glycan
structures on EPHA2 but also site-specific O-glycosylation can regulate EPHA2 activity. EFFECTS OF EPHA2 KNOCKDOWN ON GASTRIC CANCER CELLS IN VITRO AND IN VIVO To know the in vitro effects
of EPHA2 knockdown on gastric cancer cells, we analyzed cell viability, migration, and invasion using MTT, transwell migration, and Matrigel invasion assays, respectively. EPHA2 was knocked
down with two independent siRNAs against _EPHA2_ in AGS and MKN45 cells, which was confirmed by western blotting (Fig. 5a). Results showed that EPHA2 knockdown did not significantly affect
cell viability (Fig. 5b). By contrast, EPHA2 knockdown suppressed cell migration (Fig. 5c) and invasion (Fig. 5d). To assess the in vivo effect of EPHA2 on gastric cancer cells, EPHA2
knockdown AGS and MKN45 cells were xenografted in NOD/SCID mice via subcutaneous or peritoneal injection. The stable knockdown of EPHA2 with shRNA in AGS and MKN45 cells was confirmed by
western blotting (Fig. 5e). We found that EPHA2 knockdown did not significantly affect tumor sizes and tumor weights in NOD/SCID mice subcutaneously injected with AGS (Fig. 5f) or MKN45
cells (Fig. 5g). By contrast, in the tail vein injection model, EPHA2 knockdown reduced the number of lung metastatic nodules of AGS cells (Fig. 5h). Moreover, in the peritoneal injection
model, EPHA2 knockdown significantly decreased the number of MKN45 tumor nodules and total tumor weights in the peritoneal cavity of NOD/SCID mice (Fig. 5i, j). These results suggest that
EPHA2 knockdown inhibits invasive behaviors of gastric cancer cells in vitro and in vivo. C1GALT1 MEDIATES ITS PRO-MIGRATORY EFFECT ON GASTRIC CANCER CELLS THROUGH EPHA2 Soluble Ephrin A1
can promote metastasis in several cancer types [38, 39, 42]. We found that C1GALT1 modified O-glycans on EPHA2 and regulated EPHA2 phosphorylation. In addition, the effects of C1GALT1
knockdown on cell invasiveness in vitro and in vivo were phenocopied by EPHA2 knockdown in gastric cancer cells. To further confirm that C1GALT1 mediated its pro-migratory effect at least
partly through EPHA2, we analyzed EPHA2 phosphorylation and cell migration in C1GALT1 knockdown, EPHA2 knockdown, or double knockdown gastric cancer cells. Similar as EPHA2 knockdown,
C1GALT1 knockdown also decreased Ephrin A1-Fc induced phosphorylation of EPHA2 at Y588 in both AGS and MKN45 cells (Fig. 6a, Supplementary Fig. S8). It has been reported that Ephrin A1 can
induce phosphorylation of STAT3, AKT, ERK, FAK, or Src in several cell types [43, 44]. Therefore, we analyzed these molecules in gastric cancer cells. We found that, among them, only ERK
phosphorylation was induced by Ephrin A1-Fc in AGS cells. Furthermore, both C1GALT1 knockdown and EPHA2 knockdown exerted an inhibitory effect on ERK phosphorylation in AGS cells
(Supplementary Fig. S9). No significant changes in the phosphorylation of other molecules were observed. Although the downstream signaling pathways of Ephrin A1-EPHA2 in gastric cancer cells
remain unclear, the effects of C1GALT1 and EPHA2 on phosphorylation of these molecules, including EPHA2, STAT3, AKT, ERK, FAK, and Src, were similar. To investigate whether the effect of
C1GALT1 siRNAs on cell migration was reduced in EPHA2 knockdown cells, stable EPHA2 knockdown AGS and MKN cells were transiently transfected with two independent C1GALT1 siRNAs. EPHA2 and/or
C1GALT1 knockdown were confirmed by western blotting (Fig. 6b). Transwell migration assays showed that C1GALT1 knockdown or EPHA2 knockdown significantly inhibited cell migration in both
AGS and MKN45 cells (Fig. 6c). However, in EPHA2 knockdown cells, C1GALT1 siRNAs were unable to decrease cell migration (Fig. 6c). To investigate whether Ephrin A1-Fc-induced migration could
be suppressed by C1GALT1 knockdown or C1GALT1 knockout in gastric cancer cells, cell migration of AGS and MKN45 cells stimulated with Ephrin A1-Fc was analyzed. Results of transwell
migration assays showed that Ephrin A1-Fc increased migration of AGS (Fig. 6d) and MKN45 cells (Fig. 6e). We confirmed that the Fc of human IgG did not affect cell migration (Supplementary
Fig. S10). Moreover, C1GALT1 knockdown or knockout significantly inhibited Ephrin A1-Fc mediated migration and blocked the effect of Ephrin A1-Fc on AGS (Fig. 6d) and MKN45 cells (Fig. 6e),
respectively. Next, we examined whether binding of Ephrin A1-Fc to surfaces of gastric cancer cells was affected by C1GALT1. Results from flow cytometry showed that C1GALT1 knockdown or
knockout significantly decreased Ephrin A1-Fc binding to AGS and MKN45 cells, respectively (Fig. 6f). These results suggest that C1GALT1 mediates its pro-migratory effect on gastric cancer
cells at least partly through EPHA2. FUNCTIONAL PATHWAYS AFFECTED BY C1GALT1 KNOCKDOWN To better understand the mechanism by which C1GALT1 regulates gastric adenocarcinoma progression, we
evaluated global gene expression in control and C1GALT1 knockdown AGS cells. Volcano plot analysis revealed 1491 probes in C1GALT1 knockdown AGS cells that were significantly altered by
twofold or more versus the control (Fig. 7a). Functional enrichment and network analysis showed that C1GALT1 knockdown in AGS cells affected several functional pathways, including cell cycle
checkpoint, immune effector process, microtubule cytoskeleton organization, and regulation of cell division (Fig. 7b). The differential gene expression of the microarray results was
validated through quantitative RT-PCR (Fig. 7c and Supplementary Table S2). To validate the functional pathway of microarray data affected by C1GALT1, we analyzed cell cycle through flow
cytometry. C1GALT1 knockdown indeed affected the cell cycle (Supplementary Fig. S11). Consistent with our in vitro and in vivo data, these results suggest that C1GALT1 regulates the
expression of genes related to several cancer malignant behaviors in gastric cancer cells. DISCUSSION In this study, we found that C1GALT1 was significantly overexpressed in gastric
adenocarcinomas; and that C1GALT1 high expression correlated with adverse clinicopathologic features and is an independent prognostic factor for poor overall survival. Knockdown or knockout
of C1GALT1 inhibited malignant properties of gastric cancer cells in vitro and suppressed subcutaneous and peritoneal tumor growth in vivo. Importantly, we identified that O-glycosylation is
a critical regulatory factor for EPHA2 activity and signaling. Silencing of C1GALT1 decreased tyrosine phosphorylation of EPHA2, inhibited binding of Ephrin A1 to cell surfaces, and
suppressed soluble Ephrin A1-induced migration in gastric cancer cells. We also found that the effect of C1GALT1 knockout on cell invasiveness and peritoneal tumor growth of gastric cancer
cells was phenocopied by EPHA2 knockdown. These results suggest that C1GALT1 promotes gastric cancer cell invasiveness at least partly through activation of EPHA2. Our findings open novel
insights into the role of O-glycosylation in EPHA2 functions and highlight C1GALT1 as a potential diagnostic and therapeutic target for gastric cancer. RTK signaling pathways are crucial for
malignant transformation of cancers [45]. Genomic alterations in RTKs, including EGFR, HER2, FGFR2, and MET, were reported to occur in ~37% of gastric cancer patients [46]. At present,
anti-HER2 therapy plays a crucial role in gastric cancer treatment [18]. We showed that silencing of C1GALT1 decreases O-glycosylation and phosphorylation of EGFR and HER2. Moreover,
C1GALT1-mediated cell viability was significantly suppressed by lapatinib. These results suggest that C1GALT1 promotes cell viability at least partly through the activation of EGFR and HER2,
although other pathways are involved. EPH receptors constitute the largest family of RTKs and control a diverse range of developmental processes [21]. However, the role of GalNAc-type
O-glycosylation in EPH receptors has been largely ignored. We therefore focused on this issue. Our data showed that C1GALT1 depletion increased, whereas C1GALT1 overexpression decreased VVA
binding to EPHA2. C1GALT1 knockdown or knockout decreased soluble Ephrin A1-mediated phosphorylation of Y588 on EPHA2. These results suggest that EPHA2 carries GalNAc-type O-glycans and
altered O-glycan structures can regulate EPHA2 activity. Our data showed that EPHA2 is the major receptor for Ephrin A1 and silencing C1GALT1 significantly decreases Ephrin A1-Fc binding to
the cells. Importantly, the effects of C1GALT1 knockout on cell migration, invasion, and peritoneal tumor growth were phenocopied by EPHA2 knockdown. These results suggest that silencing of
C1GALT1 modifies O-glycan structures on EPHA2 to decrease Ephrin A1-Fc binding to EPHA2 and in turn suppresses cell migration. Henrik Clausen’s team identified O-glycosites at S277 and T429
of EPHA2 using proteomics approaches [41]. To further determine the role of site-specific O-glycosylation in EPHA2 phosphorylation, we mutated serine or threonine to alanine to block the
initiation of O-glycosylation. The results showed that lack of O-glycosylation on either S277 or T429 dramatically enhanced phosphorylation of Y588 on EPHA2, suggesting that site-specific
O-glycosylation can also determine EPHA2 activity. It will be of great interest to further investigate which GalNAc transferases (GALNTs) initiate O-glycosylation on specific O-glycosites to
regulate EPHA2 functions. Our data showed that C1GALT1 knockdown also decreased phospho-EPHA1 levels in AGS cells treated with Ephrin A1-Fc. Henrik Clausen’s glycoproteomic data indicated
that EPHB4 is decorated with GalNAc-type O-glycans [41]. Therefore, it is possible that several EPH receptors are O-glycosylated and their functions can be modulated by O-glycosylation. In
addition to EPHA2 and EPHA1, we found that soluble Ephrin A1-Fc can activate EGFR, but not MET. Consistently, it has been reported that progranulin activates EPHA2 and concurrently enhances
EGFR signaling [47]. These findings support that EPHA2 can crosstalk with other RTKs, such as EGFR, to mediate its functions and imply that targeting EPHA2 could be beneficial for other
RTK-dependent diseases, especially cancers. Although the effectiveness of 5-FU monotherapy is limited due to drug resistance, it is still used as the standard first-line chemotherapy for
metastatic gastric cancer [48]. In a previous study, glycosylation was highly involved in the acquisition of multidrug resistance at specific positions and through its changes in secreted
glycoproteins [35]. We previously indicated that C1GALT1 knockdown suppresses the MUC1-C/β-catenin signaling pathway in breast cancer cells [49]. Blocking MUC1-C function was revealed to be
effective when combined with taxol and doxorubicin to treat breast cancer [50]. Our data consistently provide evidence that silencing C1GALT1 enhances the cytotoxicity of the
chemotherapeutic drug 5-FU in gastric cancer cells through potentiating the apoptotic death response. These findings suggest that the combination of C1GALT1 inhibition and 5-FU may be a
promising therapy strategy to overcome the resistance of 5-FU in gastric cancer treatment. In our study, a functional enrichment and network analysis indicated that C1GALT1 regulate several
functional pathways. Our results from the clinical analyses and in vitro phenotypic assays support that these functional pathways can be regulated by C1GALT1. For example, the in vitro
finding that C1GALT1 knockdown affects cell cycle and cell viability is consistent with the function map showing altered cell cycle checkpoints and cell divisions. C1GALT1 knockdown inhibits
gastric cancer cell migration and invasion, which is associated with altered microtubule cytoskeleton organization. Noteworthily, the microtubule organization was also indicated to be
related to lymph node metastasis, tumor stage, and poor outcomes of patients with gastric cancer [51], which are associated with C1GALT1 high expression. Microarray data suggested that
C1GALT1 modulates genes associated with the immune effector process. Because the targeting of immune checkpoints seems promising in the treatment of several cancers [2], the regulatory role
of C1GALT1 in immune responses warrants further investigation. MATERIALS AND METHODS PATIENT SAMPLES Twenty-five patients who had undergone surgery at National Taiwan University Hospital
were selected for this study. Written consent was obtained from the patients; the hospital’s Institutional Review Board approved this study (IRB No: 201604068RIND). Subsequently,
paraffin-embedded tissue blocks of gastric adenocarcinomas and their surrounding nontumor tissues were collected. IMMUNOHISTOCHEMISTRY Tissue microarray of gastric adenocarcinoma from 111
patients was purchased (HStm-Ade178Sur-01, US Biomax, Inc., MD, USA) for immunohistochemical staining. The tissue microarray was incubated with an anti-C1GALT1 antibody (1:100, Santa Cruz
Biotechnology, CA, USA) at 4 °C for 16 h. Super SensitiveTM Link-Label IHC Detection System (BioGenex, CA, USA) was used and signals were visualized through a 3,3-diaminobenzidine (DAB)
liquid substrate system (Sigma, MO, USA). The tissues were counterstained with hematoxylin and mounted with UltraKitt (J.T. Baker, Deventer, Holland). Negative controls were performed
through replacing the primary antibody with a control IgG at the same concentration. CELL LINES AND CELL CULTURE The human gastric cancer cell lines AGS and MKN45 were a gift from Dr
Chiung-Nien Chen (National Taiwan University Hospital, Taiwan). AGS and MKN45 cell lines were authenticated using short tandem repeat (STR) profiling analysis. Gastric cancer cell lines
NCI-N87, SNU-1, and KATO3 were a gift from Dr I-Rue Lai (National Taiwan University Hospital, Taiwan), who recently purchased them from Bioresources Collection & Research Center
(Hsinchu, Taiwan). HEK293FT cells were purchased from Thermo Fisher Scientific. Cell lines were maintained in complete medium containing 10% fetal bovine serum (FBS) (Life Technologies,
Burlington, Canada) and 1% penicillin/streptomycin (P/S; Gibco, Invitrogen™, Thermo Fisher Scientific), and cultured at 37 °C in air with 5% CO2. Dulbecco’s Modified Eagle Medium (DMEM)
(Thermo Fisher Scientific, Yokohama, Japan) was used for HEK293FT cells. RPMI 1640 medium (GE healthcare, Chicago, USA) was used for AGS, MKN45, NCI-N87, KATO3, and SNU-1 cells. CDNA
SYNTHESIS AND REAL-TIME RT-PCR Total RNA was isolated using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. Two micrograms of total RNA were used in a 20 μL reverse
transcription reaction using High-Capacity cDNA Reverse Transcription Kits (AB, USC) for cDNA synthesis. For real-time PCR, QuantStudio 3 Real-Time PCR system (Thermo) was used. The
real-time PCR reactions were performed in 20 μL volume containing 1 μL cDNA, 10 μL SensiFAST SYBER Lo-ROX Mix (BIOLINE), and primer pairs. The following primer pairs were used: _C1GALT1_,
_CDK1_, _K1F20A_, _F1F2C_, _BUB1_, _GBP1_, _PRF1_, _OAS2_, _BST2_, and _GAPDH_ (Supplementary Table S1). TRANSFECTION AND PLASMID CONSTRUCTION For transient knockdown, three independent
C1GALT1-specific siRNAs (Invitrogen), two independent EPHA2-specific siRNAs (Dharmacon), and nontargeting siRNA (Invitrogen; Dharmacon), were used to transfect gastric cancer cells through
Lipofectamine RNAiMAX (Invitrogen) with a final concentration of 10 nM for two days. The siRNAs against C1GALT1 were si-C1GALT1-1: 5′-UUAGUAUACGUUCAGGUAAGGUAGG-3′, si-C1GALT1-2:
5′-UUAUGUUGGCUAGAAUCUGCAUUGA-3′, and si-C1GALT1-3: 5′-CCUACCUUACCUGAACGUAUACUAA-3′. The siRNAs against EPHA2 were si-EPHA2-1: 5′-UGAAUGACAUGCCGAUCUA-3′, and si-EPHA2-2:
5′-GAAGUUCACUACCGAGAUC-3′. The nontargeting siRNAs (si-Control) were 5′-CAACCUCAGCCAUGUCGACUGGUUU-3′. For stable knockdown, shC1GALT1/pLKO (TRCN35411), shEPHA2/pLKO (TRCN6403), and
nontargeting control (TRC025) were obtained from National RNAi Core Facility (Academia Sinica, Taipei, Taiwan). For stable overexpression, C1GALT1/pcDNA3.1A [11] or control empty pcDNA3.1A
plasmid were used to transfect AGS cells using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s protocol. WESTERN BLOT ANALYSIS Proteins were separated on an 8% SDS-PAGE and
transferred onto a PVDF membrane. After blocking with 5% bovine serum albumin (BSA; Bio-Rad, CA, USA) for 1 h at room temperature, membranes were incubated with primary antibodies at 4 °C
overnight. Antibodies against C1GALT1, GAPDH, Ephrin A1, and FAK were purchased from Santa Cruz Biotechnology. Antibodies against EGFR, p-EGFR, HER2, p-HER2, p-AKT, EPHA2, p-EPHA2, p-ERK,
ERK, p-STAT3, STAT3, and p-FAK were purchased from Cell Signaling Technology (MA, USA). Antibodies against AKT, p-Src, and Src were purchased from GeneTex Inc. (CA, USA). The membranes were
then incubated with horseradish peroxidase-conjugated secondary antibodies, and proteins were detected using ECL reagents (GE Healthcare Life Sciences). MTT ASSAY Gastric cancer cells (1.5 ×
103) in 100 μL of complete RPMI were seeded in 96-well plates for 16 h; subsequently, 10 μL of 5 mg/mL 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide solution (MTT; Sigma)
was added to each well for the indicated times and incubated at 37 °C for 3 h, and the MTT formazan crystals were dissolved with 100 μL 10% SDS containing 0.01 N HCl. The resultant optical
density was measured spectrophotometrically at the dual wavelengths of 550 and 630 nm. TRANSWELL MIGRATION AND MATRIGEL INVASION ASSAYS Cell migration and invasion assays were evaluated
using transwell (Corning, NY, USA) or Matrigel-coated (BD Biosciences, CA, USA) transwell chamber, respectively. Each transwell chamber contained a membrane of pore size 8 μm. AGS (3 × 104),
MKN45 and SNU-1 cells (2 × 105) in 0.25 mL serum-free RPMI were seeded into the transwell or Matrigel-coated transwell chamber, following which the chambers were put into 24-well plates.
After incubating for 24 h or 48 h, the cells were fixed and stained with 0.5% (w/v) crystal violet (Sigma) containing 20% (v/v) methanol. The migrated and invaded cells from three random
fields were counted under a microscope. PHOSPHO-RECEPTOR TYROSINE KINASE ARRAY ASSAY A human phospho-RTK array kit including 49 RTKs was purchased from R&D Systems (MN, USA). AGS cells
were serum-starved for 24 h and stimulated with 10% FBS for 10 min or Ephrin A1-Fc (R&D, MN, USA) for 3 min. Cells were lysed, and 500 μg of protein lysates were subjected to western
blotting according to the manufacturer’s protocol. LECTIN PULL-DOWN ASSAY To analyze changes in O-glycans, 300 μg of total cell lysates were treated with or without neuraminidase (Sigma) to
remove sialic acids and then incubated overnight with _Vicia Villosa_ lectin (VVA) or peanut agglutinin (PNA) agarose beads (Vector Laboratories, CA, USA) at 4 °C. After washing with PBS
twice, the pulled-down proteins were subjected to western blot analysis. FLOW CYTOMETRY Cells (1 × 105) were resuspended in 100 μL PBS with 1% BSA and then stained with VVA-FITC, PNA-FITC
(Vector Laboratories), Ephrin A1-Fc, or anti-EPHA2 primary antibody (R&D, MN, USA) on ice for 30 min. FITC-conjugated secondary antibody was used. After washing twice, the fluorescence
intensity was analyzed using a flow cytometer (BD LSR-II; BD Pharmingen). For apoptosis assay, cells were treated with DMSO solvent or 5-fluorouracil (5-FU) and stained with annexin V-FITC
and propidium iodide (PI). Samples not containing the fluorescent reagents were used as negative controls. CDNA MICROARRAY ANALYSIS To determine the C1GALT1-regulated gene expression profile
in AGS cells, C1GALT1 was knocked down using _C1GALT1_-specific siRNA (Invitrogen™, Thermo Fisher Scientific) in AGS cells, and nontargeting siRNA was used as the control. Total RNA was
extracted from AGS cells using a GeneJET RNA Purification Kit (Thermo Scientific) and was subjected to cDNA microarray analysis (Agilent-072363 SurePrint G3 Human GE v3 8 × 60 K Microarray
039494) and gene ontology enrichment analysis as described previously [15]. The selected genes with differential expression were validated through real-time RT-PCR analysis. Microarray data
were deposited in the GEO database (accession number GSE90672). The raw data are normalized using the quantile method and the differential expressed genes were determined using the R package
‘limma’. The over-representative Gene Ontology (GO) terms of the differential expressed genes were determined using the Fisher’s exact test. The over-representative GO terms were depicted
as a network via Cytoscape and EnrichmentMap. KNOCKOUT OF C1GALT1 IN MKN45 CELLS USING CRISPR/CAS9 SYSTEM CRISPR/Cas9 system was used to knock out C1GALT1 in MKN45 cells. Small guide (sg)
RNA for targeting C1GALT1 was designed according to database prediction (http://crispr.mit.edu/). The target sequence of sgC1GALT1 is 5′-GCAACACTTTGTTACAACGC-3′. Knockout of C1GALT1 in the
genome was confirmed by DNA sequencing. IN VIVO XENOGRAFT ANIMAL MODELS For in vivo tumor growth analysis, 3 × 106 of MKN45 cells or 107 of AGS cells were mixed with Matrigel™ Basement
Membrane Matrix (Corning Incorporated, Corning, NY, USA) and subcutaneously injected into five-week-old male nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice (National
Laboratory Animal Center, Taiwan). AGS cells (2 × 106) were used for tail vein injection, and the lungs were paraffin-embedded for hematoxylin and eosin (H&E) staining. For peritoneal
injection, 107 of MKN45 cells were used. Animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee IACUC) of College of Medicine, National Taiwan
University. STATISTICAL ANALYSIS Statistical analyses were performed using R 3.1.2 for Mac OS X and GraphPad Prism 6 for Mac OS X. The correlations between C1GALT1 expression and
clinicopathologic characteristics were tested using a chi-square test. Student’s _t_ test was used to compare differences between the two experimental groups. Univariate comparisons of the
parameters were made using the nonparametric chi-square test or the Fisher and Mann–Whitney _U_ tests. Overall survival was compared through log-rank comparisons for time-to-event data using
Kaplan–Meier methods. Student’s _t_ test was used to analyze in vitro and in vivo experiments. Two-sided _P_ < 0.05 was considered statistically significant. REFERENCES * Siegel RLMK,
Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. PubMed Google Scholar * Tran PN, Sarkissian S, Chao J, Klempner SJ. PD-1 and PD-L1 as emerging therapeutic targets in
gastric cancer: current evidence. Gastrointest Cancer Targets Ther. 2017;7:1–11. Article Google Scholar * Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic
targets. Nat Rev Cancer. 2005;5:526–42. Article CAS PubMed Google Scholar * Tran DT, Ten Hagen KG. Mucin-type O-glycosylation during development. J Biol Chem. 2013;288:6921–9. Article
CAS PubMed PubMed Central Google Scholar * Ten Hagen KG, Fritz TA, Tabak LA. All in the family: the UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases. Glycobiology.
2003;13:1R–16R. Article CAS PubMed Google Scholar * Ju T, Cummings RD. A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc
Natl Acad Sci USA. 2002;99:16613–8. Article CAS PubMed PubMed Central Google Scholar * Tian E, Ten Hagen KG. Recent insights into the biological roles of mucin-type O-glycosylation.
Glycoconj J. 2009;26:325–34. Article CAS PubMed Google Scholar * Gill DJ, Tham KM, Chia J, Wang SC, Steentoft C, Clausen H, et al. Initiation of GalNAc-type O-glycosylation in the
endoplasmic reticulum promotes cancer cell invasiveness. Proc Natl Acad Sci USA. 2013;110:E3152–61. Article PubMed PubMed Central Google Scholar * Kudo T, Sato T, Hagiwara K, Kozuma Y,
Yamaguchi T, Ikehara Y, et al. C1galt1-deficient mice exhibit thrombocytopenia due to abnormal terminal differentiation of megakaryocytes. Blood. 2013;122:1649–57. Article CAS PubMed
Google Scholar * Hung JS, Huang J, Lin YC, Huang MJ, Lee PH, Lai HS, et al. C1GALT1 overexpression promotes the invasive behavior of colon cancer cells through modifying O-glycosylation of
FGFR2. Oncotarget. 2014;5:2096–106. PubMed PubMed Central Google Scholar * Wu YM, Liu CH, Huang MJ, Lai HS, Lee PH, Hu RH, et al. C1GALT1 enhances proliferation of hepatocellular
carcinoma cells via modulating MET glycosylation and dimerization. Cancer Res. 2013;73:5580–90. Article CAS PubMed Google Scholar * Duarte HO, Freitas D, Gomes C, Gomes J, Magalhaes A,
Reis CA. Mucin-type _O_-glycosylation in gastric carcinogenesis. Biomolecules. 2016;6:33. * Liu SY, Shun CT, Hung KY, Juan HF, Hsu CL, Huang MC, et al. Mucin glycosylating enzyme GALNT2
suppresses malignancy in gastric adenocarcinoma by reducing MET phosphorylation. Oncotarget. 2016;7:11251–62. PubMed PubMed Central Google Scholar * Wu YM, Liu CH, Hu RH, Huang MJ, Lee
JJ, Chen CH, et al. Mucin glycosylating enzyme GALNT2 regulates the malignant character of hepatocellular carcinoma by modifying the EGF receptor. Cancer Res. 2011;71:7270–9. Article CAS
PubMed Google Scholar * Huang MJ, Hu RH, Chou CH, Hsu CL, Liu YW, Huang J, et al. Knockdown of GALNT1 suppresses malignant phenotype of hepatocellular carcinoma by suppressing EGFR
signaling. Oncotarget. 2015;6:5650–65. PubMed PubMed Central Google Scholar * Yao HP, Zhou YQ, Zhang R, Wang MH. MSP-RON signalling in cancer: pathogenesis and therapeutic potential. Nat
Rev Cancer. 2013;13:466–81. Article CAS PubMed Google Scholar * Peng Z, Zhu Y, Wang Q, Gao J, Li Y, Li Y, et al. Prognostic significance of MET amplification and expression in gastric
cancer: a systematic review with meta-analysis. PloS One. 2014;9:e84502. Article CAS PubMed PubMed Central Google Scholar * Gravalos C, Jimeno A. HER2 in gastric cancer: a new
prognostic factor and a novel therapeutic target. Ann Oncol. 2008;19:1523–9. Article CAS PubMed Google Scholar * Bradley CA, Salto-Tellez M, Laurent-Puig P, Bardelli A, Rolfo C,
Tabernero J, et al. Targeting c-MET in gastrointestinal tumours: rationale, opportunities and challenges. Nat Rev Clin Oncol. 2017;14:562–76. Article CAS PubMed Google Scholar * Sierra
JC, Asim M, Verriere TG, Piazuelo MB, Suarez G, Romero-Gallo J, et al. Epidermal growth factor receptor inhibition downregulates Helicobacter pylori-induced epithelial inflammatory
responses, DNA damage and gastric carcinogenesis. Gut. 2018;67:1247–60. Article CAS PubMed Google Scholar * Kania A, Klein R. Mechanisms of ephrin-Eph signalling in development,
physiology and disease. Nat Rev Mol Cell Biol. 2016;17:240–56. Article CAS PubMed Google Scholar * Xi HQ, Wu XS, Wei B, Chen L. Eph receptors and ephrins as targets for cancer therapy. J
Cell Mol Med. 2012;16:2894–909. Article CAS PubMed PubMed Central Google Scholar * Vaught D, Brantley-Sieders DM, Chen J. Eph receptors in breast cancer: roles in tumor promotion and
tumor suppression. Breast Cancer Res. 2008;10:217. Article CAS PubMed PubMed Central Google Scholar * Herath NI, Boyd AW. The role of Eph receptors and ephrin ligands in colorectal
cancer. Int J Cancer. 2010;126:2003–11. CAS PubMed Google Scholar * Lisle JE, Mertens-Walker I, Rutkowski R, Herington AC, Stephenson SA. Eph receptors and their ligands: promising
molecular biomarkers and therapeutic targets in prostate cancer. Biochim Biophys Acta. 2013;1835:243–57. CAS PubMed Google Scholar * Pasquale EB. Eph receptors and ephrins in cancer:
bidirectional signalling and beyond. Nat Rev Cancer. 2010;10:165–80. Article CAS PubMed PubMed Central Google Scholar * Boyd AW, Bartlett PF, Lackmann M. Therapeutic targeting of EPH
receptors and their ligands. Nat Rev Drug Disco. 2014;13:39–62. Article CAS Google Scholar * Yuan WJ, Ge J, Chen ZK, Wu SB, Shen H, Yang P, et al. Over-expression of EphA2 and EphrinA-1
in human gastric adenocarcinoma and its prognostic value for postoperative patients. Dig Dis Sci. 2009;54:2410–7. Article CAS PubMed Google Scholar * Nakamura R, Kataoka H, Sato N,
Kanamori M, Ihara M, Igarashi H, et al. EPHA2/EFNA1 expression in human gastric cancer. Cancer Sci. 2005;96:42–47. Article CAS PubMed Google Scholar * Huang J, He Y, McLeod HL, Xie Y,
Xiao D, Hu H, et al. miR-302b inhibits tumorigenesis by targeting EphA2 via Wnt/ beta-catenin/EMT signaling cascade in gastric cancer. BMC Cancer. 2017;17:886. Article CAS PubMed PubMed
Central Google Scholar * Wen Q, Chen Z, Chen Z, Chen J, Wang R, Huang C, et al. EphA2 affects the sensitivity of oxaliplatin by inducing EMT in oxaliplatin-resistant gastric cancer cells.
Oncotarget. 2017;8:47998–8011. PubMed PubMed Central Google Scholar * Yuan W, Chen Z, Chen Z, Wu S, Guo J, Ge J, et al. Silencing of EphA2 inhibits invasion of human gastric cancer
SGC-7901 cells in vitro and in vivo. Neoplasma. 2012;59:105–13. Article CAS PubMed Google Scholar * Washington K. 7th edition of the AJCC cancer staging manual: stomach. Ann Surgical
Oncol. 2010;17:3077–9. Article Google Scholar * Goseki N, Takizawa T, Koike M. Differences in the mode of the extension of gastric cancer classified by histological type: new histological
classification of gastric carcinoma. Gut. 1992;33:606–12. Article CAS PubMed PubMed Central Google Scholar * Wu J, Qin H, Li T, Cheng K, Dong J, Tian M, et al. Characterization of
site-specific glycosylation of secreted proteins associated with multi-drug resistance of gastric cancer. Oncotarget. 2016;7:25315–27. PubMed PubMed Central Google Scholar * Lin MC, Chien
PH, Wu HY, Chen ST, Juan HF, Lou PJ, et al. C1GALT1 predicts poor prognosis and is a potential therapeutic target in head and neck cancer. Oncogene. 2018;37:5780–93. * Ho WL, Chou CH, Jeng
YM, Lu MY, Yang YL, Jou ST, et al. GALNT2 suppresses malignant phenotypes through IGF-1 receptor and predicts favorable prognosis in neuroblastoma. Oncotarget. 2014;5:12247–59. PubMed
PubMed Central Google Scholar * Alford S, Watson-Hurthig A, Scott N, Carette A, Lorimer H, Bazowski J, et al. Soluble ephrin a1 is necessary for the growth of HeLa and SK-BR3 cells. Cancer
Cell Int. 2010;10:41. Article CAS PubMed PubMed Central Google Scholar * Chu M, Zhang C. Inhibition of angiogenesis by leflunomide via targeting the soluble ephrin-A1/EphA2 system in
bladder cancer. Sci Rep. 2018;8:1539. Article CAS PubMed PubMed Central Google Scholar * Cui XD, Lee MJ, Yu GR, Kim IH, Yu HC, Song EY, et al. EFNA1 ligand and its receptor EphA2:
potential biomarkers for hepatocellular carcinoma. Int J Cancer. 2010;126:940–9. CAS PubMed Google Scholar * Campos D, Freitas D, Gomes J, Magalhaes A, Steentoft C, Gomes C, et al.
Probing the O-glycoproteome of gastric cancer cell lines for biomarker discovery. Mol Cell Proteom. 2015;14:1616–29. Article CAS Google Scholar * Ieguchi K, Tomita T, Omori T, Komatsu A,
Deguchi A, Masuda J, et al. ADAM12-cleaved ephrin-A1 contributes to lung metastasis. Oncogene. 2014;33:2179–90. Article CAS PubMed Google Scholar * Wykosky J, Debinski W. The EphA2
receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol Cancer Res. 2008;6:1795–806. Article CAS PubMed PubMed Central Google Scholar * Beauchamp A,
Debinski W. Ephs and ephrins in cancer: ephrin-A1 signalling. Semin Cell Dev Biol. 2012;23:109–15. Article CAS PubMed Google Scholar * Blume-Jensen P, Hunter T. Oncogenic kinase
signalling. Nature. 2001;411:355–65. Article CAS PubMed Google Scholar * Deng N, Goh LK, Wang H, Das K, Tao J, Tan IB, et al. A comprehensive survey of genomic alterations in gastric
cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut. 2012;61:673–84. Article CAS PubMed Google Scholar * Neill T,
Buraschi S, Goyal A, Sharpe C, Natkanski E, Schaefer L, et al. EphA2 is a functional receptor for the growth factor progranulin. J Cell Biol. 2016;215:687–703. Article CAS PubMed PubMed
Central Google Scholar * Ohtsu A, Shimada Y, Shirao K, Boku N, Hyodo I, Saito H, et al. Randomized phase III trial of fluorouracil alone versus fluorouracil plus cisplatin versus uracil
and tegafur plus mitomycin in patients with unresectable, advanced gastric cancer: The Japan Clinical Oncology Group Study (JCOG9205). J Clin Oncol. 2003;21:54–59. Article CAS PubMed
Google Scholar * Chou CH, Huang MJ, Chen CH, Shyu MK, Huang J, Hung JS, et al. Up-regulation of C1GALT1 promotes breast cancer cell growth through MUC1-C signaling pathway. Oncotarget.
2015;6:6123–35. PubMed PubMed Central Google Scholar * Uchida Y, Raina D, Kharbanda S, Kufe D. Inhibition of the MUC1-C oncoprotein is synergistic with cytotoxic agents in the treatment
of breast cancer cells. Cancer Biol Ther. 2013;14:127–34. Article CAS PubMed PubMed Central Google Scholar * Fife CM, McCarroll JA, Kavallaris M. Movers and shakers: cell cytoskeleton
in cancer metastasis. Br J Pharmacol. 2014;171:5507–23. Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS The authors thank Dr Shu-Yun Tung (Academia
Sinica, Taiwan) for performing global gene expression analysis and Dr Chiung-Nien Chen and Dr I-Rue Lai (National Taiwan University Hospital, Taiwan) for providing gastric cancer cell
lines. This study was supported by National Taiwan University Hospital (109-049) to PCL, and Ministry of Science and Technology, Taiwan (108-2320-B-002-064-MY3) to MCH. AUTHOR INFORMATION
AUTHORS AND AFFILIATIONS * Department of Surgery, National Taiwan University Hospital, Taipei, Taiwan Po-Chu Lee, Ting-Chun Kuo, John Huang, Ji-Shiang Hung & Po-Huang Lee * Department of
Traumatology, National Taiwan University Hospital, Taipei, Taiwan Po-Chu Lee & Ting-Chun Kuo * Graduate Institute of Clinical Medicine, College of Medicine, National Taiwan University,
Taipei, Taiwan Po-Chu Lee * Graduate Institute of Anatomy and Cell Biology, College of Medicine, National Taiwan University, Taipei, Taiwan Syue-Ting Chen, Ting-Chun Kuo, Tzu-Chi Lin &
Min-Chuan Huang * Department of Otolaryngology, National Taiwan University Hospital, Taipei, Taiwan Mei-Chun Lin * Department of Medical Research, National Taiwan University Hospital,
Taipei, Taiwan Chia-Lang Hsu * Department of Life Science, National Taiwan University, Taipei, Taiwan Hsueh-Fen Juan * Department of Surgery, E-DA Hospital, Kaohsiung City, Taiwan Po-Huang
Lee Authors * Po-Chu Lee View author publications You can also search for this author inPubMed Google Scholar * Syue-Ting Chen View author publications You can also search for this author
inPubMed Google Scholar * Ting-Chun Kuo View author publications You can also search for this author inPubMed Google Scholar * Tzu-Chi Lin View author publications You can also search for
this author inPubMed Google Scholar * Mei-Chun Lin View author publications You can also search for this author inPubMed Google Scholar * John Huang View author publications You can also
search for this author inPubMed Google Scholar * Ji-Shiang Hung View author publications You can also search for this author inPubMed Google Scholar * Chia-Lang Hsu View author publications
You can also search for this author inPubMed Google Scholar * Hsueh-Fen Juan View author publications You can also search for this author inPubMed Google Scholar * Po-Huang Lee View author
publications You can also search for this author inPubMed Google Scholar * Min-Chuan Huang View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
PCL collected clinical samples, analyzed data, and wrote the manuscript. STC, TCK, TCL, and MCL conducted the experiments and analyzed the data. CLH and HFJ performed the microarray data
analysis. JH, JSH, and PHL contributed to the project’s conception. MCH conceived the project, supervised all experiments, and revised the manuscript. All authors approved the final version
of the article including the authorship list. CORRESPONDING AUTHOR Correspondence to Min-Chuan Huang. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare that they have no conflict
of interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY TABLES SUPPLEMENTARY FIGURES RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which
permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to
the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless
indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or
exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints
and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Lee, PC., Chen, ST., Kuo, TC. _et al._ C1GALT1 is associated with poor survival and promotes soluble Ephrin A1-mediated cell migration
through activation of EPHA2 in gastric cancer. _Oncogene_ 39, 2724–2740 (2020). https://doi.org/10.1038/s41388-020-1178-7 Download citation * Received: 08 April 2019 * Revised: 13 January
2020 * Accepted: 21 January 2020 * Published: 31 January 2020 * Issue Date: 26 March 2020 * DOI: https://doi.org/10.1038/s41388-020-1178-7 SHARE THIS ARTICLE Anyone you share the following
link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature
SharedIt content-sharing initiative





