- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Microbial interaction with the host through sensing receptors, including SIGNR1, sustains intestinal homeostasis against pathogenic inflammation. The newly discovered commensal
_Propionibacterium_ strain, P. UF1, regulates the intestinal immunity against pathogen challenge. However, the molecular events driving intestinal phagocytic cell response, including colonic
dendritic cells (DCs), by this bacterium are still elusive. Here, we demonstrate that the glycosylation of bacterial large surface layer protein A (LspA) by protein _O_-mannosyltransferase
1 (Pmt1) regulates the interaction with SIGNR1, resulting in the control of DC transcriptomic and metabolomic machineries. Programmed DCs promote protective T cell response to intestinal
_Listeria_ infection and resist chemically induced colitis in mice. Thus, our findings may highlight a novel molecular mechanism by which commensal surface glycosylation interacting with
SIGNR1 directs the intestinal homeostasis to potentially protect the host against proinflammatory signals inducing colonic tissue damage. You have full access to this article via your
institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS ILC3S REGULATE THE GUT MICROBIOTA VIA HOST INTESTINAL GALACTOSYLATION TO LIMIT PATHOGEN INFECTION IN MICE Article 17 February
2025 PGLYRP1-MEDIATED INTRACELLULAR PEPTIDOGLYCAN DETECTION PROMOTES INTESTINAL MUCOSAL PROTECTION Article Open access 21 February 2025 RATIONAL DESIGN OF A MICROBIAL CONSORTIUM OF MUCOSAL
SUGAR UTILIZERS REDUCES _CLOSTRIDIODES DIFFICILE_ COLONIZATION Article Open access 09 October 2020 INTRODUCTION Commensal bacteria, via their surface layer (S-layer) gene products, and the
gastrointestinal phagocytic cells expressing sensing receptors (e.g., SIGNR1) synergistically interact to fine-tune the T cell signaling that is critical for protecting the host against
pathogenic inflammation exerted by intestinal infections.1,2 In this process, bacterial S-layer macromolecules along with induced metabolites transduce critical signals via cognate receptors
into these cells that profoundly control the host homeostasis to protect against tissue damage.3,4 Although the bacterial S-layer proteins display a similar architecture composed of a
peptidoglycan layer decorated with proteins and polysaccharides, various modifications, particularly glycosylation, exhibit strain-specific properties that differentially modify the host
immune physiology.5 Disruption of mutualistic interactions of the commensal’s S-layer with the host triggers deleterious signals that may manifest in pathogenic inflammation potentially
impairing the intestinal barrier function.6 Thus, understanding how host intestinal immunity is regulated through the recognition of these well-structured bacterial gene products by their
cognate receptors7 to coordinate protective immune responses is currently of particular therapeutic significance and requires further mechanistic investigations.8 _Propionibacterium_ strain,
P. UF1, is a newly discovered commensal bacterium isolated from the gut microbiota of premature infants fed human breast milk.9 This bacterium increases the frequency of colonic Th17 and
Treg cells9 involved in mucosal barrier repair and regulation of the intestinal inflammation.10 Induced bacteria-specific Th17 cell differentiation requires the bacterial dihydrolipoamide
acetyltransferase (DlaT), an enzymatic component of the pyruvate dehydrogenase complex.9 Chromosomal deletion of _dlaT_ gene impairs the regulation of protective Th17 cell response to
intestinal and systemic _Listeria monocytogenes (L. m)_ infection.9,11 Furthermore, P. UF1 regulates the neonatal T cells against necrotizing enterocolitis (NEC)-like injury in mice9 and
enhances the neonatal protective T cells against intestinal pathogen infection over time.12 However, the bacterial effector mechanisms potentially instructing the function of colonic DCs to
possibly control protective T cell immunity remain largely unknown. Here, we demonstrate that the glycosylation of bacterial LspA interacting with SIGNR1 is a pivotal factor, which
transcriptionally and metabolically programs colonic DCs, leading to protective T cell activation in steady state and during intestinal infection. Further, glycosylated LspA-SIGNR1
interaction critically protects mice against colitis-induced intestinal barrier injury. Errors in the bacterial glycosylation significantly disrupt the intestinal homeostasis, manifesting in
an inflammatory condition resulting in pathogen persistence and colonic tissue damage. Thus, this finding highlights the critical relevance of the glycosylated LspA in programming DC
immunophysiology to mitigate pathogenic inflammation and the induced colitogenic potential in mice. RESULTS GLYCOSYLATION OF LSPA BY PMT1 Knowing the significance of bacterial S-layer
complexes in communicating with host cells,13 we sought to investigate the functional relevance of P. UF1 S-layer proteins potentially involved in the regulation of colonic DC function. One
of the S-layer proteins of P. UF1 is LspA, which contains six N-terminal LGFP repeats [L-G-X-P-X(7-8)-D/N-G] involved in cell membrane anchoring and a C-terminal N-acetylglucosaminidase-like
domain, potentially implicated in bacterial cell wall metabolism (Supplementary Fig. 1a). Phylogenetic analysis demonstrated that LspA was highly conserved in P. UF1 and closely related
_Propionibacterium_ strains. Moreover, LspA homologs were also found in evolutionarily distantly related bacterial species, including _Bifidobacterium_ and _Geodermatophilus_ (Supplementary
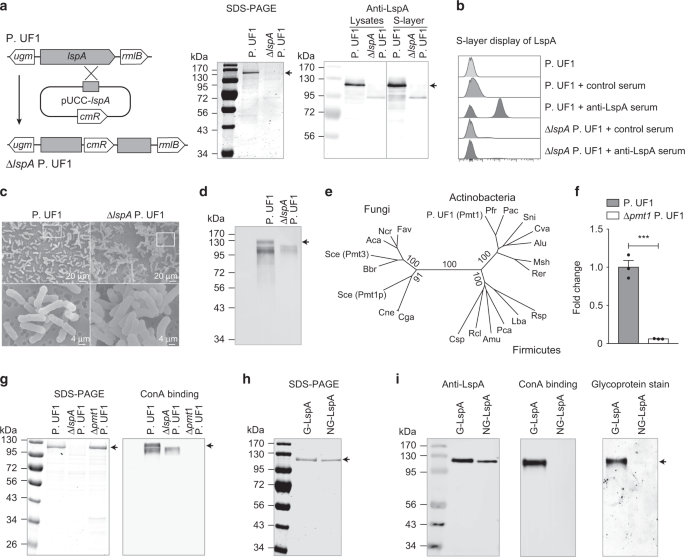
Fig. 1b). Thus, to elucidate the functional significance of LspA within P. UF1 molecular machinery, the _lspA_ gene was deleted from the bacterial chromosome, resulting in Δ_lspA_ P. UF1
(Fig. 1a, b). Δ_lspA_ P. UF1 demonstrated enhanced bacterial clusters and autoagglutination (Fig. 1c), suggesting the critical involvement of this protein in bacterial S-layer structures.
Further, deletion of LspA significantly affected the bacterial transcriptomic and metabolomic signaling, including differential metabolic pathways involved in peptidoglycan biosynthesis,
amino and nucleotide sugar metabolism, fructose and mannose metabolism (Supplementary Fig. 2a). The analyzed metabolites involved in protein glycosylation (e.g., GDP-mannose and mannose
1-phosphate), along with those important for cell wall metabolism (e.g., GlcNAc-6-phosphate and UDP-GlcNAc), were significantly deregulated within Δ_lspA_ P. UF1 compared to P. UF1
(Supplementary Fig. 2b). RNA-Seq analysis further documented differentially expressed genes implicated in bacterial mannosylation and nucleotide sugar metabolism, including
phosphatidylinositol mannosyltransferase _pimA_14 and GDP-mannose-dependent alpha-mannosyltransferase _mgtA_15 (Supplementary Fig. 2c). Thus, these data emphasize the importance of LspA in
the regulation of glycan metabolism that may fundamentally impact the bacterial S-layer glycosylation. The bacterial S-layer proteins are generally glycosylated for their noncovalent
anchoring to the cell surface and interactions with environmental factors and host immune cells.5 Data demonstrated that the S-layer of P. UF1 reacted with concanavalin A (ConA), a
mannose/glucose-binding lectin, while LspA deficiency resulted in the loss of ConA binding (Fig. 1d), suggesting that LspA may be glycosylated. Therefore, we investigated the
glycosyltransferases responsible for adding glycol moieties to the bacterial S-layer using genome-wide bioinformatic analysis. Pmt1, a potential member of protein _O_-mannosyltransferase
family responsible for mannose transfer to serine and threonine residues of proteins in yeast16, was identified in P. UF1 genome. Further analysis demonstrated that Pmt1 homologs fell into
separate and loosely related groups of bacteria, including Actinobacteria (Fig. 1e). The _pmt1_ gene was then deleted in P. UF1 to assess the status of LspA glycosylation (Fig. 1f). Although
Δ_pmt1_ P. UF1 showed similar S-layer protein patterns when compared to P. UF1, no binding to ConA was observed for S-layer proteins isolated from Δ_pmt1_ P. UF1 (Fig. 1g). To underscore
the role of Pmt1 in the glycosylation of LspA, this protein was overproduced by Δ_lspA_ P. UF1 and Δ_pmt1_ P. UF1 strains to isolate and purify the glycosylated LspA (G-LspA) and
non-glycosylated LspA (NG-LspA), respectively (Fig. 1h). While both G-LspA and NG-LspA were recognized by anti-LspA serum antibodies, only purified G-LspA bound to ConA and illuminated
staining for glycoprotein (Fig. 1i). Thus, Pmt1 is critically required for the glycosylation of LspA. _O_-MANNOSYLATED LSPA INTERACTION WITH SIGNR1 To elaborate on the nature of LspA
glycosylation, the purified G-LspA and NG-LspA proteins were treated with PNGase F to release any N-glycans, permethylated and analyzed by MALDI-MS. Here, no _N_-linked glycans were detected
in either of the LspA proteins (Supplementary Fig. 1c). The _O_-linked glycans were released by β-elimination procedure and permethylated prior to MALDI-MS analysis. Signals corresponding
to Hex1-Hex6 were observed in the G-LspA protein (Fig. 2a), but not in the NG-LspA (Supplementary Fig. 1d). Furthermore, glycan compositional analysis demonstrated that mannose (Man) was the
major monosaccharide of G-LspA, with a retention time of 10.9 min (Fig. 2b). In contrast, NG-LspA showed no traces of Man (Supplementary Fig. 1e). Note that a minor peak of glucose was also
detected in both samples. However, glucose, as a very common contaminant, could be derived from reagents and detected as a free and minor glucose peak in HPAEC analysis. Moreover, glycomic
analysis of released oligosaccharides demonstrated that Man3 was the major glycan in the G-LspA, comprising 77% of the total glycans. While Man2 and Man4 oligosaccharides were minor glycans,
only traces of Man1, Man5 and Man6 were detected in the G-LspA (Fig. 2c). The purified G-LspA protein was then digested with trypsin and elastase, resulting in peptides with >75%
coverage (Supplementary Fig. 1a). LC-MS/MS analysis of the enriched peptides revealed seven _O_-glycopeptides at the N-terminus of LspA (Fig. 2d). In addition to the 41 threonine/serine
residues involved in Man attachment, adjacent proline and alanine residues that may facilitate local conformational changes for protein _O_-glycosylation17 were also found in all the
glycopeptides (Fig. 2d). Further, GC-MS analysis was performed to investigate the glycosyl linkages and positions of released _O_-glycans. Data demonstrated that Man oligosaccharides of LspA
were short linear chains interconnected via (1 → 6)-linkage and (1 → 2)-linkage (Fig. 2e). Man(1 → 6)Man(1 → 2)Man was the major trisaccharide component in the G-LspA, while disaccharide
Man(1 → 6)Man and tetrasaccharide Man(1 → 6)Man(1 → 2)Man(1 → 2)Man comprised a small percentage (Fig. 2e). These data indicate that LspA is a mannosylated S-layer glycoprotein with linear
short-chain _O_-glycans. SIGNR1 expressed by myeloid DCs recognizes characteristic molecular patterns with complex mannose and fucose structures in bacteria and fungi.18 Recently, we
observed the binding of P. UF1 to SIGNR1, but not SIGNR3.9 To precisely delineate the role of G-LspA binding to SIGNR1 in regulating DCs to subsequently initiate T cell commitment, the
interaction of G-LspA with SIGNR1 was biochemically investigated. Here, G-LspA and NG-LspA proteins were first separated by SDS-PAGE, transferred to PVDF membrane, and then incubated with
SIGNR1-hFc fusion protein. This protein binding complex was analyzed by subsequent incubation with anti-human Fc secondary antibody. Data demonstrated that the purified G-LspA bound
specifically to SIGNR1-hFc, and this binding was abolished in the presence of EDTA (Fig. 2f). Further, SIGNR1 interaction with G-LspA was assessed by ELISA showing G-LspA binding to
SIGNR1-hFc (Fig. 2g), but not to Dectin-1-hFc used as a control fusion protein (Fig. 2h). This binding was completely blocked by pre-incubation of SIGNR1-hFc with anti-SIGNR1 antibody, or
with the competitive ligand zymosan that is composed of β-glucan, α-mannan and mannosyl proteins (Fig. 2g). In contrast, no binding was observed for NG-LspA using similar assays (Fig. 2g).
Furthermore, the G-LspA exhibited a dose-dependent binding with SIGNR1-hFc using protein concentrations ranging from 0.08 μg/ml to 10 μg/ml with a _K__d_ value of 2.617 μg/ml G-LspA (Fig.
2i). In contrast, NG-LspA did not react with SIGNR1-hFc, even with higher protein concentrations up to 20 μg/ml (Fig. 2i), highlighting the specificity of SIGNR1 binding to glycosylated
LspA. ACTIVATION OF COLONIC DCS BY GLYCOSYLATED LSPA IN STEADY STATE SIGNR1 was majorly expressed by colonic CD11chi MHCIIhi DCs (Supplementary 3a, b). To shed light on the relevance of LspA
glycosylation interacting with colonic DCs, CD11chi MHCIIhi CD11b+ F4/80_−_ DCs were FACS sorted (Supplementary Fig. 4a, b) from mice gavaged with P. UF1 or Δ_lspA_ P. UF1 to analyze their
transcriptome by RNA-Seq. Data demonstrated the modulation of costimulatory molecules (_Cd40_, _Cd80_, _Cd86_, and _Tnfsf4_) in colonic DCs by P. UF1 (Fig. 3a). NF-κB signaling (_Casp4_,
_Traf1_, _Tnfrsf1b_, _Mapk6_, _Nfkbie_, and _Nfkbiz_), cytokine/chemokine transcripts (e.g., _Il1b, Il12b, Cxcl1_, and _Cxcl2_), and antigen presentation-related genes (e.g., _Serpinb9_,
_Rab8b_) were also significantly augmented in DCs derived from mice gavaged with P. UF1 compared to Δ_lspA_ P. UF1. In contrast, DCs derived from Δ_lspA_ P. UF1-gavaged mice showed
activation of _Sod3_, _Rhoh_ and _Klf2_, which may instruct functional suppression in these cells. Migrating DCs constitutively express genes with regulatory functions.19 Accordingly, DCs
derived from mice gavaged with P. UF1 had elevated expression of genes, such as _Cd274_, _Spred1_, _Etv3_, _Tnfnip2_, _Stat3_, _Stat4_, and _Stat5a_. These cells were also enriched with
genes implicated in DC development (_Edn1, Cish_), migration (_Nrp2_, _Ccr10_, _Eps8_), and differentiation (_Pdk1_, _Hilpda_), while DCs of Δ_lspA_ P. UF1-gavaged mice exhibited increased
quantities of genes suppressing cellular regulatory functions (e.g., _Cyr61_, _Sdc1_). In addition, transcription factor _Irf4_ controlling Th17 cell cytokine machinery,20 cell cycle
inhibitor _Cdkn1a_ involved in Treg cell formation,21 and T cell-attracting chemokines _Ccl17_ and _Ccl22_, were all significantly activated in DCs of P.UF1-gavaged mice. In contrast,
Δ_lspA_ P. UF1 enhanced the DC expression of _Cd55_ and _Gilz_ genes associated with suppression of T cell function.22,23 Gene set enrichment analysis (GSEA) demonstrated that gene sets for
metabolic activities [e.g., glycolysis and oxidative phosphorylation (OXPHOS)] and DC activation, including activation of innate responses, regulation of I-κB/NF-κB signaling, cytokine
receptor activity and cell migration, were enriched in DCs derived from mice gavaged with P. UF1 (Fig. 3b). In contrast, glycosylated LspA deficiency abrogated cellular protein responses in
DCs, resulting in enhanced endoplasmic reticulum (ER) stress-associated activities (Fig. 3b), which were associated with increased transcripts of unfolded protein response (UPR)-related
genes _Xbp1_, Derl3, and _Edem2_ in these cells (Fig. 3a). Further, P. UF1-induced DC activation (e.g., _Cd80_, _Cd86_, _Il1b_, and _Il12b_) was abrogated due to _Signr1_ deficiency (Fig.
3c). Moreover, loss of LspA glycosylation (Δ_lspA_ P. UF1-gavaged _Signr1__+/+_ mice) mirrored the deficiency of SIGNR1 (P. UF1-gavaged _Signr1__−_/_−_ mice), suggesting a role of
glycosylated LspA-SIGNR1 interaction in DC activation. Having shown that LspA deficiency impacted the transcriptome of DCs, we asked whether LspA deficiency would also influence the
regulation of T cell polarization in steady state. Thus, C57BL/6 mice were gavaged with P. UF1 or Δ_lspA_ P. UF1 to analyze colonic DC and T cell responses. Data demonstrated that Δ_lspA_ P.
UF1 compared to P. UF1 significantly decreased DC and T cell responses, including IL-10+ Th17 cells and 10+ Tregs (Supplementary Fig. 5a, b). Notably, no difference in intestinal bacterial
colonization was observed in mice gavaged with P. UF1 or Δ_lspA_ P. UF1, as both strains transiently colonized conventional mice (Supplementary Fig. 5c), suggesting that glycosylated LspA is
not involved in bacterial colonization, but instructs DC activation to polarize T cells. PROGRAMMING TRANSCRIPTOMIC AND METABOLOMIC DC-MACHINERIES DURING INTESTINAL INFECTION Intestinal DCs
rapidly respond to invading pathogens.24 However, these cells may be functionally conditioned by pathological surrounding signals induced by intestinal infections.25 To elaborate on the
bacterial glycosylated LspA influencing DC transcriptome, colonic MHCIIhi CD11chi CD11b+ F4/80_−_ DCs were FACS sorted from mice gavaged with P. UF1 or Δ_lspA_ P. UF1 and then orally
infected with Δ_actA L. m_. RNA-Seq analysis demonstrated that P. UF1 controlled the expression of genes associated with DC activation (e.g., _Cstl_, _Tsc1_) and suppression of TGF-β
signaling (e.g., _Tgbfi_, _Eaf2_, and _Cited2_) (Fig. 4a). Further, transcripts of _Tnip3_ regulating NF-κB activation and _Dok-2_ suppressing Ras-Erk signaling were stimulated in colonic
DCs isolated from mice gavaged with P. UF1. Importantly, _Cd209b_ (_Signr1_), whose activation highly depends on the mannosylation of glycans, was upregulated in DCs derived from mice
gavaged with P. UF1. In contrast, Toll-like receptors (_Tlr3_, _Tlr4_, and _Tlr12_) associated with proinflammatory response were upregulated in DCs of mice gavaged with Δ_lspA_ P. UF1 (Fig.
4a, b). _Mx1_ and _Irf1_, selectively activated during TLR-induced DC stimulation,26 were also enriched in DCs derived from Δ_lspA_ P. UF1-gavaged mice. Consistently, a proinflammatory
status was readily observed in DCs of mice gavaged with Δ_lspA_ P. UF1. Accordingly, a set of proinflammatory genes (e.g., _Ifng_, _Tnfsf15_, and _Ifi204_), several genes associated with
cell apoptosis (e.g., _Daxx_, _Nab2_, and _Fbxw7_), and stress-associated activities were activated in DCs derived from mice gavaged with Δ_lspA_ P. UF1 (Fig. 4a, b). In addition, GSEA
demonstrated enhanced mitochondrial biogenesis in DCs isolated from P. UF1-gavaged mice, as indicated by enriched gene sets for OXPHOS, TCA cycle and respiratory chain, and NADH
dehydrogenase complex (Fig. 4c). Moreover, pathways regulating apoptosis and protein localization to ER were also enhanced in these cells. In contrast, DCs from Δ_lspA_ P. UF1-gavaged mice
exhibited dysregulated protein folding and ER stress-associated activities (Fig. 4c), consistent with increased transcripts of the UPR-related genes _Xbp1_ and _Derl3_ in these cells (Fig.
4a, b). Inflammatory DC response is associated with declined mitochondrial activity promoted by TLR signaling.27 Having demonstrated that glycosylated LspA controlled TLR-activation and
enriched gene sets for OXPHOS (Fig. 4), we further assessed whether P. UF1 expressing glycosylated LspA would maintain mitochondrial respiration during intestinal infection. Thus, colonic
DCs were enriched by magnetic beads from mice gavaged with P. UF1 or Δ_lspA_ P. UF1 and orally infected with Δ_actA L.m_ to analyze the real-time changes in the rate of extracellular
acidification (ECAR), a measurement of glycolysis, and the mitochondrial rate of oxygen consumption (OCR). While no difference in the ECAR was observed, OCR was significantly decreased in
enriched DCs derived from mice gavaged with Δ_lspA_ P. UF1 compared to DCs derived from P. UF1-gavaged mice (Fig. 5a). These data support the notion that glycosylated LspA may regulate DC
metabolic function during intestinal infection. It was recently demonstrated that DC function can be impacted by cellular metabolic factors that facilitate the biosynthetic and bioenergetic
needs of these cells.28 To elaborate on this notion, colonic MHCIIhi CD11chi CD11b+ F4/80_−_ DCs were FACS sorted from mice gavaged with P. UF1 or Δ_lspA_ P. UF1 and orally infected with
Δ_actA L. m_ to analyze the metabolomic activity of these cells. Here, a distinct metabolome was identified in DCs derived from P. UF1-gavaged mice compared to those derived from Δ_lspA_ P.
UF1-gavaged mice (Fig. 5b). Metabolic pathways, including arginine and proline metabolism, purine metabolism and _de novo_ fatty acid biosynthesis, were differentially activated in DCs
isolated from the aforementioned groups of mice (Fig. 5c). Putatively annotated eicosatrienoic acid, known as an anti-inflammatory metabolite, 4-aminobutanal indicative of anti-inflammatory
putrescine metabolism, and proline involved in suppressing reactive oxygen species (ROS),29 were markedly accumulated in DCs from P. UF1-gavaged mice compared to the other group (Fig. 5d).
In contrast, putative metabolites associated with proinflammation and energy starvation [e.g., methylimidazoleacetic acid and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR)] and cell
apoptosis-inducing deoxyadenosine were significantly enriched in DCs derived from Δ_lspA_ P. UF1-gavaged mice (Fig. 5d). Thus, these data specifically demonstrate the requirement of
glycosylated LspA expressed by P. UF1 that programs the regulation of DC function during _Listeria_-induced inflammation. REGULATING PROTECTIVE T CELL RESPONSE BY P. UF1 EXPRESSING
GLYCOSYLATED LSPA To further elucidate the functional relevance of glycosylated LspA in modulating colonic homeostasis during intestinal infection, C57BL/6 mice were gavaged with P. UF1,
Δ_lspA_ P. UF1 or PBS, and then orally infected with Δ_actA L. m_. Notably, P. UF1, compared to Δ_lspA_ P. UF1, regulated proinflammatory responses by controlling the frequencies and cell
counts of IL-1β+, IL-6+, and IL-12/23p40+ DCs (Fig. 6a). However, such induced DC regulation was abolished in _Signr1__−/−_ mice gavaged with P. UF1, Δ_lspA_ P. UF1 or PBS and subsequently
infected with Δ_actA L. m_ (Fig. 6a), denoting a potential role of glycosylated LspA-SIGNR1 interaction in controlling inflammatory DC response. Further, tuned DC response was associated
with T cell regulation, leading to an increased percentage and number of Th17 and IL-10+ Treg cells in mice gavaged with P. UF1 compared to Δ_lspA_ P. UF1 or PBS (Fig. 6b). Loss of
protective immunity in _Signr1__+/+_ mice gavaged with Δ_lspA_ P. UF1 resulted in delayed Δ_actA L. m_ clearance compared to mice gavaged with P. UF1 (Fig. 6c). In addition, transcripts of
proinflammatory molecules such as _Il6_, _Il12b_, _Ifng_, _Ccl5_, and _Cxcl2_ were significantly decreased in colonic tissues of _Signr1__+/+_ mice gavaged with P. UF1 compared to those
gavaged with Δ_lspA_ P. UF1 or PBS (Fig. 6d). In contrast, the protective T cell immunity was blunted in _Signr1__−/−_ mice gavaged with P. UF1, Δ_lspA_ P. UF1 or PBS and then infected with
Δ_actA L. m_ (Fig. 6b), and no difference in pathogen clearance was observed in these groups of mice (Fig. 6c). Furthermore, data demonstrated that deficiency in bacterial glycosylated LspA
did not induce any changes in the gut microbial composition of mice gavaged with Δ_lspA_ P. UF1 compared to P. UF1 during Δ_actA L. m_ infection (Supplementary Fig. 6a, b). Together, these
results indicate the crucial role of glycosylated LspA-SIGNR1 interaction in regulating colonic DCs to orchestrate protective T cell response to intestinal _Listeria_ infection. AMELIORATING
DEXTRAN SULFATE SODIUM (DSS)-INDUCED COLITIS BY P. UF1 EXPRESSING GLYCOSYLATED LSPA IL-17A and IL-10+ Tregs contribute to protection against acute intestinal colitis.30,31,32 To further
investigate the protective role of glycosylated LspA expressed by P. UF1 in chemically induced colitis, groups of mice were gavaged with P. UF1, Δ_lspA_ P. UF1 or PBS, and the experimental
colonic tissue damage was induced by 3% DSS. Data demonstrated that P. UF1, compared to other groups of mice, significantly reduced the disease severity, as indicated by the reduction of
weight loss, diarrhea and fecal blood scores (Fig. 7a, b), and increased colon length (Fig. 7c). Furthermore, severe signs of inflammation-induced thickening of the bowel wall and loss of
regular blood vessel structures were observed in the colons of PBS- and Δ_lspA_ P. UF1-gavaged mice but not in P. UF1-gavaged mice (Fig. 7d). Histological analysis demonstrated severe tissue
destruction, characterized by extensive segmental to diffuse mucosal epithelium and crypt loss with associated mucosal and submucosal inflammation in the colonic tissues of mice gavaged
with PBS or Δ_lspA_ P. UF1, while DSS-induced colitis was substantially mitigated in mice gavaged with P. UF1 (Fig. 7e, f). Furthermore, obtained data also demonstrated that DSS-treated mice
gavaged with P. UF1 were markedly protected from leaky gut when compared with other groups, as documented by FITC-dextran assay (Fig. 7g). Consistently, enhanced transcripts of tight
junction proteins (_Cldn2_, _Cldn3_, _Cldn7_, _Cldn8_, and _ZO-3_) in colonic tissues of P. UF1-gavaged mice were observed compared to the other groups (Fig. 6h). Thus, glycosylated LspA
expressed by P. UF1 contributes to the maintenance of intestinal barrier integrity. To further elucidate the protective role of glycosylated LspA-SIGNR1 signaling in DSS-induced colitis,
_Signr1__−/−_ mice were gavaged with P. UF1, Δ_lspA_ P. UF1 or PBS and treated with DSS. Once again, lack of SIGNR1 interaction with glycosylated LspA resulted in no improvement of
colitogenic disease progression in any of the groups of mice (Fig. 7a–f). Here, the disruption of glycosylated LspA-SIGNR1 interaction abolished the regulation of intestinal permeability
(Fig. 7g) and blunted the expression of tight junction proteins in _Signr1__−/−_ mice (Fig. 7i). Collectively, these data suggest that glycosylated LspA interacting with SIGNR1 sustains gut
homeostasis to protect against detrimental signals inducing tissue damage. DISCUSSION The maintenance of gut homeostasis requires a rigidly balanced dialog between the resident microbes and
the host.1 This can be established through the interaction of a variety of bacterial gene products with their cognate receptors expressed by phagocytic cells.6 Disruption of such elegantly
induced communication may result in pathogenic inflammation and intestinal tissue damage.33 In this study, we demonstrate a protective mechanism at the intestinal interface whereupon
glycosylated LspA interacting with SIGNR1 dictates DC response that in turn regulates protective T cells against intestinal pathogen infection and chemically induced colitis. There has
recently been a growing interest in the bacterial S-layer proteins with multiple regulatory properties.34,35 Accordingly, the composition of the S-layer proteins of _Propionibacterium_
species is remarkably variable, and the functions of bacterial S-layer proteins are strain-dependent.36 For instance, surface layer protein B (SlpB), highly expressed by some of _P.
freudenreichii_ strains, facilitates bacterial adhesion to epithelial cells.37 While no SlpB homolog is found in P. UF1, LspA constitutes the major extractable S-layer protein in this
bacterium. The high protein expression may also suggest that LspA, particularly in P. UF1, is a valuable factor for not only supporting the S-layer structure of this bacterium but could also
be important for fine-tuning intestinal immunity and may serve as an elegant vehicle for mucosal vaccine and therapeutic approaches.38 Although LspA is conserved, the levels of protein
expression vary dramatically in different _Propionibacterium_ strains,35 indicating potentially differential gene regulation in these bacterial strains. With this notion in mind and the
unique expression pattern of LspA by P. UF1, we were prompted to further elaborate on its physiological characteristics and its potential implication in regulating intestinal immunity. Here,
we demonstrate that deletion of _lspA_ leads to fundamental changes in pathways associated with S-layer carbohydrate metabolisms, including mannosylation, suggesting the critical role of
LspA in maintaining the S-layer glycosylation profile. Indeed, ConA binding assays comparing S-layer proteins isolated from P. UF1 and Δ_lspA_ P. UF1 show that glycosylation of other
potential glycoproteins is also likely impaired by LspA deficiency. Further, changes in S-layer glycosylation may directly impact the bacterial cell-cell interactions, leading to enhanced
autoagglutination due to LspA deficiency in P. UF1, possibly as a result of altered cell surface hydrophobicity. This is consistent with the observations that surface glycosylation, such as
flagella glycosylation, is highly associated with autoagglutination, which is an important step for microcolony formation on the intestinal epithelial cells that contributes to intestinal
colonization.39,40 In this study, we also clearly demonstrate that LspA is an _O_-mannosylated glycoprotein, uniquely representing the first glycoprotein characterized in _Propionibacteria_.
Furthermore, the glycosylation of LspA critically requires Pmt1 activity. This feature is reminiscent of protein glycosylation in _Streptomyces_ and _Mycobacterium_, whereupon the
_O_-glycosylation of surface lipoproteins or phosphate-binding protein PstS, are highly dependent on the membrane-associated lipoprotein Pmt.41 Thus, the Pmt-mediated protein
_O_-mannosylation seems to be a general pathway in actinomycetes. In fungi and yeasts, up to seven Pmt family members have been identified thus far, and the homomeric and/or heteromeric
interactions among Pmt members cooperatively initiate the protein glycosylation, resulting in the biosynthesis of diversified glycan structures.42 However, Pmt1 seems to be the only protein
glycosyltransferase in P. UF1, and no other protein glycosylation is observed in the S-layer proteins isolated from Δ_pmt1_ P. UF1. Furthermore, the linear short-chain mannose is found to be
the major glycan structure of bacterial LspA; thus, concluding that Pmt1 is the crucial enzyme responsible for modifying S-layer proteins, particularly LspA, with simple mannoses in P. UF1.
Recognition of microbial gene products by sensing receptors, including C-type lectins, is essential for translating the nature of microbes into gene-transcriptional and metabolic programs
that may initiate the regulation of DC function to prime T cell polarization.43 SIGNR1, a murine homolog of human DC-SIGN, conditions intestinal DCs for the induction of oral tolerance44 and
plays a key role in host defense against pathogen infection.45 However, further rigid molecular studies are still required to mechanistically elucidate the gene and metabolic programs that
are modulated by the interactions of this sensing receptor with the bacterial surface glycosylation. Here, we demonstrate that LspA glycosylation is required to be recognized by its
receptor, SIGNR1, leading to the regulation of DC activation in steady state and during pathogen-induced inflammation. Accordingly, in steady state P. UF1, via its glycosylated LspA, seems
to optimally induce the activation of colonic DCs when compared with its counterpart, Δ_lspA_ P. UF1. Mainly, P. UF1 regulates the expression of costimulatory, cytokine and antigen
presentation molecules (e.g., _Cd40_, _Il1b, Serpinb9_, _Rab8b,_ and _Bcl3_) in colonic DCs while glycosylated LspA deficiency results in the activation of suppressive molecules (e.g.,
_Sod3_, _Rhoh_ and _Klf2, Cyr61_, _Sdc1_) that may dysfunction colonic DCs to properly induce T cell differentiation. More importantly, P. UF1 decorated with glycosylated LspA regulates DCs
that control T cell response to pathogen infections. Here, regulatory signaling appears to be activated by LspA glycosylation, as _Tgfbi_, _Eaf2_, and _Cited2_ genes associated with
suppression of TGF-β signaling are tuned down in DCs derived from mice gavaged with P. UF1 compared to Δ_lspA_ P. UF1. Further, proinflammatory genes (e.g., _Ifng_ and _Ifi204_) are also
downregulated in DCs derived from mice gavaged with P. UF1. Consistently, anti-inflammatory polyunsaturated fatty acid eicosatrienoic acid is highly enriched in DCs of P. UF1-gavaged mice,
while proinflammatory metabolites, such as methylimidazoleacetic acid and AICAR, are significantly elevated in DCs of Δ_lspA_ P. UF1-gavaged mice during _Listeria_ infection. In addition,
DCs derived from Δ_lspA_ P. UF1-gavaged mice exhibit significant quantities of genes associated with cell apoptosis and stress response, consistent with decreased cellular proline levels
that are important for protecting against various cellular stresses.29 This may emphasize the impaired DC metabolism induced by glycosylated LspA deficiency, which is further supported by
reduced mitochondrial respiration and enriched metabolite AICAR that serves as a metabolic activator responding to energy starvation of the cells. Collectively, these data suggest that LspA
glycosylation may be an important factor that modulates colonic DC response via transcriptomic and metabolic reprogramming. Proinflammatory DC regulation may impact T cell polarization.7
Indeed, dysregulated cellular pathways in DCs induced by Δ_lspA_ P. UF1 significantly reduce the protective T cell immunity and correspondingly manifest in uncontrolled intestinal pathogen
infection, resulting in delayed pathogen clearance and enhanced intestinal inflammation. In addition, no protection is seen in mice when either SIGNR1 or its ligand, glycosylated LspA, is
deficient, indicating the critical role of the glycosylated LspA-SIGNR1 axis in regulating colonic DC response that dictates protective T cell response against pathogen infection. We have
previously shown that P. UF1 induces DlaT-specific Th17 cells, which are indispensable for protection against _Listeria_ infection.9,11,12 While DlaT expression is not impacted by _lspA_
deletion (Supplementary Fig. 2c), induced Th17 cells notably require a regulated cytokine environment initiated by DCs through glycosylated LspA interacting with SIGNR1. Interestingly,
glycosylated LspA deficiency does not impact the phylum of gut microbiota in bacterially-gavaged groups of mice and then infected with _Listeria_, suggesting that glycosylated LspA may not
contribute to any changes in the intestinal bacterial community. Yet, do changes in bacterial metabolome due to glycosylated LspA deficiency contribute to reduced regulation of DCs during
intestinal infection? Although critical metabolic changes are observed within Δ_lspA_ P. UF1, we posit that these metabolites may be mostly restricted to intracellular bacterial metabolic
networks in response to altered S-layer glycosylation and may not directly impact the immune cells. Nonetheless, this important notion still requires further rigid investigations. Ample data
demonstrate that IL-17A serves as a protective factor against DSS-induced colitis by maintaining intestinal tight junctions and promoting epithelial repair.31,46 Here, Th17 cells, together
with IL-10+ Tregs, regulated by colonic SIGNR1+ DCs interacting with glycosylated LspA, may be required for ameliorating DSS-induced colitis, all of which potentially result in the
maintenance of tight junction expression that controls the intestinal permeability and the mitigation of chemically induced inflammation and intestinal tissue damage. In summary, our data
demonstrate the molecular mechanism through which DC functions can be fine-tuned via bacterial glycosylated LspA interacting with SIGNR1. Such a regulated DC response is pivotal in priming
protective T cell response to intestinal infection and plays a critical role in mitigating DSS-induced colitis in mice. Induced immune regulatory processes involving the fine-tuned
receptor-ligand interaction, mainly glycosylated LspA-SIGNR1, elicits functional gut homeostasis during the intestinal inflammatory condition. Thus, consistent with this finding,
glycosylation of a bacterial gene product such as LspA may be an important feature for a future formulated prebiotic, conjugated vaccines and therapeutic targets that not only induce the
regulation of innate and protective T cells against intestinal pathogen challenges, but may also prevent induced pathogenic inflammation that triggers tissue damage and the progression of
intestinal proinflammatory diseases (e.g., colitis) in affected patients.47,48 Thus, a deeper understanding of beneficial bacterial gene products influencing the regulation of gut
homeostasis may pave the way for the development of new pre or postbiotic therapeutic strategies to potentially treat inflammatory bowel disease (IBD) or colon cancer.49,50 Finally, our work
uniquely illustrates the significance of the mannosylated ligand, bacterial LspA, and its critical binding to SIGNR1 that sustains the optimal activation and regulation of colonic DCs in
intestinal steady state and during inflammatory condition. Conclusively, shedding light on the relevance of a sensing receptor, SIGNR1 and its human homolog, DC-SIGN, may reveal the critical
innate factor in resisting detrimental signals inducing tissue damage that manifest in IBD. Thus, activating critical signals involving SIGNR1 (DC-SIGN) in health and human diseases may
advance our vision to improve and develop therapeutic platforms mitigating IBD, which increasingly affects more than 3.5 million worldwide.51 MATERIALS AND METHODS MICE C57BL/6 mice (6–9
weeks old) were obtained from Jackson Laboratory and maintained under specific pathogen-free, _Helicobacter_-free conditions. _Signr1__−/−_ mice were provided by Huang Shau-Ku (Johns Hopkins
University School of Medicine, Baltimore, Maryland, USA). All animal studies were approved by the Animal Care and Use Committee of the University of Florida under the protocol number
201708484. Mice were maintained in accordance with the Animal Welfare Act and the Public Health Policy on Humane Care. BACTERIAL ADMINISTRATION In steady state, _Signr1__+/+_ and
_Signr1__−/−_ mice were gavaged with P. UF1 or Δ_lspA_ P. UF1 (109 CFU/mouse/100 μl) every 3 days over the course of 12 days, and mice were euthanized on day 14 to isolate colonic immune
cells. During Δ_actA L. m_ infection, _Signr1__+/+_ and _Signr1__−/−_ mice were gavaged with P. UF1, Δ_lspA_ P. UF1 (109 CFU/mouse/100 μl) or PBS on days −7, −4, −1, and 2. Mice were denied
food for 4 h on day 0 and then orally infected with 100 μl PBS containing 50 mg/ml CaCO3 and approximately 109 CFU of Δ_actA L. m_. Fecal samples were collected on days 1–4, and mice were
sacrificed on day 7. Pathogen loads were determined by plating serial dilutions of fecal samples on BHI agar supplemented with 200 μg/ml streptomycin. CELL ISOLATION AND FLOW CYTOMETRY
Density gradient centrifugation using Percoll was performed to isolate lamina propria cells from mouse colon. Briefly, tissues were collected and fecal contents in the colon were carefully
removed. Colons were opened longitudinally and cleaned with cold PBS and then shaken in PBS containing 20 mM Hepes and 10 mM EDTA for 30 min at 37 °C. Tissues were cut into small pieces and
incubated with digestion solution [RPMI 1640 containing 10% FBS (Thermo Fisher Scientific, Waltham, MA), 0.4% β-mercaptoethanol, 400 U/ml collagenase VIII (Sigma Aldrich, St. Louis, MO) and
100 μg/ml DNase I (Sigma Aldrich, St. Louis, MO)] for 1.5 h at 37 °C in a 5% CO2 incubator. Digested tissues were filtered through 100 μm cell strainer (Genesee Scientific, San Diego, CA),
and cells were resuspended in 5 ml of 40% Percoll (Sigma Aldrich, St. Louis, MO) and overlaid on 5 ml of 80% Percoll. Cells in the interphase were collected after gradient centrifugation
(1258 g, 25 min, 25 °C). Flow cytometry was performed as described previously9 with some modifications. Isolated cells were stimulated with 50 ng/ml PMA (Sigma Aldrich, St. Louis, MO) and
500 ng/ml ionomycin calcium salt (Sigma Aldrich, St. Louis, MO) for 4 h and 5 μg/ml brefeldin A (BioLegend, San Diego, CA) was added 2 h before cells were harvested. Stimulation was
performed in IMDM medium (Sigma Aldrich, St. Louis, MO) containing 10% FBS, 1% penicillin/streptomycin, 0.4% β-mercaptoethanol, 10 mM Hepes and 2 mM L-Glutamine. Dead cells were detected
using LIVE/DEAD® Fixable Blue Dead Cell Stain Kit (Thermo Fisher Scientific, Waltham, MA), followed by incubation with Mouse Fc Blocking Reagent (Miltenyi Biotec, Auburn, CA). Cells were
first stained for cell-surface markers and then resuspended in fixation/permeabilization solution [Cytofix/Cytoperm kit (BD Biosciences, San Diego, CA) for DC cytokine analysis, or Foxp3
Transcription Factor Staining Buffer Set (eBiosciences, San Diego, CA) for T cell analysis]. Cells were stained with following fluorescent antibodies: eVolve 655-CD45 (catalog 86-0451-42),
eFlour506-CD11c (catalog 69-0114-80), PE-MHCII (catalog 12-5321-82), PE/Cy5-F4/80 (catalog 15-4801-80), APC-SIGNR1 (catalog 17-2093-82), PE/Cy5-CD8 (catalog 15-0081-83), PE/Cy7-Pro-IL-1β
(catalog 25-7114-82)/rat IgG1 κ, FITC-IL-6 (catalog 11-7061-82)/rat IgG1 κ, eFlour 450-IL-12/23p40 (catalog 48-7123-82)/mouse IgG1 κ, and eFlour450-FoxP3 (catalog 48-5773-82)/rat IgG2a κ
from Thermo Fisher Scientific; APC/Cy7-CD11b (catalog 101226), PerCP/Cy5.5-CD64 (catalog 139308), APC/Cy7-CD3 (catalog 100330), Brilliant Violet 605-CD4 (catalog 100548), PE-IL-17A (catalog
506904)/rat IgG1 κ, and FITC-IL-10 (catalog 505006)/rat IgG2b κ from BioLegend. Data were collected by an LSR II Fortessa (BD Biosciences, San Jose, CA) and analyzed with FlowJo software
(version 10) (TreeStar, Ashland, OR). After dead and doublet cell exclusion, and the subsequent CD45+ selection, DCs were defined as CD11chi MHCIIhi CD11b+ F4/80_−_ (Supplementary Fig. 7a)
and T cells as CD3+ CD8_−_ CD4+ (Supplementary Fig. 7b). DC SORTING Colonic cell suspensions were prepared from mice gavaged with P. UF1 or Δ_lspA_ P. UF1 in steady state or during
intestinal Δ_actA L. m_ infection. Cells were labeled with a cocktail of fluorescent antibodies specific for: PE/Cy7-CD11c (BioLegend, catalog 117318), PE-MHCII (Thermo Fisher Scientific,
catalog 12-5321-82), APC-CD11b (BioLegend, catalog 101212), FITC-F4/80 (Thermo Fisher Scientific, catalog 11-4801-82). Dead cells were identified and excluded using LIVE/DEAD Fixable Violet
Dead Cell Stain (Thermo Fisher Scientific, Waltham, MA). CD11chi MHCIIhi CD11b+ F4/80_−_ DCs (Supplementary Fig. 4a) were isolated using a SONY SH800S Cell Sorter (Sony, Tokyo, Japan). The
purity of sorted cells analyzed by flow cytometry was determined to be >98% (Supplementary 4b). Subsequently, cell pellets were resuspended in RLT plus buffer (Qiagen, Germantown, MD) for
RNA extraction or snap-frozen in liquid nitrogen for metabolomic analysis. RNA-SEQ ANALYSIS Total RNA was extracted from about 1 × 104 CD11chi MHCIIhi CD11b+ F4/80_−_ DCs isolated from each
individual mouse using an RNeasy Plus Micro Kit (Qiagen, Germantown, MD). cDNA was generated using a SMART-Seq HT kit (Takara Bio Inc., Mountain View, CA) and RNA-Seq libraries were
constructed using a Nextera XT DNA Library Preparation Kit (Illumina, Inc., San Diego, CA). Barcoded samples were sequenced on an Illumina HiSeq instrument (Illumina, Inc., San Diego, CA) at
the University of Florida ICBR NextGen DNA Sequencing Core Facility. The sequencing reads were mapped to the _Mus musculus_ genome (NCBI GRCm38/mm10) using STAR aligner (v2.6.0), and count
table was generated using SubReads featureCounts (v1.6.0). Significantly altered genes (RPKM ≥ 1, FDR _P_ < 0.05, fold change ≥ 1.5) were identified by DESeq2.
Regularized-log-transformation of count data was performed for heatmap plotting. Gene set enrichment analysis (GSEA) was performed in the javaGSEA (v3.0) using GO, Hallmark, KEGG and
REACTOME database. EXTRACELLULAR FLUX ANALYSIS Colonic DCs were isolated from mice gavaged with P. UF or Δ_lspA_ P. UF1 and orally infected with Δ_actA L. m_ using a Pan-DC Enrichment Kit
(StemCell Technologies, Vancouver, Canada). For real-time analysis of extracellular acidification rate (ECAR) and oxygen consumption rate (OCR), DCs were analyzed using an XFe96
Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA) as described.52 Briefly, enriched colonic DCs (3 × 105 cells/well, pooled from 3 mice) were analyzed in non-buffered
RPMI medium supplemented with 2.5 μM dextrose, 2 nM glutamine, and 1 μM sodium pyruvate. ECAR and OCR were analyzed in response to 1 μM oligomycin, 1.25 μM fluoro-carbonyl cyanide
phenylhydrazone (FCCP), 1 μM rotenone and 1 μM antimycin A. HIGH-RESOLUTION METABOLOMICS ANALYSIS FACS sorted colonic CD11chi MHCIIhi CD11b+ F4/80_−_ DCs were isolated from mice gavaged with
P. UF1 or Δ_lspA_ P. UF1 and orally infected with Δ_actA L_. _m_. Note: colonic cells of 2 mice were combined to obtain 60,000–80,000 DCs/sample. After vortex and incubation with
acetonitrile-water (2:1) at 4 °C for 30 min, DC samples were centrifuged and the supernatants were analyzed by LC-MS. Each sample was run in triplicates on an Orbitrap Fusion Tribrid Mass
spectrometer with the resolution of 120,000 (Thermo Fisher, San Diego, CA), with dual chromatography using a 5 min C18 reversed-phase chromatography in negative electrospray ionization (ESI)
mode and a 5 min HILIC chromatography in positive ESI mode over a mass-to-charge ratio (_m_/_z_) range of 85–1250. Student’s _t_-test was performed between treatment groups. Subsequently,
metabolic pathway analysis was performed by _Mummichog_ software (v2.0) with default parameters. 786 significant metabolite features in negative mode and 1234 metabolite features in positive
mode were used as input to _Mummichog_. The pathways represented by at least two significant metabolites and enriched at _P_ < 0.05 in positive mode (HILIC column) are presented.
DSS-INDUCED COLITIS _Signr1__+/+_ and _Signr__1−/−_ mice were treated with 3% DSS in drinking water for 5 consecutive days (made fresh every 2–3 days) to induce colitis. Mice were monitored
for disease progression through day 12 (_Signr1__+/+_ mice) or day 10 (_Signr1__−/−_ mice as they are more susceptible to induced colitis) after DSS treatment. For bacterial administration,
mice were orally gavaged with P. UF1, Δ_lspA_ P. UF1 or PBS on days −7, −4, −1, 2, 5, and 8 for a total of 6 gavages. Colitis severity was determined by histopathology. Tissues were fixed,
sectioned, and stained with hematoxylin and eosin (Histology Tech Services, Gainesville, FL). Stained sections were evaluated by a boarded veterinary pathologist. Macroscopic damage in the
colons of DSS-treated mice gavaged with P. UF1, Δ_lspA_ P. UF1 or PBS was visualized with a Multi-Purpose Rigid Telescope attached to a TELE PACK X (Karl Storz-Endoscope, Germany), as
described previously.13 FITC-DEXTRAN GUT PERMEABILITY ASSAY DSS-treated mice were orally gavaged with FITC-dextran 4000 (Sigma-Aldrich, St. Louis, MO), a nonmetabolizable macromolecule that
is used as a permeability probe. All mice were orally gavaged with FITC-dextran (0.6 mg/g mouse weight) and sacrificed 4 h later for serum harvest. Fluorescent intensity in the serum was
measured using a microplate reader (BioTek, Winooski, VT) with an excitation wavelength of 485 nm and an emission wavelength of 519 nm. FITC-dextran concentrations in the mouse sera were
determined from standard curves generated by serial dilution of FITC-dextran. Serum from mice that were not gavaged with the permeability tracer was used as a control and subtracted from the
tested samples. STATISTICAL ANALYSIS Statistical analyses were performed using GraphPad Prism v7.0. Prior to statistical analysis, normality was tested using the Shapiro-Wilk normality
test. Where the groups follow a Gaussian distribution, parametric analyses were performed (2-tailed unpaired _t_ test for 2 variables or one-way ANOVA followed by Tukey’s post-test for 3
variables). Where the groups did not follow a Gaussian distribution, nonparametric analyses were performed (Mann–Whitney _U_ test for 2 variables or Kruskal–Wallis test followed by Dunn’s
post-test for 3 variables). _P_ values lower than 0.05 were considered as significant: *_P_ < 0.05, **_P_ < 0.01, ***_P_ < 0.001, ****_P_ < 0.0001. REFERENCES * Littman, D. R.
& Pamer, E. G. Role of the commensal microbiota in normal and pathogenic host immune responses. _Cell Host Microbe_ 10, 311–323 (2011). Article CAS PubMed PubMed Central Google
Scholar * Lathrop, S. K. et al. Peripheral education of the immune system by colonic commensal microbiota. _Nature_ 478, 250–254 (2011). Article CAS PubMed PubMed Central Google Scholar
* Lebeer, S., Vanderleyden, J. & De Keersmaecker, S. C. Host interactions of probiotic bacterial surface molecules: comparison with commensals and pathogens. _Nat. Rev. Microbiol_ 8,
171–184 (2010). Article CAS PubMed Google Scholar * Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. _Cell_ 124, 783–801 (2006). Article CAS PubMed
Google Scholar * Fagan, R. P. & Fairweather, N. F. Biogenesis and functions of bacterial S-layers. _Nat. Rev. Microbiol_ 12, 211–222 (2014). Article CAS PubMed Google Scholar *
Thaiss, C. A., Zmora, N., Levy, M. & Elinav, E. The microbiome and innate immunity. _Nature_ 535, 65–74 (2016). Article CAS PubMed Google Scholar * Banchereau, J. & Steinman, R.
M. Dendritic cells and the control of immunity. _Nature_ 392, 245–252 (1998). Article CAS PubMed Google Scholar * Medzhitov, R. Recognition of microorganisms and activation of the immune
response. _Nature_ 449, 819–826 (2007). Article CAS PubMed Google Scholar * Colliou, N. et al. Commensal _Propionibacterium_ strain UF1 mitigates intestinal inflammation via Th17 cell
regulation. _J. Clin. Invest._ 127, 3970–3986 (2017). Article PubMed PubMed Central Google Scholar * Littman, D. R. & Rudensky, A. Y. Th17 and regulatory T cells in mediating and
restraining inflammation. _Cell_ 140, 845–858 (2010). Article CAS PubMed Google Scholar * Colliou, N. et al. Regulation of Th17 cells by P. UF1 against systemic _Listeria monocytogenes_
infection. _Gut Microbes._ 279–287 (2018). Article CAS PubMed PubMed Central Google Scholar * Ge, Y. et al. Neonatal intestinal immune regulation by the commensal bacterium, P. UF1.
_Mucosal Immunol._ 12, 434–444 (2019). Article CAS PubMed PubMed Central Google Scholar * Lightfoot, Y. L. et al. SIGNR3-dependent immune regulation by _Lactobacillus acidophilus_
surface layer protein A in colitis. _EMBO J._ 34, 881–895 (2015). Article CAS PubMed PubMed Central Google Scholar * Kordulakova, J. et al. Definition of the first mannosylation step in
phosphatidylinositol mannoside synthesis PimA is essential for growth of mycobacteria. _J. Biol. Chem._ 277, 31335–31344 (2002). Article CAS PubMed Google Scholar * Tatituri, R. V. et
al. Inactivation of _Corynebacterium glutamicum_ NCgl0452 and the role of MgtA in the biosynthesis of a novel mannosylated glycolipid involved in lipomannan biosynthesis. _J. Biol. Chem._
282, 4561–4572 (2007). Article CAS PubMed Google Scholar * Gentzsch, M. & Tanner, W. The PMT gene family: protein _O_-glycosylation in _Saccharomyces cerevisiae_ is vital. _EMBO J._
15, 5752–5759 (1996). Article CAS PubMed PubMed Central Google Scholar * Herrmann, J. L., OGaora, P., Gallagher, A., Thole, J. E. R. & Young, D. B. Bacterial glycoproteins: A link
between glycosylation and proteolytic cleavage of a 19 kDa antigen from _Mycobacterium tuberculosis_. _EMBO J._ 15, 3547–3554 (1996). Article CAS PubMed PubMed Central Google Scholar *
Sancho, D. & Reis e Sousa, C. Signaling by myeloid C-type lectin receptors in immunity and homeostasis. _Annu. Rev. Immunol._ 30, 491–529 (2012). Article CAS PubMed PubMed Central
Google Scholar * Bekiaris, V., Persson, E. K. & Agace, W. W. Intestinal dendritic cells in the regulation of mucosal immunity. _Immunol. Rev._ 260, 86–101 (2014). Article CAS PubMed
Google Scholar * Schlitzer, A. et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. _Immunity_ 38, 970–983 (2013).
Article CAS PubMed PubMed Central Google Scholar * Price, J. G. et al. CDKN1A regulates Langerhans cell survival and promotes Treg cell generation upon exposure to ionizing irradiation.
_Nat. Immunol._ 16, 1060–1068 (2015). Article CAS PubMed PubMed Central Google Scholar * Cohen, N. et al. GILZ expression in human dendritic cells redirects their maturation and
prevents antigen-specific T lymphocyte response. _Blood_ 107, 2037–2044 (2006). Article CAS PubMed Google Scholar * Liu, J. N. et al. The complement inhibitory protein DAF (CD55)
suppresses T cell immunity in vivo. _J. Exp. Med._ 201, 567–577 (2005). Article CAS PubMed PubMed Central Google Scholar * Niess, J. H. & Reinecker, H. C. Lamina propria dendritic
cells in the physiology and pathology of the gastrointestinal tract. _Curr. Opin. Gastroenterol._ 21, 687–691 (2005). Article PubMed Google Scholar * Rescigno, M. & Di Sabatino, A.
Dendritic cells in intestinal homeostasis and disease. _J. Clin. Invest_. 119, 2441–2450 (2009). Article CAS PubMed PubMed Central Google Scholar * Dalod, M., Chelbi, R., Malissen, B.
& Lawrence, T. Dendritic cell maturation: functional specialization through signaling specificity and transcriptional programming. _EMBO J._ 33, 1104–1116 (2014). Article CAS PubMed
PubMed Central Google Scholar * Everts, B. et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. _Blood_ 120, 1422–1431 (2012). Article CAS
PubMed PubMed Central Google Scholar * Pearce, E. J. & Everts, B. Dendritic cell metabolism. _Nat. Rev. Immunol._ 15, 18–29 (2015). Article CAS PubMed PubMed Central Google
Scholar * Sahu, N. et al. Proline starvation induces unresolved ER stress and hinders mTORC1-dependent tumorigenesis. _Cell Metab._ 24, 753–761 (2016). Article CAS PubMed Google Scholar
* Tang, C. et al. Suppression of IL-17F, but not of IL-17A, provides protection against colitis by inducing Treg cells through modification of the intestinal microbiota. _Nat. Immunol._
19, 755–765 (2018). Article CAS PubMed Google Scholar * Lee, J. S. et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. _Immunity_ 43, 727–738
(2015). Article CAS PubMed PubMed Central Google Scholar * Verma, R. et al. Cell surface polysaccharides of _Bifidobacterium bifidum_ induce the generation of Foxp3(+) regulatory T
cells. _Sci. Immunol_. 3, eaat6975 (2018). Article PubMed Google Scholar * Kamada, N., Seo, S. U., Chen, G. Y. & Nunez, G. Role of the gut microbiota in immunity and inflammatory
disease. _Nat. Rev. Immunol._ 13, 321–335 (2013). Article CAS PubMed Google Scholar * Deutsch, S. M. et al. Identification of proteins involved in the anti-inflammatory properties of
_Propionibacterium freudenreichii_ by means of a multi-strain study. _Sci. Rep._ 7, 46409 (2017). Article CAS PubMed PubMed Central Google Scholar * Le Marechal, C. et al. Surface
proteins of _Propionibacterium freudenreichii_ are involved in its anti-inflammatory properties. _J. Proteom._ 113, 447–461 (2015). Article CAS Google Scholar * do Carmo, F. L. R. et al.
Extractable bacterial surface proteins in probiotic-host interaction. _Front. Microbiol._ 9, 645 (2018). Article PubMed PubMed Central Google Scholar * do Carmo, F. L. R. et al.
_Propionibacterium freudenreichii_ surface protein SlpB is involved in adhesion to intestinal HT-29 cells. _Front. Microbiol._ 8, 1033 (2017). Article PubMed PubMed Central Google Scholar
* Michon, C., Langella, P., Eijsink, V. G., Mathiesen, G. & Chatel, J. M. Display of recombinant proteins at the surface of lactic acid bacteria: strategies and applications. _Micro.
Cell Fact._ 15, 70 (2016). Article CAS Google Scholar * Ewing, C. P., Andreishcheva, E. & Guerry, P. Functional characterization of flagellin glycosylation in _Campylobacter jejuni_
81-176. _J. Bacteriol._ 191, 7086–7093 (2009). Article CAS PubMed PubMed Central Google Scholar * Nothaft, H. & Szymanski, C. M. Protein glycosylation in bacteria: sweeter than
ever. _Nat. Rev. Microbiol._ 8, 765–778 (2010). Article CAS PubMed Google Scholar * Liu, C. F. et al. Bacterial protein-_O_-mannosylating enzyme is crucial for virulence of
_Mycobacterium tuberculosis_. _Proc. Natl Acad. Sci. USA_ 110, 6560–6565 (2013). Article CAS PubMed PubMed Central Google Scholar * Girrbach, V. & Strahl, S. Members of the
evolutionarily conserved PMT family of protein _O_-mannosyltransferases form distinct protein complexes among themselves. _J. Biol. Chem._ 278, 12554–12562 (2003). Article CAS PubMed
Google Scholar * Geijtenbeek, T. B. & Gringhuis, S. I. C-type lectin receptors in the control of T helper cell differentiation. _Nat. Rev. Immunol._ 16, 433–448 (2016). Article CAS
PubMed Google Scholar * Zhou, Y. et al. Oral tolerance to food-induced systemic anaphylaxis mediated by the C-type lectin SIGNR1. _Nat. Med._ 16, 1128–1133 (2010). Article CAS PubMed
PubMed Central Google Scholar * Lanoue, A. et al. SIGN-R1 contributes to protection against lethal pneumococcal infection in mice. _J. Exp. Med._ 200, 1383–1393 (2004). Article CAS
PubMed PubMed Central Google Scholar * Song, X. et al. Growth factor FGF2 cooperates with interleukin-17 to repair intestinal epithelial damage. _Immunity_ 43, 488–501 (2015). Article
CAS PubMed Google Scholar * Kreisman, L. S. & Cobb, B. A. Infection, inflammation and host carbohydrates: a Glyco-Evasion Hypothesis. _Glycobiology_ 22, 1019–1030 (2012). Article CAS
PubMed PubMed Central Google Scholar * Dewald, J. H., Colomb, F., Bobowski-Gerard, M., Groux-Degroote, S., Delannoy, P. Role of cytokine-induced glycosylation changes in regulating cell
interactions and cell signaling in inflammatory diseases and cancer. _Cells_ 5, 43 (2016). Article PubMed Central CAS Google Scholar * Gevers, D. et al. The treatment-naive microbiome
in new-onset Crohn’s disease. _Cell Host Microbe_ 15, 382–392 (2014). Article CAS PubMed PubMed Central Google Scholar * Shreiner, A. B., Kao, J. Y. & Young, V. B. The gut
microbiome in health and in disease. _Curr. Opin. Gastroenterol._ 31, 69–75 (2015). Article CAS PubMed PubMed Central Google Scholar * Kaplan, G. G. The global burden of IBD: from 2015
to 2025. _Nat. Rev. Gastroenterol. Hepatol._ 12, 720–727 (2015). Article PubMed Google Scholar * Li, W. et al. Targeting T cell activation and lupus autoimmune phenotypes by inhibiting
glucose transporters. _Front Immunol._ 10, 833 (2019). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by NIH R01 DK109560
(to M.M.). Glycomic and glycoproteomic analyses were supported in part by NIH 1S10OD018530 and P41GM10349010 grants (to P.A.) at the Complex Carbohydrate Research Center. AUTHOR INFORMATION
AUTHORS AND AFFILIATIONS * Department of Infectious Diseases and Immunology, University of Florida, Gainesville, FL, 32611, USA Yong Ge, Minghao Gong, Mojgan Zadeh, Jing Li & Mansour
Mohamadzadeh * Division of Gastroenterology, Hepatology and Nutrition, Department of Medicine, University of Florida, Gainesville, FL, 32611, USA Yong Ge, Minghao Gong, Mojgan Zadeh &
Mansour Mohamadzadeh * Department of Comparative, Diagnostic and Population Medicine, University of Florida, Gainesville, FL, 32611, USA Jeffrey R. Abbott * Department of Pathology,
Immunology and Laboratory Medicine, University of Florida, Gainesville, FL, 32611, USA Wei Li & Laurence Morel * Complex Carbohydrate Research Center, University of Georgia, Athens, GA,
30602, USA Roberto Sonon, Nitin T. Supekar & Parastoo Azadi * Division of Pulmonary, Allergy, Critical Care and Sleep Medicine, Department of Medicine, Emory University School of
Medicine, Atlanta, GA, 30322, USA Yating Wang, Dean P. Jones & Shuzhao Li Authors * Yong Ge View author publications You can also search for this author inPubMed Google Scholar * Minghao
Gong View author publications You can also search for this author inPubMed Google Scholar * Mojgan Zadeh View author publications You can also search for this author inPubMed Google Scholar
* Jing Li View author publications You can also search for this author inPubMed Google Scholar * Jeffrey R. Abbott View author publications You can also search for this author inPubMed
Google Scholar * Wei Li View author publications You can also search for this author inPubMed Google Scholar * Laurence Morel View author publications You can also search for this author
inPubMed Google Scholar * Roberto Sonon View author publications You can also search for this author inPubMed Google Scholar * Nitin T. Supekar View author publications You can also search
for this author inPubMed Google Scholar * Parastoo Azadi View author publications You can also search for this author inPubMed Google Scholar * Yating Wang View author publications You can
also search for this author inPubMed Google Scholar * Dean P. Jones View author publications You can also search for this author inPubMed Google Scholar * Shuzhao Li View author publications
You can also search for this author inPubMed Google Scholar * Mansour Mohamadzadeh View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS M.M.
directed the experiments, which were executed by Y.G., M.G., M.Z., and J.L. Y.G. and M.Z. performed animal experiments; Y.G. performed flow cytometry analysis, genetic and biochemical
assays. Y.G. and J.L. constructed RNA-Seq libraries. M.G. analyzed RNA-Seq data. J.R.A. evaluated and scored all colonic tissue sections. W.L. and L.M. performed extracellular flux analysis.
R.S., N.T.S., and P.A. analyzed glycomics and glycoproteomics. M.G., Y.W., D.P.J. and S.L. performed, analyzed, and directed metabolomic studies. Y.G. and M.M. wrote the paper.
CORRESPONDING AUTHOR Correspondence to Mansour Mohamadzadeh. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION RIGHTS AND
PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ge, Y., Gong, M., Zadeh, M. _et al._ Regulating colonic dendritic cells by commensal glycosylated large surface
layer protein A to sustain gut homeostasis against pathogenic inflammation. _Mucosal Immunol_ 13, 34–46 (2020). https://doi.org/10.1038/s41385-019-0210-0 Download citation * Received: 08
July 2019 * Revised: 30 August 2019 * Accepted: 23 September 2019 * Published: 16 October 2019 * Issue Date: January 2020 * DOI: https://doi.org/10.1038/s41385-019-0210-0 SHARE THIS ARTICLE
Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided
by the Springer Nature SharedIt content-sharing initiative








