- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Alcoholic liver disease (ALD) is one of the fastest-growing concerns worldwide. In addition to bacterial endotoxins in the portal circulation, recent lines of evidence have
suggested that sterile inflammation caused by a wide range of stimuli induces alcoholic liver injury, in which damage-associated molecular patterns (DAMPs) play critical roles in inducing de
novo lipogenesis and inflammation through the activation of cellular pattern recognition receptors such as Toll-like receptors in non-parenchymal cells. Interestingly, alcohol-mediated
metabolic, neurological, and immune stresses stimulate the generation of DAMPs that are released not only in the liver, but also in other organs, such as adipose tissue, intestine, and bone
marrow. Thus, diverse DAMPs, including retinoic acids, proteins, lipids, microRNAs, mitochondrial DNA, and mitochondrial double-stranded RNA, contribute to a broad spectrum of ALD through
the production of multiple pro-inflammatory cytokines, chemokines, and ligands in non-parenchymal cells, such as Kupffer cells, hepatic stellate cells, and various immune cells. Therefore,
this review summarizes recent studies on the identification and understanding of DAMPs, their receptors, and cross-talk between the liver and other organs, and highlights successful
therapeutic targets and potential strategies in drug development that can be used to combat ALD. SIMILAR CONTENT BEING VIEWED BY OTHERS IMMUNOLOGICAL MECHANISMS AND THERAPEUTIC TARGETS OF
FATTY LIVER DISEASES Article Open access 02 December 2020 IMMUNOLOGICAL MECHANISMS AND EMERGING THERAPEUTIC TARGETS IN ALCOHOL-ASSOCIATED LIVER DISEASE Article Open access 21 May 2025
NON-ALCOHOLIC FATTY LIVER DISEASE: THE INTERPLAY BETWEEN METABOLISM, MICROBES AND IMMUNITY Article 20 December 2021 INTRODUCTION Chronic alcohol consumption is the third highest health risk
in the world. The World Health Organization has reported that the abuse of alcohol kills up to three million people per year globally; it accounts for ~5% of the global disease burden in
20181,2. Alcoholic liver disease (ALD) progresses from a mild form of alcoholic fatty liver to severe forms, such as alcoholic steatohepatitis, alcoholic hepatitis, alcoholic
fibrosis/cirrhosis, and hepatocellular carcinoma (HCC)2,3. In general, three inflammatory pathways primarily trigger ALD, wherein a change in intestinal microbiome composition increases the
amount of pathogen-associated molecular patterns (PAMPs) that further mediate activation of Kupffer cells through pattern recognition receptors (PRRs). Moreover, damage to hepatocytes by
alcohol metabolites generates reactive oxygen species (ROS), lipid-originated metabolites (retinoic acid and endocannabinoids), and damage-associated molecular patterns (DAMPs) to stimulate
inflammatory signals through Toll-like receptors (TLRs), nuclear/neuronal receptors, and the inflammasome2,4. Furthermore, inter-organ cross-talk contributes to ALD by delivering DAMPs or
migrating inflammatory cells to the liver. Through these processes, hepatocytes and non-parenchymal cells produce pro-inflammatory cytokines and chemokines to recruit additional immune
cells, such as neutrophils and macrophages. In addition, diverse types of hepatic lymphocytes that are activated by PAMPs, DAMPs, and cytokines promote liver injury by producing interferon
(IFN)-γ, interleukin (IL)-22, IL-17, etc5. Recent emerging data on sterile inflammation may bring to light potential therapeutic targets for ALD. Thus, this review describes the current
state of understanding concerning the pathophysiological mechanisms of DAMP- and PAMP-mediated inflammation and organ cross-talk in ALD. In addition, we briefly summarize lists of DAMPs and
PAMPs in ALD (Table 1). DAMPS IN ALD ALCOHOL METABOLISM AND DAMPS In the early stage of alcohol consumption, alcohol is metabolized to acetaldehyde by alcohol dehydrogenase, which is further
converted to acetate by acetaldehyde dehydrogenase6; however, chronic exposure results in alcohol metabolism occurring mainly through cytochrome P450 2E1 (CYP2E1)7. In the late stage,
alcohol metabolism-induced oxidative stresses cause damage to hepatocytes, in which DAMPs, including high mobility group box-1 (HMGB1), mitochondrial DNA (mtDNA), mitochondrial
double-stranded RNA (mtdsRNA), microRNAs (miRNAs), ATP, and several metabolites are generated and released from the injured hepatocytes, leading to sterile inflammation in ALD8,9,10. In
addition to recognizing TLRs, DAMPs amplify liver injury by stimulation of the inflammasome, which activates caspase-1 and secretes the pro-inflammatory cytokines, such as IL-1β and IL-18,
which play important roles in ALD5,11,12. EXTRACELLULAR VESICLES (EVS), MIRNA, MTDSRNA, AND MTDNA IN ALD EVs, such as exosomes, microvesicles, and apoptotic bodies, play important roles in
cell-to-cell communication by delivering hepatic DAMPs to target cells13. Recent studies in mice and patients with alcoholic steatohepatitis (ASH) have demonstrated that many miRNAs are not
only generated within the cells but can also be transferred into other target cells via EVs14. For instance, miR-27a from alcohol-exposed monocytes can program naive monocytes to polarize
into M2 macrophages15. In addition, mtdsRNA can be generated by the inhibition of restricting enzymes, such as mitochondrial RNA helicase SUV3 and polynucleotide phosphorylase (PNPase)16. We
recently found that accumulation of mtdsRNA is induced in ethanol-exposed hepatocytes through downregulated expression of PNPase, and exosomal delivery of mtdsRNA to Kupffer cells leads to
an augmentation of IL-17 production in hepatic γδ T cells and the severity of acute ALD in a TLR3-dependent manner17. In RNA-sequencing analysis, most mitochondrial mRNAs were enhanced in
ethanol-exposed exosomes, whereas mitochondrial DNAs were mainly enriched in microvesicles after treating hepatocytes with ethanol in vitro17. Another study strongly suggested that
hepatocyte mtdsRNA could act as self-ligands to TLR3, which could aggravate not only ALD but also other liver diseases17,18. In contrast, poly I:C-mediated multiple activation of TLR3 in
Kupffer cells and hepatic stellate cells (HSCs) attenuates hepatic steatosis and inflammation via IL-10 production19. Thus, the roles of TLR3 should be investigated carefully in further
studies. Similarly, previous studies have reported that nuclear DNA and mtDNA of damaged hepatocytes contribute to ALD. For example, EVs deliver hepatocyte-derived apoptotic DNA and mtDNA to
HSCs, neutrophils or tumor cells, consequently accelerating liver fibrosis, alcoholic hepatitis, and HCC, respectively, through TLR99,20,21. In addition to nucleic acids, CD40 ligands and
heat shock protein 90 present in EVs stimulate the production of TNF-α and IL-1β in macrophages in mice and patients with alcoholic hepatitis, in vitro and in vivo22,23. Therefore, EVs could
be therapeutic targets for ALD. GLUTAMATE AND ENDOCANNABINOIDS IN ALD During the last decade, studies have demonstrated that the bidirectional cross-talk between hepatocytes and HSCs
contributes to alcoholic steatosis, in which a line of neurological responses between hepatocytes and HSCs suggests the presence of a metabolic synapse24,25. Alcoholic steatosis is mediated
by activation of the endocannabinoid-induced CB1 receptor (CB1R) in hepatocytes, in which the metabotropic glutamate receptor 5 (mGluR5) in HSCs interacts with hepatocyte-derived glutamate
and generates the endocannabinoid, 2-arachidonoylglycerol (2-AG), after alcohol exposure25. Chronic alcohol exposure induces CYP2E1-mediated oxidative stress, which suppresses the methionine
cycle and transsulfuration system, and decreases cysteine concentrations hepatocytes, thereby lowering the levels of the antioxidant molecule glutathione (GSH)26. Moreover, to compensate
for the GSH shortage, hepatocytes take up extracellular cystine by exchanging it with glutamate through the xCT transporter, whereas HSCs generate 2-AG through mGluR5-mediated diacylglycerol
lipase-β activation25. The produced 2-AG, in turn, stimulates hepatic CB1R to induce de novo lipogenesis by upregulating sterol regulatory element-binding protein 1c (SREBP1c) and
downregulating AMP-activated protein kinase in hepatocytes, leading to fat accumulation. Furthermore, we found that chronic alcohol consumption increases glutamate generation from
glutamic-γ-semialdehyde through upregulation of ALDH4A1 expression in perivenous hepatocytes25, suggesting that the glutamate released from damaged hepatocytes might be a type of DAMP.
Therefore, a functional metabolic synapse between hepatocytes and HSCs may be a critical therapeutic target for ALD. RETINOIC ACIDS IN ALD The liver is a representative organ for storage and
metabolism of retinol (vitamin A)27. In particular, quiescent HSCs store ~80% of total liver retinols as retinyl palmitate in fat droplets, whereas activated HSCs lose their retinols by
releasing or metabolizing retinols into retinoids, including retinoic acids (RAs), in response to liver injury2,27. In HSCs, a class III ADH3 enzyme plays a critical role in retinol
metabolism, and the metabolized RAs bind to retinoic acid receptors (RAR-α/β/γ) and retinoic X receptors (RXR-α/β/γ) and regulate gene expression2,28. Although the exact underlying mechanism
by which HSCs lose or metabolize retinols in ALD is still unclear, HSCs do not induce liver fibrosis after chronic ethanol consumption in mice2. It is probable that activated HSCs express
retinoic acid early inducible gene 1 (RAE1), which is a specific ligand for natural killer group 2 member D (NKG2D) in natural killer (NK) cells, and then hepatic NK cells specifically kill
the activated HSCs by producing IFN-γ in response to the RAE1-NKG2D interaction at an early stage of ALD2,29. In contrast, in an advanced stage of liver disease, activated HSCs show
resistance to NK cell cytotoxicity via 9-cis forms of retinoid-mediated production of transforming growth factor (TGF)-β1 and the expression of suppressor of cytokine signaling protein 1
(SOCS1)29,30,31. In addition, a study reported that retinol metabolism in HSCs inhibits the recruitment of regulatory T cells (Tregs), whereas blocking retinol metabolism mitigates
Concanavalin A-induced hepatitis through increased migration of Tregs in mice32. As mentioned above, liver diseases including ALD stimulate retinol metabolism of HSCs, and retinoids trigger
transcription of diverse genes through their nuclear receptors, inducing beneficial or detrimental functions related to liver diseases. However, to implicate retinols as DAMPs in ALD, the
exact molecular signaling pathways of retinol metabolism in HSCs and functions in the cells should be clearly investigated, and then these molecules might be therapeutic targets for ALD in
the future. PAMPS IN ALD The liver–gut axis is a critical pathway for ALD because it is a central player in the response to gut bacteria-originated PAMPs, as well as nutrients received
through the portal vein. In this regard, ALD does not represent a true sterile inflammatory liver disease. Thus, we briefly address the role of PAMPs in ALD here. Alcohol consumption affects
multiple defense barriers, including chemical, physical, and immune factors in the gut4. Impairment of this barrier owing to acute, binge, or chronic alcohol intake increases blood levels
of bacterial components and their products (e.g., PAMPs) in animal models and humans33,34. Among bacterial PAMPs, lipopolysaccharide (LPS) and bacterial DNA interact with TLR4 and TLR9,
respectively, and produce pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α and IL-1β through NF-κB in innate immune cells, including Kupffer cells. Similarly, these PAMPs and
inflammatory mediators, such as TNF-α, IL-6, and CCL2, are increased in the sera of humans after alcohol exposure and exposure to LPS33,34. In addition, alcohol intake increases serum
levels of lipoteichoic acid (LTA) and flagellin, and it sensitizes LTA and flagellin to TLR2 and TLR5, respectively, leading to enhanced TNF-α production and liver injury in a mouse model of
ALD35. More intriguingly, non-alcoholic fatty liver disease (NAFLD) is also triggered by sterile inflammatory responses, and a recent interesting study in humans suggests that the presence
of _Klebsiella pneumoniae_ in the gut microbiome might be one of the causes of NAFLD because of its ability to produce alcohol, thereby increasing the alcohol concentration in the blood36.
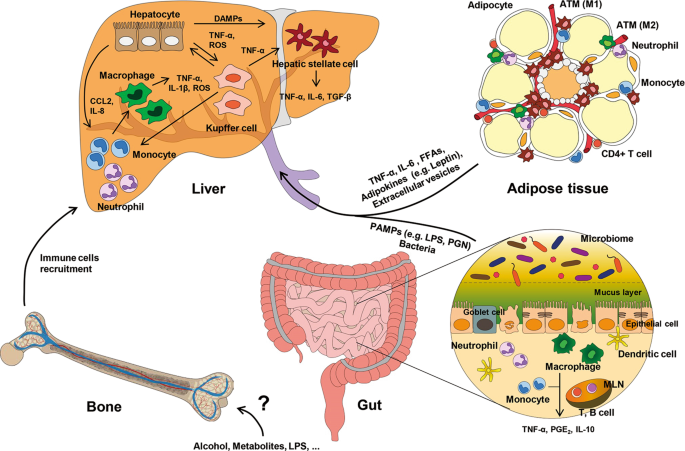
INTER-ORGAN CROSS-TALK IN ALD Several lines of evidence have reported cross-talk between the liver and other organs, such as adipose tissue, the gut, and bone marrow (BM, Fig. 1). For
example, metabolic and genetic changes in adipocytes and enterocytes affect steatosis and inflammation of the liver. In addition, altered gene expression in hepatocytes and non-parenchymal
cells in the liver influences adipogenesis, lipolysis, and inflammation of adipose tissue, the gut, or BM. THE ADIPOSE TISSUE-LIVER AXIS IN ALD METABOLIC EFFECTS OF ALCOHOL IN ADIPOSE TISSUE
Anatomically, adipose tissue consists of visceral adipose tissue (VAT) and subcutaneous adipose tissue37. VAT is mainly present within the abdominal cavity, and visceral fat venous blood is
drained directly into the liver through the portal vein. Thus, abnormal metabolic pathways and inflammation in VAT are implicated in the pathogenesis of metabolic syndromes, including
obesity, diabetes, atherosclerosis, and NAFLD38. White adipocytes, which account for most adipocytes, function to store energy as triglycerides in large lipid droplets and release adipokines
and cytokines for metabolic and endocrine activities. In contrast, brown adipocytes contribute to thermogenesis through the high expression of uncoupling protein 1 in mitochondria39. Recent
studies have suggested that chronic alcohol consumption is inversely correlated with fat accumulation in adipose tissue. In mice and rats, chronic alcohol exposure stimulates adipose
triglyceride lipase (ATGL)-mediated lipolysis in adipose tissue, leading to the release of free fatty acids (FFAs) and a decrease in the epididymal adipose tissue mass and adipocyte size40.
Moreover, binge drinking or chronic alcohol consumption impairs insulin sensitivity, thus resulting in increased lipolysis and decreased lipogenesis41. The expression of lipogenic enzymes,
peroxisome proliferator-activated receptor-γ, and CCAAT enhancer-binding protein α, is also downregulated in white adipose tissue after chronic alcohol intake40. Interestingly, alcohol can
be metabolized in adipose tissue in humans and rodents owing to the expression of CYP2E1 and ALDH42. In adipose tissue, increased CYP2E1 expression owing to chronic alcohol intake decreases
the GSH/GSSG ratio, induces oxidative stress through ROS production, and inhibits adiponectin secretion42. Leptin (an energy expenditure hormone) is also known to be related to alcohol
intake, but its expression depends on the amount and duration of alcohol consumption in ALD patients and a rodent model43,44. INFLAMMATION IN ADIPOSE TISSUE Chronic alcohol consumption
stimulates the secretion of several cytokines and chemokines, thereby inducing inflammation in adipose tissue. In alcoholic patients, the expression of pro-inflammatory cytokines (e.g., IL-6
and TNF-α) and chemokines (e.g., CCL2), is upregulated in adipose tissue45,46. IL-6 expression increases in all stages of ALD, whereas increased expression of TNF-α is only observed in
patients with alcoholic steatosis and alcoholic hepatitis37. Rodent models also show increased expression of inflammatory mediators, such as TNF-α, IL-6, CCL2, and IFN-γ, in adipose
tissue46. In adipose tissue inflammation, the number of macrophages is increased (up to 4–50%), in which adipose tissue macrophages (ATMs) are divided into M1 and M2 types47. M1 macrophages
exist as crown-like structures around dying adipocytes and classically express inflammatory cytokines, such as TNF-α, IL-6, and inducible nitric oxide synthase (iNOS), whereas the M2 type is
an alternatively activated cell type that produces anti-inflammatory cytokines, such as IL-10, IL-4, and arginase 1. In a normal state, most ATMs exist as the M2 type; however, in a state
of alcohol consumption, these cells shift to the M1 type, expressing CD11c and producing inflammatory cytokines or causing infiltration of M1 macrophages in a CCL2-dependent manner46. In
addition, binge alcohol intake increases the infiltration of neutrophils with neutrophil-attracting chemokines, thereby inducing tissue damage in mice48. In contrast, IL-10 levels in adipose
tissue are increased in acute alcoholic hepatitis, suggesting that some cells possess anti-inflammatory functions that regulate the immune system49. ADIPOCYTE DEATH Adipocyte death is
induced by chronic inflammation and oxidative stress. The ATMs surround dead adipocytes in crown-like structures, and they produce pro-inflammatory cytokines such as TNF-α and IL-6 to
facilitate phagocytosis or scavenging of adipocytes50. Moreover, it has been reported that chronic ethanol consumption increases Bid-mediated apoptosis in adipocytes, whereas CYP2E1
deficiency results in decreased expression of TNF-α, IL-6, and CCL2 and reduced adipocyte apoptosis in mice46, suggesting that adipose tissue inflammation is dependent on CYP2E1-mediated
alcohol metabolism. Thus, CYP2E1 might be a good therapeutic target in adipose tissue. CROSS-TALK BETWEEN THE LIVER AND ADIPOSE TISSUE Increased amounts of circulating FFAs caused by
lipolysis in adipocytes contribute to lysosomal destabilization in hepatocytes, leading to TNF-α production and hepatic de novo lipogenesis through transcription of SREBP1c51. Moreover,
among adipokines, adiponectin in humans and rodents contributes to not only the oxidization of fatty acids in hepatocytes but also the reduction in TNF-α and IL-10 production in Kupffer
cells, thereby alleviating liver steatosis and inflammation52. The leptin from adipose tissue induces hepatic inflammation and fibrogenic responses by activating HSCs and Kupffer cells, and
Kupffer cells increase TNF-α production through the P38 and JNK pathways53,54. In chronic alcohol consumption, as observed in alcoholic patients and mouse models, the production of
adiponectin is decreased, but leptin production is increased, resulting in damage to hepatocytes by high concentrations of TNF-α55. Furthermore, adipose tissue influences alcoholic liver
injury by modulating the cargo of the secreted EVs trafficking to the liver via the secretome56. THE GUT–LIVER AXIS IN ALD GUT MICROBIOTA AND THE IMMUNE SYSTEM In addition to energy
absorption, the gut has a well-established immune system that plays a role in homeostasis. Mechanochemically, the surface of enterocytes in the small intestine consists of microvilli coated
with a glycocalyx of mucin, which produces diverse enzymes for the defense against antigens or pathogens from the lumen57. The intestinal lamina propria consists of various types of immune
cells and lymphatic tissues. For example, gut-associated lymphoid tissue and mesenteric lymph nodes, including Peyer’s patches, are composed of numerous T cells and B cells, and naive
lymphocytes are primed by pathogens or antigen-presenting cells that are external to the immune response58. Several subsets of dendritic cells regulate T-cell stimulation and suppression by
recognizing pathogens through TLR signaling; in addition, macrophages are involved in the phagocytosis of dead cells and induce proliferation of regulatory T cells by producing IL-1057,59.
Interestingly, the gut microbiome coexists with its host; there are over 100 trillion microorganisms in the human gastrointestinal tract, and the microbiome not only comprises bacteria, but
also fungi, archaea, protists, and viruses60. The microbiome is established by the influence of environmental conditions and food consumption after birth and is important for immune and
metabolic homeostasis of the host. However, the composition of the microbiome changes in ALD61. METABOLIC EFFECTS OF ALCOHOL ON GUT MICROBIOTA AND IMMUNITY Ingested alcohol is absorbed and
diffused in the gastrointestinal tract, where the stomach and the proximal small intestine are responsible for ~20% and 70% of its absorption, respectively62. Among alcohol dehydrogenases
(ADHs), ADH1A is expressed in the small intestine and enables alcohol metabolism, whereas the other isoenzymes have a role in vitamin A (retinol) metabolism, which is essential for
intestinal epithelial proliferation or differentiation63. However, high concentrations of alcohol and chronic absorption are metabolized by CYP2E1, and oxidative stress and byproducts caused
by alcohol metabolism alter the intestinal tight junction proteins (e.g., occludin and zonula occludens-1) and adherent junction proteins (e.g., β-catenin and E-cadherin), which
interconnect the epithelial cells that have a role in intestinal barrier integrity63,64,65. In addition, CYP2E1-mediated alcohol metabolism increases intestinal permeability by inducing the
expression of circadian clock proteins such as circadian locomotor output cycles kaput and period circadian clock 266. Furthermore, chronic alcohol consumption destroys the intestinal
epithelial barrier, leading to changes, such as overgrowth and dysbiosis, in the intestinal microbiome of rodents and patients. Alcohol-mediated dysbiosis increases the levels of
unconjugated bile acids in the gut, which reduces farnesoid X receptor activity and fibroblast growth factor (FGF)-15 expression in enterocytes, leading to increased bile acid concentration
in the blood through upregulated CYP7A1 expression in hepatocytes67. However, treatment with the FXR agonist, fexaramine, or overexpression of FGF-15 leads to recovery of the gut barrier and
attenuates ALD67. Overgrowth of gram-negative bacteria owing to alcohol consumption induces endotoxin production and release in the blood of rodents and human patients68. In addition, LPS
enhances intestinal permeability by producing nitric oxide by autocrine signaling, thereby activating myosin light chain kinase and the expression of TLR4 and CD14 in enterocytes69,70. In
alcohol-fed mice, bacteria of the phyla _Verrucomicrobia_ and _Bacteroidetes_ increase, whereas those of the phylum _Firmicutes_ decrease71. In alcoholic patients, _Proteobacteria_ and
_Firmicute_ phyla increase; however, this increase depends on the stage of liver disease61,72. A recent study reported that the numbers of cytolysin-positive _Enterococcus faecalis_
correlate with the severity of alcoholic hepatitis and mortality73. Interestingly, intestinal overgrowth of _K. pneumoniae_ causes fatty liver disease because these bacteria produce alcohol,
in the absence of alcohol consumption74. Moreover, populations of fungi, as well as bacteria, are increased in the gut owing to alcohol consumption and translocate to the liver to induce
inflammation through the β-glucan-CLEC7A axis75. INFLAMMATION IN THE GUT Increasing intestinal permeability leads to translocation of bacteria and PAMPs to the portal blood and exposure to
the intestinal immune system, wherein they stimulate myeloid cells and induce systemic inflammation76. However, in ALD, the exact underlying mechanisms of gut inflammation by alcohol are
still unclear. In mice with chronic exposure to alcohol, the expression of pro-inflammatory mediators, such as TNF-α, IL-1β, IL-6, and iNOS, increases in the distal ileum, and the
anti-inflammatory cytokine IL-11 also increases significantly77. In an “alcohol combined with burn injury” model, intestinal macrophages or monocytes increase the production of TNF-α,
prostaglandin E2, and IL-10 and decrease the expression of MHC class II and antigen presentation78. In addition, in this model, increased IL-18 has a critical role in the recruitment and
activation of neutrophils in the damaged intestines of rats79. Regarding adaptive immune responses, acute alcohol administration to mice depletes T cells and B cells in the MLN80,81. IL-12,
which has key roles in Th1 differentiation and IFN-γ production, is reduced in rat T cells after alcohol intoxication and burn injury82. CROSS-TALK BETWEEN THE LIVER AND GUT In ALD,
gut-liver cross-talk is mainly caused by increased gut permeability, leading to the entry of PAMPs (e.g., LPS) into the liver through the portal circulation. LPS binds to TLR4 in combination
with CD14, MD-2, and LPS-binding protein, and its signal is delivered by the recruitment of adapter molecules, such as MyD88 and TRIF, in Kupffer cells and macrophages83. MyD88-mediated
NF-kB activation produces pro-inflammatory cytokines (e.g., TNF-α, IL-6, and IL-1β) and the chemokine CCL2, whereas the TRIF pathway induces the production of type-I interferons84. Thus,
both TLR4 and CD14 are considered therapeutic targets for ALD. Moreover, TLR4 is expressed not only in immune cells but also in hepatocytes and HSCs. In hepatocytes, TLR4-LPS activates the
NF-kB pathway and pro-inflammatory signaling, leading to increased expression of SREBP1c that further results in steatosis or hepatic injury55. In response to LPS, HSCs release
pro-inflammatory cytokines (e.g., TNF-α, IL-6, and IL-8), chemokines (e.g., CCL2, ICAM-1, RANTES, and CCR5) and adhesion molecules84. Furthermore, TNF-α production in Kupffer cells and the
recruitment of immune cells by LPS/TLR4 activate HSCs and induce liver fibrosis by producing TGF-β and extracellular matrix85. THE BM–LIVER AXIS IN ALD CROSS-TALK BETWEEN THE LIVER AND BM In
addition to tissue-resident immune cells, such as Kupffer cells, most inflammatory cells are derived from the BM in alcoholic inflammation of adipose tissue, the gut, and the liver. The BM
is thought to be an immunoregulatory organ that has a role not only in hematopoiesis but also in immune responses86. Thus, inflammatory cells mature and proliferate in the BM and egress into
the bloodstream by the gradients of cytokines and chemokines, such as CXCL12-CXCR4, CXCL1-CXCR2, and CCL2-CCR287,88. Clinical and experimental studies have shown that autologous or
allogenic BM cell transplantation is effective for liver cirrhosis and fibrosis in patients and mice, respectively, in which migrated BM cells improve impaired liver functions in patients or
stimulate IL-10 production in Gr1+CD11b+ cells in mice89,90. However, autologous BM cell transplantation did not show beneficial effects in liver function or IL-10 production in a patient
with alcoholic cirrhosis90, suggesting that chronic alcohol consumption might affect BM cells. Recently, granulocyte colony-stimulating factor (G-CSF), a glycoprotein that differentiates and
matures stem cells into granulocytes in the BM, has emerged as a treatment candidate for alcoholic hepatitis in several countries91,92,93,94. Clinical and experimental studies have shown
that G-CSF treatment improves alcoholic hepatitis by migrating CD34+ hematopoietic stem cells and stimulating hepatocyte regeneration in patients and mice, suggesting a novel therapeutic
effect of G-CSF on alcoholic hepatitis91,95. In contrast, the effects of alcohol intake on the BM have yet to be clearly elucidated. Intriguingly, our recent study suggests that BM-derived
monocytes can be differentiated into F4/80high Kupffer-like cells in a CX3CR1-dependent manner and have pro-inflammatory functions in ALD96. Similarly, other studies have demonstrated that
monocytes are recruited by liver HSCs and liver sinusoidal endothelial cells, and they differentiate into monocyte-derived Kupffer cells through the NOTCH-BMP pathway in certain
situations97,98. These studies suggest that BM-derived macrophages may have a role in ALD through the differentiation of Kupffer-like cells. Although the exact functions of such macrophages
are not yet clear, they may have various functions depending on multiple subtypes of macrophages in the BM and various conditions in ALD. Given that alcohol metabolism occurs within the BM,
clarification of alcohol-metabolizing cells and their roles in ALD are likely to be of vital importance in the treatment of ALD. Consequently, further studies on this subject are of the
utmost importance. FUTURE PERSPECTIVES Although the best way to prevent further progression of ALD is to abstain from consuming alcohol, current therapies for ALD focus on the stages of
disease severity and mostly depend on steroid treatments. However, we are still struggling to use such treatment strategies in ALD patients who consume high amounts of alcohol and are
resistant to medication. Thus, there is an urgent need to develop alternative approaches to treat ALD. Generally, alcohol metabolism is considered to occur in the liver, but recent studies
have demonstrated that other organs, including adipose tissue and intestine, can metabolize alcohol partially owing to the expression of ADHs and CYP2E1. In addition, mechanical pathways
related to oxidative stress-mediated inflammation and injury are well known to occur in adipose tissue and the gut. Furthermore, organ cross-talk is triggered by the entry of inflammatory
cytokines and molecules, such as adipokines, DAMPs, and PAMPs, as well as the migration of pro-inflammatory cells into the liver, which further exacerbates ALD by activating hepatic immune
cells and inducing hepatocyte injury. As a result, the entire paradigm of ALD originates not only due to liver injury but also due to cross-talk with other organs. Therefore, using a single
factor in a specific organ as a therapeutic target may be ineffective, and combination therapy aimed at multiple organs is likely required for the treatment of ALD. REFERENCES * Bajaj, J. S.
Alcohol, liver disease and the gut microbiota. _Nat. Rev. Gastroenterol. Hepatol._ 16, 235–246 (2019). Article PubMed Google Scholar * Seo, W. & Jeong, W. I. Hepatic non-parenchymal
cells: master regulators of alcoholic liver disease? _World J. Gastroenterol._ 22, 1348–1356 (2016). Article CAS PubMed PubMed Central Google Scholar * Suh, Y. G. & Jeong, W. I.
Hepatic stellate cells and innate immunity in alcoholic liver disease. _World J. Gastroenterol._ 17, 2543–2551 (2011). Article PubMed PubMed Central Google Scholar * Szabo, G. Gut-liver
axis in alcoholic liver disease. _Gastroenterology_ 148, 30–36 (2015). Article CAS PubMed Google Scholar * Gao, B., Ahmad, M. F., Nagy, L. E. & Tsukamoto, H. Inflammatory pathways in
alcoholic steatohepatitis. _J. Hepatol._ 70, 249–259 (2019). Article PubMed PubMed Central Google Scholar * Mueller, S. et al. Carcinogenic etheno DNA adducts in alcoholic liver
disease: correlation with cytochrome P-4502E1 and fibrosis. _Alcohol Clin. Exp. Res._ 42, 252–259 (2018). Article CAS PubMed Google Scholar * Lu, Y. & Cederbaum, A. I. CYP2E1 and
oxidative liver injury by alcohol. _Free Radic. Biol. Med._ 44, 723–738 (2008). Article CAS PubMed Google Scholar * Ge, X. et al. High mobility group box-1 (HMGB1) participates in the
pathogenesis of alcoholic liver disease (ALD). _J. Biol. Chem._ 289, 22672–22691 (2014). Article CAS PubMed PubMed Central Google Scholar * Cai, Y. et al. Mitochondrial DNA-enriched
microparticles promote acute-on-chronic alcoholic neutrophilia and hepatotoxicity. _JCI Insight_ 2, e92634 (2017). Article PubMed Central Google Scholar * Iracheta-Vellve, A. et al.
Inhibition of sterile danger signals, uric acid and ATP, prevents inflammasome activation and protects from alcoholic steatohepatitis in mice. _J. Hepatol._ 63, 1147–1155 (2015). Article
CAS PubMed PubMed Central Google Scholar * Schroder, K. & Tschopp, J. The inflammasomes. _Cell_ 140, 821–832 (2010). Article CAS PubMed Google Scholar * Petrasek, J. et al. IL-1
receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. _J. Clin. Invest_ 122, 3476–3489 (2012). Article CAS PubMed PubMed Central Google Scholar *
Hirsova, P. et al. Extracellular vesicles in liver pathobiology: small particles with big impact. _Hepatology_ 64, 2219–2233 (2016). Article PubMed Google Scholar * Eguchi, A. et al.
Extracellular vesicles released by hepatocytes from gastric infusion model of alcoholic liver disease contain a MicroRNA barcode that can be detected in blood. _Hepatology_ 65, 475–490
(2017). Article CAS PubMed Google Scholar * Saha, B., Momen-Heravi, F., Kodys, K. & Szabo, G. MicroRNA cargo of extracellular vesicles from alcohol-exposed monocytes signals naive
monocytes to differentiate into M2 macrophages. _J. Biol. Chem._ 291, 149–159 (2016). Article CAS PubMed Google Scholar * Dhir, A. et al. Mitochondrial double-stranded RNA triggers
antiviral signalling in humans. _Nature_ 560, 238–242 (2018). Article CAS PubMed PubMed Central Google Scholar * Lee, J. H. et al. Mitochondrial double-stranded RNA in exosome promotes
interleukin-17 production through toll-like receptor 3 in alcoholic liver injury. _Hepatology_ (2019) [Ahead of print]. * Seo, W. et al. Exosome-mediated activation of toll-like receptor 3
in stellate cells stimulates interleukin-17 production by gammadelta T cells in liver fibrosis. _Hepatology_ 64, 616–631 (2016). Article CAS PubMed Google Scholar * Byun, J. S., Suh, Y.
G., Yi, H. S., Lee, Y. S. & Jeong, W. I. Activation of toll-like receptor 3 attenuates alcoholic liver injury by stimulating Kupffer cells and stellate cells to produce interleukin-10 in
mice. _J. Hepatol._ 58, 342–349 (2013). Article CAS PubMed Google Scholar * Watanabe, A. et al. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor
9. _Hepatology_ 46, 1509–1518 (2007). Article CAS PubMed Google Scholar * Seo, W. et al. ALDH2 deficiency promotes alcohol-associated liver cancer by activating oncogenic pathways via
oxidized DNA-enriched extracellular vesicles. _J. Hepatol._ 71, 1000–1011 (2019). Article CAS PubMed PubMed Central Google Scholar * Saha, B. et al. Extracellular vesicles from mice
with alcoholic liver disease carry a distinct protein cargo and induce macrophage activation through heat shock protein 90. _Hepatology_ 67, 1986–2000 (2018). Article CAS PubMed Google
Scholar * Verma, V. K. et al. Alcohol stimulates macrophage activation through caspase-dependent hepatocyte derived release of CD40L containing extracellular vesicles. _J. Hepatol._ 64,
651–660 (2016). Article CAS PubMed Google Scholar * Jeong, W. I. et al. Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty
liver. _Cell Metab._ 7, 227–235 (2008). Article CAS PubMed Google Scholar * Choi, W. M. et al. Glutamate signaling in hepatic stellate cells drives alcoholic steatosis. _Cell Metab._ 30,
877–889 e877 (2019). Article CAS PubMed PubMed Central Google Scholar * Tsukamoto, H. & Lu, S. C. Current concepts in the pathogenesis of alcoholic liver injury. _FASEB J._ 15,
1335–1349 (2001). Article CAS PubMed Google Scholar * Lee, Y. S. & Jeong, W. I. Retinoic acids and hepatic stellate cells in liver disease. _J. Gastroenterol. Hepatol._ 27, 75–79
(2012). Article CAS PubMed Google Scholar * Yi, H. S. et al. Alcohol dehydrogenase III exacerbates liver fibrosis by enhancing stellate cell activation and suppressing natural killer
cells in mice. _Hepatology_ 60, 1044–1053 (2014). Article CAS PubMed Google Scholar * Jeong, W. I., Park, O., Radaeva, S. & Gao, B. STAT1 inhibits liver fibrosis in mice by
inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. _Hepatology_ 44, 1441–1451 (2006). Article CAS PubMed Google Scholar * Jeong, W. I., Park, O. & Gao, B.
Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. _Gastroenterology_ 134, 248–258 (2008). Article CAS
PubMed Google Scholar * Jeong, W. I. et al. Suppression of innate immunity (natural killer cell/interferon-gamma) in the advanced stages of liver fibrosis in mice. _Hepatology_ 53,
1342–1351 (2011). Article CAS PubMed Google Scholar * Lee, Y. S. et al. Blockade of retinol metabolism protects T cell-induced hepatitis by increasing migration of regulatory T cells.
_Mol. Cells_ 38, 998–1006 (2015). Article CAS PubMed PubMed Central Google Scholar * Bala, S., Marcos, M., Gattu, A., Catalano, D. & Szabo, G. Acute binge drinking increases serum
endotoxin and bacterial DNA levels in healthy individuals. _PLoS ONE_ 9, e96864 (2014). Article PubMed PubMed Central Google Scholar * Michelena, J. et al. Systemic inflammatory response
and serum lipopolysaccharide levels predict multiple organ failure and death in alcoholic hepatitis. _Hepatology_ 62, 762–772 (2015). Article CAS PubMed Google Scholar * Gustot, T. et
al. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. _Hepatology_ 43, 989–1000 (2006). Article CAS PubMed Google Scholar * Yuan, J. et
al. Fatty liver disease caused by high-alcohol-producing Klebsiella pneumoniae. _Cell Metab._ 30, 1172 (2019). Article CAS PubMed Google Scholar * Parker, R., Kim, S. J. & Gao, B.
Alcohol, adipose tissue and liver disease: mechanistic links and clinical considerations. _Nat. Rev. Gastroenterol. Hepatol._ 15, 50–59 (2018). Article CAS PubMed Google Scholar *
Kusminski, C. M., Bickel, P. E. & Scherer, P. E. Targeting adipose tissue in the treatment of obesity-associated diabetes. _Nat. Rev. Drug Discov._ 15, 639–660 (2016). Article CAS
PubMed Google Scholar * Fedorenko, A., Lishko, P. V. & Kirichok, Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. _Cell_ 151, 400–413 (2012). Article
CAS PubMed PubMed Central Google Scholar * Zhong, W. et al. Chronic alcohol exposure stimulates adipose tissue lipolysis in mice: role of reverse triglyceride transport in the
pathogenesis of alcoholic steatosis. _Am. J. Pathol._ 180, 998–1007 (2012). Article CAS PubMed PubMed Central Google Scholar * Lindtner, C. et al. Binge drinking induces whole-body
insulin resistance by impairing hypothalamic insulin action. _Sci. Transl. Med._ 5, 170ra114 (2013). Article CAS Google Scholar * Tang, H. et al. Ethanol-induced oxidative stress via the
CYP2E1 pathway disrupts adiponectin secretion from adipocytes. _Alcohol Clin. Exp. Res._ 36, 214–222 (2012). Article CAS PubMed Google Scholar * Obradovic, T. & Meadows, G. G.
Chronic ethanol consumption increases plasma leptin levels and alters leptin receptors in the hypothalamus and the perigonadal fat of C57BL/6 mice. _Alcohol Clin. Exp. Res._ 26, 255–262
(2002). Article CAS PubMed Google Scholar * Kalafateli, M. et al. Adipokines levels are associated with the severity of liver disease in patients with alcoholic cirrhosis. _World J.
Gastroenterol._ 21, 3020–3029 (2015). Article CAS PubMed PubMed Central Google Scholar * Voican, C. S. et al. Alcohol withdrawal alleviates adipose tissue inflammation in patients with
alcoholic liver disease. _Liver Int._ 35, 967–978 (2015). Article CAS PubMed Google Scholar * Sebastian, B. M. et al. Identification of a cytochrome P4502E1/Bid/C1q-dependent axis
mediating inflammation in adipose tissue after chronic ethanol feeding to mice. _J. Biol. Chem._ 286, 35989–35997 (2011). Article CAS PubMed PubMed Central Google Scholar * Weisberg, S.
P. et al. Obesity is associated with macrophage accumulation in adipose tissue. _J. Clin. Invest_ 112, 1796–1808 (2003). Article CAS PubMed PubMed Central Google Scholar * Qin, Y. et
al. Adipose inflammation and macrophage infiltration after binge ethanol and burn injury. _Alcohol Clin. Exp. Res._ 38, 204–213 (2014). Article CAS PubMed Google Scholar * Naveau, S. et
al. Harmful effect of adipose tissue on liver lesions in patients with alcoholic liver disease. _J. Hepatol._ 52, 895–902 (2010). Article CAS PubMed Google Scholar * Russo, L. &
Lumeng, C. N. Properties and functions of adipose tissue macrophages in obesity. _Immunology_ 155, 407–417 (2018). Article CAS PubMed PubMed Central Google Scholar * Feldstein, A. E. et
al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. _Hepatology_ 40, 185–194 (2004). Article CAS PubMed Google Scholar *
Mandal, P., Pritchard, M. T. & Nagy, L. E. Anti-inflammatory pathways and alcoholic liver disease: role of an adiponectin/interleukin-10/heme oxygenase-1 pathway. _World J.
Gastroenterol._ 16, 1330–1336 (2010). Article CAS PubMed PubMed Central Google Scholar * Ikejima, K. et al. Leptin augments inflammatory and profibrogenic responses in the murine liver
induced by hepatotoxic chemicals. _Hepatology_ 34, 288–297 (2001). Article CAS PubMed Google Scholar * Shen, J., Sakaida, I., Uchida, K., Terai, S. & Okita, K. Leptin enhances
TNF-alpha production via p38 and JNK MAPK in LPS-stimulated Kupffer cells. _Life Sci._ 77, 1502–1515 (2005). Article CAS PubMed Google Scholar * Nagy, L. E. The role of innate immunity
in alcoholic liver disease. _Alcohol Res._ 37, 237–250 (2015). PubMed PubMed Central Google Scholar * McCullough, R. L. et al. Anaphylatoxin receptors C3aR and C5aR1 are important factors
that influence the impact of ethanol on the adipose secretome. _Front. Immunol._ 9, 2133 (2018). Article PubMed PubMed Central CAS Google Scholar * Mowat, A. M. & Agace, W. W.
Regional specialization within the intestinal immune system. _Nat. Rev. Immunol._ 14, 667–685 (2014). Article CAS PubMed Google Scholar * Habtezion, A., Nguyen, L. P., Hadeiba, H. &
Butcher, E. C. Leukocyte trafficking to the small intestine and colon. _Gastroenterology_ 150, 340–354 (2016). Article CAS PubMed Google Scholar * Okumura, R. & Takeda, K.
Maintenance of gut homeostasis by the mucosal immune system. _Proc. Jpn. Acad. Ser. B Phys. Biol. Sci._ 92, 423–435 (2016). Article CAS PubMed PubMed Central Google Scholar * Thursby,
E. & Juge, N. Introduction to the human gut microbiota. _Biochem J._ 474, 1823–1836 (2017). Article CAS PubMed Google Scholar * Mutlu, E. A. et al. Colonic microbiome is altered in
alcoholism. _Am. J. Physiol. Gastrointest. Liver Physiol._ 302, G966–G978 (2012). Article CAS PubMed PubMed Central Google Scholar * Levitt, M. D. et al. Use of measurements of ethanol
absorption from stomach and intestine to assess human ethanol metabolism. _Am. J. Physiol._ 273, G951–G957 (1997). CAS PubMed Google Scholar * Elamin, E. E., Masclee, A. A., Dekker, J.
& Jonkers, D. M. Ethanol metabolism and its effects on the intestinal epithelial barrier. _Nutr. Rev._ 71, 483–499 (2013). Article PubMed Google Scholar * Forsyth, C. B., Voigt, R. M.
& Keshavarzian, A. Intestinal CYP2E1: a mediator of alcohol-induced gut leakiness. _Redox Biol._ 3, 40–46 (2014). Article CAS PubMed PubMed Central Google Scholar * Cho, Y. E. et
al. Fructose promotes leaky gut, endotoxemia, and liver fibrosis through ethanol-inducible cytochrome P450-2E1-mediated oxidative and nitrative stress. _Hepatology_ (2019) [Ahead of print].
* Forsyth, C. B. et al. Role for intestinal CYP2E1 in alcohol-induced circadian gene-mediated intestinal hyperpermeability. _Am. J. Physiol. Gastrointest. Liver Physiol._ 305, G185–G195
(2013). Article CAS PubMed PubMed Central Google Scholar * Tripathi, A. et al. The gut-liver axis and the intersection with the microbiome. _Nat. Rev. Gastroenterol. Hepatol._ 15,
397–411 (2018). Article CAS PubMed PubMed Central Google Scholar * Parlesak, A., Schafer, C., Schutz, T., Bode, J. C. & Bode, C. Increased intestinal permeability to macromolecules
and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. _J. Hepatol._ 32, 742–747 (2000). Article CAS PubMed Google Scholar * Gu, L.
et al. Berberine ameliorates intestinal epithelial tight-junction damage and down-regulates myosin light chain kinase pathways in a mouse model of endotoxinemia. _J. Infect. Dis._ 203,
1602–1612 (2011). Article CAS PubMed Google Scholar * Guo, S., Al-Sadi, R., Said, H. M. & Ma, T. Y. Lipopolysaccharide causes an increase in intestinal tight junction permeability in
vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. _Am. J. Pathol._ 182, 375–387 (2013). Article CAS PubMed PubMed Central Google Scholar
* Yan, A. W. et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. _Hepatology_ 53, 96–105 (2011). Article CAS PubMed Google Scholar * Chen, Y. et al.
Characterization of fecal microbial communities in patients with liver cirrhosis. _Hepatology_ 54, 562–572 (2011). Article PubMed Google Scholar * Duan, Y. et al. Bacteriophage targeting
of gut bacterium attenuates alcoholic liver disease. _Nature_ 575, 505–511 (2019). Article CAS PubMed PubMed Central Google Scholar * Yuan, J. et al. Fatty liver disease caused by
high-alcohol-producing Klebsiella pneumoniae. _Cell Metab._ 30, 675–688 e677 (2019). Article CAS PubMed Google Scholar * Yang, A. M. et al. Intestinal fungi contribute to development of
alcoholic liver disease. _J. Clin. Invest_ 127, 2829–2841 (2017). Article PubMed PubMed Central Google Scholar * Leclercq, S., De Saeger, C., Delzenne, N., de Timary, P. & Starkel,
P. Role of inflammatory pathways, blood mononuclear cells, and gut-derived bacterial products in alcohol dependence. _Biol. Psychiatry_ 76, 725–733 (2014). Article CAS PubMed Google
Scholar * Fleming, S. et al. Pro- and anti-inflammatory gene expression in the murine small intestine and liver after chronic exposure to alcohol. _Alcohol Clin. Exp. Res._ 25, 579–589
(2001). Article CAS PubMed Google Scholar * Choudhry, M. A. et al. Impaired intestinal immunity and barrier function: a cause for enhanced bacterial translocation in alcohol intoxication
and burn injury. _Alcohol_ 33, 199–208 (2004). Article CAS PubMed Google Scholar * Akhtar, S., Li, X., Chaudry, I. H. & Choudhry, M. A. Neutrophil chemokines and their role in
IL-18-mediated increase in neutrophil O2- production and intestinal edema following alcohol intoxication and burn injury. _Am. J. Physiol. Gastrointest. Liver Physiol._ 297, G340–G347
(2009). Article CAS PubMed PubMed Central Google Scholar * Sibley, D. A., Fuseler, J., Slukvin, I. & Jerrells, T. R. Ethanol-induced depletion of lymphocytes from the mesenteric
lymph nodes of C57B1/6 mice is associated with RNA but not DNA degradation. _Alcohol Clin. Exp. Res._ 19, 324–331 (1995). Article CAS PubMed Google Scholar * Choudhry, M. A., Fazal, N.,
Goto, M., Gamelli, R. L. & Sayeed, M. M. Gut-associated lymphoid T cell suppression enhances bacterial translocation in alcohol and burn injury. _Am. J. Physiol. Gastrointest. Liver
Physiol._ 282, G937–G947 (2002). Article CAS PubMed Google Scholar * Li, X., Chaudry, I. H. & Choudhry, M. A. ERK and not p38 pathway is required for IL-12 restoration of T cell IL-2
and IFN-gamma in a rodent model of alcohol intoxication and burn injury. _J. Immunol._ 183, 3955–3962 (2009). Article CAS PubMed Google Scholar * Jarvelainen, H. A. et al. Promoter
polymorphism of the CD14 endotoxin receptor gene as a risk factor for alcoholic liver disease. _Hepatology_ 33, 1148–1153 (2001). Article CAS PubMed Google Scholar * Guo, J. &
Friedman, S. L. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. _Fibrogenes. Tissue Repair_ 3, 21 (2010). Article CAS Google Scholar * Seki, E. et al. TLR4
enhances TGF-beta signaling and hepatic fibrosis. _Nat. Med_ 13, 1324–1332 (2007). Article CAS PubMed Google Scholar * Zhao, E. et al. Bone marrow and the control of immunity. _Cell Mol.
Immunol._ 9, 11–19 (2012). Article CAS PubMed Google Scholar * Shi, C. & Pamer, E. G. Monocyte recruitment during infection and inflammation. _Nat. Rev. Immunol._ 11, 762–774
(2011). Article CAS PubMed PubMed Central Google Scholar * Soehnlein, O., Steffens, S., Hidalgo, A. & Weber, C. Neutrophils as protagonists and targets in chronic inflammation.
_Nat. Rev. Immunol._ 17, 248–261 (2017). Article CAS PubMed Google Scholar * Terai, S. et al. Improved liver function in patients with liver cirrhosis after autologous bone marrow cell
infusion therapy. _Stem Cells_ 24, 2292–2298 (2006). Article CAS PubMed Google Scholar * Suh, Y. G. et al. CD11b(+) Gr1(+) bone marrow cells ameliorate liver fibrosis by producing
interleukin-10 in mice. _Hepatology_ 56, 1902–1912 (2012). Article CAS PubMed Google Scholar * Spahr, L. et al. Granulocyte-colony stimulating factor induces proliferation of hepatic
progenitors in alcoholic steatohepatitis: a randomized trial. _Hepatology_ 48, 221–229 (2008). Article CAS PubMed Google Scholar * Cho, Y. et al. Efficacy of granulocyte colony
stimulating factor in patients with severe alcoholic hepatitis with partial or null response to steroid (GRACIAH trial): study protocol for a randomized controlled trial. _Trials_ 19, 696
(2018). Article CAS PubMed PubMed Central Google Scholar * Shasthry, S. M., Sharma, M. K., Shasthry, V., Pande, A. & Sarin, S. K. Efficacy of granulocyte colony-stimulating factor
in the management of steroid-nonresponsive severe alcoholic hepatitis: a double-blind randomized controlled trial. _Hepatology_ 70, 802–811 (2019). Article CAS PubMed Google Scholar *
Singh, V. et al. Granulocyte colony-stimulating factor in severe alcoholic hepatitis: a randomized pilot study. _Am. J. Gastroenterol._ 109, 1417–1423 (2014). Article CAS PubMed Google
Scholar * Yannaki, E. et al. G-CSF-primed hematopoietic stem cells or G-CSF per se accelerate recovery and improve survival after liver injury, predominantly by promoting endogenous repair
programs. _Exp. Hematol._ 33, 108–119 (2005). Article CAS PubMed Google Scholar * Lee, Y. S. et al. CX3CR1 differentiates F4/80(low) monocytes into pro-inflammatory F4/80(high)
macrophages in the liver. _Sci. Rep._ 8, 15076 (2018). Article PubMed PubMed Central CAS Google Scholar * Bonnardel, J. et al. Stellate cells, hepatocytes, and endothelial cells imprint
the kupffer cell identity on monocytes colonizing the liver macrophage niche. _Immunity_ 51, 638–654 e639 (2019). Article CAS PubMed PubMed Central Google Scholar * Sakai, M. et al.
Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain kupffer cell identity. _Immunity_ 51, 655–670 e658 (2019). Article CAS PubMed PubMed Central
Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (MEST)
(2018R1A2A1A05077608), Korea Mouse Phenotyping Project (2014M3A9D5A01073556), and the Intelligent Synthetic Biology Center of Global Frontier Project (2011–0031955) funded by the Ministry of
Science, ICT and Future Planning. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Laboratory of Liver Research, Graduate School of Medical Science and Engineering, Korea Advanced Institute of
Science and Technology, Daejeon, Korea Young-Ri Shim & Won-Il Jeong Authors * Young-Ri Shim View author publications You can also search for this author inPubMed Google Scholar * Won-Il
Jeong View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Won-Il Jeong. ETHICS DECLARATIONS CONFLICT OF INTEREST The
authors declare that they have no conflict of interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and
institutional affiliations. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons
license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a
credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted
use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT
THIS ARTICLE CITE THIS ARTICLE Shim, YR., Jeong, WI. Recent advances of sterile inflammation and inter-organ cross-talk in alcoholic liver disease. _Exp Mol Med_ 52, 772–780 (2020).
https://doi.org/10.1038/s12276-020-0438-5 Download citation * Received: 06 February 2020 * Revised: 07 April 2020 * Accepted: 07 April 2020 * Published: 26 May 2020 * Issue Date: May 2020 *
DOI: https://doi.org/10.1038/s12276-020-0438-5 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is
not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative









