- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT In non-small cell lung cancer (NSCLC), receptor tyrosine kinases (RTKs) stand out among causal dominant oncogenes, and the ablation of RTK signaling has emerged as a novel tailored
therapeutic strategy. Nonetheless, long-term RTK inhibition leads invariably to acquired resistance, tumor recurrence and metastatic dissemination. In ALK+ cell lines, inhibition of ALK
signaling was associated with coactivation of several RTKs, whose pharmacological suppression reverted the partial resistance to ALK blockade. Remarkably, ERBB2 signaling synergized with ALK
and contributed to the neoplastic phenotype. Moreover, the engagement of wild-type epidermal growth factor receptor or MET receptors could sustain cell viability through early growth
response 1 (EGR1) and/or Erk1/2; Akt activation and EGR1 overexpression prevented cell death induced by combined ALK/RTK inhibition. Membrane expression of ERBB2 in a subset of primary naive
ALK+ NSCLC could be relevant in the clinical arena. Our data demonstrate that the neoplastic phenotype of ALK-driven NSCLC relays ‘_ab initio_’ on the concomitant activation of multiple RTK
signals via autocrine/paracrine regulatory loops. These findings suggest that molecular and functional signatures are required in _de novo_ lung cancer patients for the design of
efficacious and multi-targeted ‘patient-specific’ therapies. SIMILAR CONTENT BEING VIEWED BY OTHERS COREGULATION OF PATHWAYS IN LUNG CANCER PATIENTS WITH EGFR MUTATION: THERAPEUTIC
OPPORTUNITIES Article 09 August 2021 TRIB3-EGFR INTERACTION PROMOTES LUNG CANCER PROGRESSION AND DEFINES A THERAPEUTIC TARGET Article Open access 21 July 2020 ALK-POSITIVE LUNG CANCER: A
MOVING TARGET Article 16 February 2023 INTRODUCTION Lung tumors are the most common form of cancer (12.3%), with 1.1 million deaths worldwide, accounting for 17.8% of all cancer fatalities.
Despite major diagnostic improvements and novel tailored compounds, 90% of lung cancer patients die within 1–2 years from diagnosis.1 Molecular studies have showed that nearly all lung
cancers display multiple genetic defects2, 3, 4 and the abnormal activation of kinases is widely recognized as a frequent tumorigenic mechanism in non-small cell lung cancer (NSCLC).3, 5, 6,
7 Epidermal growth factor receptor (EGFR) and its ligands are commonly deregulated, leading to receptor tyrosine kinase (RTK) phosphorylation fostering cell proliferation, inhibition of
apoptosis and enhancing pro-angiogenesis and invasion signals.8 Tumor-acquired EGFR mutations have been documented in 5–10% of Caucasian NSCLC, often associated to unique clinico-pathologic
features.9 Specific genetic alterations of KRAS, BRAF, PIK3CA, ALK and, more recently, ROS and RET have been described in well-defined subsets of NSCLC.6, 7, 10, 11 These lesions represent
‘driver mutations’, as lung cancers depend on their constitutive activation.12, 13 Thus, molecular screening of NSCLC is mandatory to guide the most appropriate therapies (that is, small
molecule kinase inhibitors).14, 15 Cancers harboring the same genetic alterations, however, respond differently to molecular targeted therapies, suggesting coexisting mutations or
coactivation of other TKs.16 Finally, although molecularly targeted therapies (that is, gefitinib or erlotinib for EGFR and crizotinib for ALK fusions) are effective in NSCLC, intrinsic or
acquired resistances inevitably lead to recurrence and/or metastatic dissemination. This is the case of MET activation/amplification,17, 18 or acquired somatic mutations of EGFR (T790M)19 or
ALK fusions (G1269A, L1196M and ALK amplification).20, 21, 22 Chromosomal translocations involving the ALK gene have been described in 6% of NSCLC, where ALK is most frequently fused to the
echinoderm microtubule-associated protein-like 4 (_EML4_).23 As described for other ALK chimera, the EML4–ALK fusions contain a dimerization domain, which forces the ligand-independent
oligomerization of the catalytic domain of ALK, resulting in its constitutive kinase activation. ALK fusions are oncogenic ‘drivers’ imposing a full cellular transformation, and are required
for the maintenance of the neoplastic phenotype.24, 25 Nevertheless, data from phase I clinical trials of ALK-rearranged NSCLC treated with the ALK inhibitor (ALKi) crizotinib showed an
overall response rate of ∼60%, suggesting that synergizing or overcoming mechanisms may occur.26, 27 Here we provide novel insights into ALK-driven transformation in NSCLC, demonstrating
that tumor maintenance and survival of ALK+ NSCLC cells, before any therapeutic selection, relay on the concomitant activation of multiple EGFR family RTKs. These data support the hypothesis
that the degree of ALK addiction might differ ‘_ab initio_’ among different ALK+ NSCLC patients and its identification should provide a novel rationale for the design of efficacious
‘patient-specific’ treatments. RESULTS ALK FUSION PROTEINS TRANSFORM HUMAN IMMORTALIZED LUNG EPITHELIAL CELLS AND SUSTAIN THE TUMOR PHENOTYPE OF ALK+ NSCLC CELL _IN VITRO_ AND _IN VIVO_ As
it has been described that transformation of lung epithelial cells requires many hits, and that several oncogenes are unable to fully transform these elements, even in the presence of
deregulated p53 and Rb-1 pathways, we assessed the transforming potential of ALK chimera in immortalized cell line (NuLi-1) derived from human normal airway epithelium. NuLi-1 cells were
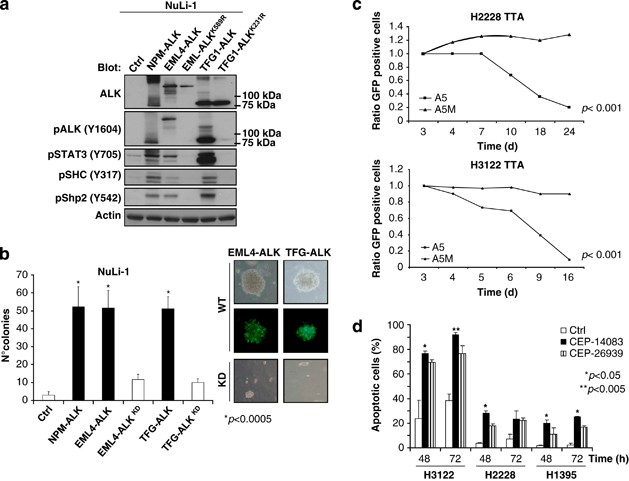
transduced with EML4–ALK variant 1 (EML4/exon13–ALK/exon20) or TFG–ALK constructs. Forced expression of EML4–ALK and TFG–ALK fusions led to constitutive phosphorylation of STAT3, SHC and
Shp2 (Figure 1a), and imposed a transformed phenotype (Figure 1b). To evaluate the biological effects of EML4–ALK, we transduced ALK-positive cell lines (H3122 and H2228) with ALK-specific
inducible short hairpin RNA (shA5) or control short hairpin RNA (shA5M) lentivirus cassettes. Alternatively, cells (ALK+ and ALK− (H1395)) were treated with ALK-selective inhibitors
(CEP-14083 and CEP-26939, from Cephalon/Teva, West Chester, PA, USA).28, 29 ALK inhibition led to a significant decrease in the phosphorylation of known ALK downstream targets (STAT3,
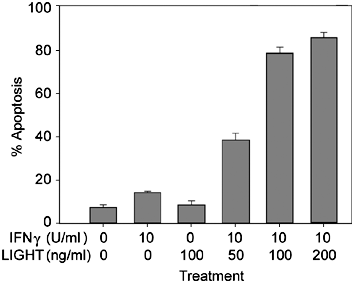
Erk1/2, Shp2 and Akt; Supplementary Figure 1A).24 As expected, ablation of ALK signaling was associated with growth impairment (Figure 1c and Supplementary Figure 1B) and a very high rate of
apoptosis for the H3122 cells (Figure 1d and Supplementary Figure 1C). Consistently, _in vivo_ treatment with CEP-26939 led to a decreased number of proliferating cells, without any
significant loss of ALK protein expression (Supplementary Figures 1D and E), and to tumor mass regression. This treatment, however, did not result in tumor eradication (Supplementary Figure
1F). SIGNALING THROUGH THE EGFR–TK PROTEINS CONTRIBUTES TO THE SURVIVAL AND GROWTH OF ALK+ NSCLC CELLS On the basis of our previous phosphoproteomics findings30 and on the different degree
of ALK oncogenic addiction of ALK+ NSCLC lines and primary tumors,31 we investigated the association of anti-ALKi29 with small anti-TK drugs currently into the clinics. Combined treatment of
H2228 cells with ALKi and with the EGFR inhibitor gefitinib, or with an anti-EGFR monoclonal antibody (cetuximab), resulted in increased cell death. Similar findings were seen in H3122
cells after single anti-ALK treatment (Figure 2a and Supplementary Figures 2A and B), possibly related to higher protein expression and phosphorylation status of EGFR in only H2228 cells
(Supplementary Figures 2C and D). Importantly, H2228 and H3122 cells were both insensitive to single treatment with gefitinib. As other members of the EGFR family are associated to RTK
inhibitor resistance, we then investigated the functional role of ERBB2 and ERBB3 in the less-responsive H2228 cells.18, 20 We observed a marked presence of ERBB2/ERBB3 and EGFR/ERBB3
heterodimers in H2228 in the absence of exogenous stimuli (Supplementary Figure 3A). Moreover, ERBB3 protein/mRNA levels were upregulated in H2228 when treated with anti-ALK small molecules
(Figure 2b). Interestingly, we detected a constitutive phosphorylation of ERBB2 that was completely abrogated upon treatment with lapatinib (Figure 2b and Supplementary Figure 3B), and the
combination of lapatinib with CEP-28122 suppressed cell growth and significantly reduced the number of viable cells as compared with CEP-28122 alone (Figure 2c). In contrast, the combined
treatment had no further effect in H3122 cells (Supplementary Figure 3C). Remarkably, a significant subset of primary ALK+ NSCLC (12/21, 57%; but ALK− NSCLC 38/180, 21%) shows a strong
immunohistochemical membrane staining for ERBB2 (Figure 2d). Overall, these data suggest that ALK inhibition might be overcome by alternative RTK pathways in ALK+ NSCLC cells.
LIGAND-MEDIATED ACTIVATION OF TYROSINE RECEPTOR KINASES RESCUES ALK KINASE INHIBITION THROUGH AKT AND ERK1/2 PATHWAYS As EGF can be secreted by lung epithelial cancer and/or stromal cells,
we investigated whether an autocrine–paracrine EGF-mediated activation of the EGFR signaling in H2228 cells could explain their partial resistance to ALK inhibition and overcome ALK
inhibition of ALK-sensitive cells. Consistent with recently published data, upon stimulation with exogenous EGF the effects associated with the abrogation of the ALK signaling (CEP-28122)
were overcome in both cell lines as documented by an increased cell viability and growth, which were restored to baseline levels (Figure 3a and Supplementary Figures 4A and B). Cells
cotreated with gefitinib and/or expressing a mutant form of the EGFR (T790, data not show) could not be rescued by the exogenous EGF (Supplementary Figure 4A).20, 32 The molecular mechanisms
operating in the EGF/EGFR-mediated rescue were first examined by the phosphorylation status of ALK and known ALK effectors. These were not affected. On the contrary, EGF stimulation could
sustain the phosphorylated status of EGFR, Akt and Erk1/2, despite an ALK inhibition (Figure 3b). Finally, an increased expression of the proapoptotic protein BIM was seen only with the dual
inhibitor cotreatment in H2228, whereas ALK inhibition alone was sufficient to induce a significant upregulation of BIM in H3122 (Figure 3b).33 Similar results were obtained by heregulin
stimulation. Upon heregulin stimulation the phosphorylation of ERBB3, Erk1/2 and Akt significantly increased (Figure 3c and Supplementary Figure 4B) and cell viability was rescued despite
ALK inhibition (Figure 3d). As hepatocyte growth factor (HGF)/Met signaling or Met overamplification can be involved in gefitinib resistance in lung cancer,18, 34 we then explored the
effects of HGF. HGF promoted a moderate proliferation and sustained cell viability upon ALK inhibition through Erk1/2 and Akt pathways (Supplementary Figures 5A and B). As expected,
crizotinib, a dual ALK–Met inhibitor, abolished the prosurvival signals triggered by HGF (Supplementary Figure 5C). As Src has a pathogenetic role in EGFR-mutated NSLCL35 and is also a
critical interactor in ALK+ anaplastic large cell lymphoma,24 we applied a selective Src kinase inhibitor-1 as a single agent (Figure 4a) or in combination with CEP-28122. The combined
treatment increased cell death only in H2228 that was partially rescued by EGF administration (Figures 4b–e). These data suggest that Src-independent pathways might be active in H2228 cells.
On the contrary, EGF did not restore cell viability of H3122 cells treated with ALK and Src–TK inhibitor. Similar findings were observed using dasatinib, a pan Src inhibitor (data not
shown). EARLY GROWTH RESPONSE 1 (_EGR1_) GENE IS UPREGULATED BY ACTIVE ALK AND EGFR, AND PROMOTES SURVIVAL To identify ALK cooperating oncogene(s) in H2228 NSCLC cells, we interrogated their
transcriptome before and after TK inhibitor treatment. Gene expression analysis showed that a single gene, early growth response 1 (_EGR1_), was significantly suppressed by the combined
ALK/EGFR treatment (fold-change >2, _P_<0.001; Supplementary Figure 6). Quantitative reverse transcription–PCR confirmed that the loss of ALK signaling or the dual treatment led to a
substantial inhibition of the EGR1 expression (Figure 5a), whereas gefitinib had no effect. Downregulation of EGR1 could be inhibited by exogenous EGF that overcame cell death as well
(Supplementary Figure 7A). Moreover, Src inhibition in association with CEP-28122 strongly reduced the level of expression of EGR1 in both cell lines (Supplementary Figure 7B). Reversely,
ectopic expression of EGR1 in H2228 cells (Supplementary Figure 7C) prevented cell growth inhibition and cell death via CEP-28122 and/or gefitinib combination (Figures 5b and c). Finally,
forced expression of EGR1 could revert BIM levels to baseline (Figure 5d and Supplementary Figure 7D), meanwhile knockdown of EGR1 in H2228 through short hairpin RNA impaired cell growth
(data not shown). Overall, these findings indicate that both ALK and EGFR signaling are required for EGR1 expression and demonstrated a central role of this transcription factor in ALK+
NSCLC cells. To identify _EGR1_-regulated genes, we investigated the transcriptional profile of H2228 cells stably transduced with EGR1 (EGR1+). This revealed a set of 780 genes linked to
EGR1 (fold-change >2, _P_<0.001). Notably, EGR1 overexpression resulted in ERBB3 mRNA upregulation that was further increased by ALK inhibition (Supplementary Figure 8). Expression
profiling of EGR1+ cells treated with ALKi and EGFR–TK inhibitor reported two clusters of genes behaving independently of ALK inhibition, as these genes showed a marked up- or downregulation
in wild-type treated cells, and no change or even an opposite behavior in EGR1+ cells (Figure 6). In particular, seven genes were upregulated by EGR1 transduction and remained significantly
upregulated despite the treatment with TK inhibitor, suggesting direct and robust regulation by this transcription factor (that is, TFF2, PPDPF, AGR2 and CCL5; Supplementary Figure 8).
DISCUSSION The discovery of specific genetic lesions in lung cancer patients has profoundly changed their clinical management. Kinases are now recognized as critical tumorigenic events in
human cancer. Constitutively active receptor and non-RTKs are excellent examples for the development of modern ‘patient-specific’ and/or ‘tumor-restricted’ therapies.36 However, clinical
responses are heterogeneous as a result of multiple mechanisms of resistance. It is believed that multi-targeted drugs may be more appropriate than highly restricted inhibitors. In NSCLC
patients, ALK is a powerful driver/oncogene;37 nevertheless, crizotinib, a dual ALK/Met inhibitor, is successful only in a subset of ALK+ patients (∼60%). Moreover, responder patients
inevitably become refractory.38 Thus, the different responses to ALKi may underlie intrinsic mechanisms that undermine ‘_ab initio_’ their efficacy and/or favor the occurrence of resistance.
Their identification provide the basis for a molecular stratification of ALK+ NSCLC patients and drive the selection of more compliant treatments.21, 39 Here we studied the level of ALK
oncogenic addiction of human ALK+ NSCLC and the cooperative role of several kinases. We show that the different responses of NSCLC cell lines to ALK are dependent on the activation of
alternative pathways in which wild-type RTK can overcome ALK signaling blockade. Using phosphoproteomics, different studies have demonstrated that lung cancer cells and primary tumors
express kinases able to jointly participate to hierarchical networks or independently signal through separate parallel pathways.5, 40 Our previous phosphomapping findings showed that ALK
inhibition is rapidly followed by the parallel activation/phosphorylation of several other TKs (EGFR, Met, fibroblast growth factor receptor, Jak1 or insulin-like growth factor receptor) in
H2228 cells, but not in H3122 cells, which are markedly ALK-addicted.30 Interestingly, the dual inhibition with gefitinib or cetuximab overcomes this resistance feedback loop and increases
cell death. The elevated levels of EGFR protein in H2228 compared with H3122, without displaying any amplification or known mutation, might explain their partial addiction to ALK and the
better responsiveness to the cotreatment. Notably, other members of the EGFR family, ERBB2 and ERBB3, can also overcome the partial resistance to ALKi, as the combination with lapatinib and
an ALKi markedly reduced ERBB2 phosphorylation and cell viability. This suggests that forced EGFR family signaling (EGFR and ERBB2), even in naive/untreated NSCLC cells, may be critical,
similar to what has been recently described for the acquired resistance to ALKi.20, 41 Nevertheless, we demonstrated that this occurs in a context of wild-type EGFR and non-mutated ALK and,
more importantly, in absence of drug selection.17, 42 Our studies are in line with those described by Prahallad _et al._43, which have documented an intrinsic resistance to vemurafenib
caused by a feedback activation of EGFR signaling in BRAF (V600E) mutant colon cancer cells. Thus, EGFR members can elicit an undermining mechanism that sustains the partial resistance seen
in some ALK+ NSCLC patients, or ultimately post-therapy resistance to ALKi.41 A greater complexity is provided by the observation that the engagement of wild-type EGFR or wild-type MET
receptors could also restore cell viability of H2228 and H3122 cells via EGR1 and/or Erk1/2 and Akt activation.32 Remarkably, in H2228 the effects of HGF on cell survival were less
significant, more likely for the partial dependence of these cells on EGFR signaling. We believe that EGFR member family expression levels or phosphorylation should be determined at
diagnosis in order to benefit the most on combined therapies. As cancer cells often trigger alternative pathways to bypass specific RTK inhibition, we further dissected other signaling
pathways to find a common node of ALK and other RTKs. In ALK+ NSCLC cells, Src kinase represents a suitable target as in ALK+ anaplastic lymphoma its ablation has relevant effects in analogy
to many human cancers.35 In H2228 cells, Src inhibitors alone or in combination with ALKi increased the percentage of apoptotic cells similarly to ALK and gefitinib, suggesting that Src
mediates ALK and EGFR signaling in NSCLC. These data are consistent with the notion that ALK and EGFR pathways functionally compensate each other. Nevertheless, other alternative signaling
pathways might be operational in H2228, as EGF partially recovered cells from death. Indeed, rescue by EGF in H3122 was completely abrogated upon ALK and Src inhibition, suggesting that
EGF/EGFR signaling triggers prosurvival responses through Src. Next, to define additional putative downstream ‘culprits’, we used gene expression profiling analyses, which showed the _EGR1_
gene as a key regulator. The cotreatment with ALKi and gefitinib downregulated the expression of a single transcript, EGR1. EGR1 is a transcription factor, known to have a
polyhedric/versatile role in human cancers,44 engaged in proapoptotic or prosurvival signals, depending on the cellular context.45 In solid tumors, including lung cancers, EGR1 is a
downstream target of EGFR through the mitogen-activated protein kinase signaling pathways.46 Indeed, its overexpression, able to sustain cell viability in presence of ALK and EGFR
inhibitors, was characterized by well-defined transcriptional signature. Among the upregulated genes, _TFF2_ (trefoil factor 2) and _AGR2_ (anterior gradient 2) seem to be relevant, as in
human cancers they contribute to tumor growth, resistance to apoptosis and cell migration.47, 48 Thus, EGR1 could represent an ideal target for the design and implementation of new
therapeutic approaches in ALK-positive NSCLC. Our data indicate that the molecular characterization of primary lung cancers is required to foresee whether any given therapeutic protocol may
be successful. The genetic heterogeneity of lung tumors implies that parallel pathways or different mechanisms of resistance may coexist ‘_ab initio_’ and that targeting a single molecule
might be ineffective. This stratification is necessary to develop new combined treatment for NSCLC patients. Targeting common molecules downstream of RTKs, such as EGR1 or Src, could bypass
intrinsic or acquired drug resistance derived from the treatment with a single RTK inhibitor. Our data support the idea that the development of less-restricted drugs or the design of new
therapeutic protocols making use of multiple TK inhibition might be more efficacious, even for the first-line treatment of NSCLC. MATERIALS AND METHODS CELL CULTURES, INHIBITORS AND GROWTH
FACTORS Human lung cancer EML4–ALK-positive cell lines, H2228 (var 2: EML4/es6–ALK/es20) and H3122 (var 1: EML4/es13–ALK/es20), were grown in RPMI medium (Lonza, Basel, Switzerland) with 10%
fetal calf serum (FCS). Human HEK-293T cells were cultured in Dulbecco’s modified Eagle’s medium with 10% FCS. Human immortalized airway epithelial cells (NuLi-1) were grown in bronchial
epithelial cell basal medium (Lonza) with 10% FCS on human collagen type-VI-coated plates (Sigma, St Louis, MO, USA). ALK inhibitors, CEP-14083, CEP-26939 and CEP-28122 (Cephalon), were used
at 300 nM/l; gefitinib (ZD1839, Iressa) at 1 μM/l; lapatinib at 2 μM/l; Src kinase inhibitor-1 (Calbiochem, Millipore Corporation, Billerica, MA, USA) at 5 μM/l; and PHA-665752 (Met
inhibitor) at 250 nM. EGF was used at 10 ng/ml, Heregulin was used at 20 ng/ml, and HGF (Sigma) was used at 50 ng/ml. CELL PROLIFERATION, CELL CYCLE ANALYSIS, CELL APOPTOSIS AND CLONOGENIC
ASSAYS In 24-well plates, 8 × 104 cells/ml were grown in duplicates. The treatment with kinase inhibitors or with growth factors was done every 24 h in 0.05% FCS. Cell growth was analyzed
using Cell Titer-GloMax assay (Promega, Fitchburg, WI, USA). For cell cycle analysis, cells were fixed for 1 h in 70% ethanol at 4 °C. After washing, cells were treated with RNase (0.25
mg/ml) and stained with propidium iodide (50 μg/ml). The sub-G0-phase fraction was calculated using the Modfit program from Becton-Dickinson (San Jose, CA, USA). Apoptosis was measured by
flow cytometry after staining with the mitochondrion-permeable voltage sensitive dye tetrametylrodamine methyl ester and fluorescence-activated cell sorting analysis. Clonogenic assay, were
performed as described.28 DNA CONSTRUCTS EGR1 cDNA in pCMV-Sport6 was purchased from ImaGenes GmbH (Berlin, Germany). EGR1 cDNA was then subcloned into the lentiviral vector pWPI for
lentivirus production. pCMV-Sport6 containing EGR1 construct was digested to obtain a _Eco_RV/_Not_I fragment, blunted with Klenow enzyme (Roche, Basilea, Switzerland) and cloned into the
_Pme_I site of the lentiviral vector pWPI. LENTIVIRUS AND SHORT HAIRPIN RNA PRODUCTION, CELL INFECTION AND CELL SORTING Lentivirus particles were obtained as described.27 For cell sorting
enrichments, EGFP-positive cells were sorted using a MoFlo High-Performance instrument (DAKO Cytomation, Glostrup, Denmark) and EGFP expression was checked by flow cytometry over time. CELL
LYSIS AND WESTERN BLOT ANALYSIS H3122 and H2228 were plated on six-well plates and treated with specific kinase inhibitors in Dulbecco’s modified Eagle’s medium with 0.05% FCS. Cells were
lysed with GST-FISH (10 mM MgCl2, 150 mM NaCl, 1% NP40, 2% Glycerol, 1 mM EDTA, 25 mM HEPES (pH 7.5)) buffer supplemented with protease inhibitors (NaF, phenylmethanesulfonylfluoride, PIC
and Na3VO4) by scraping. The list of the antibodies is provided in Supplementary Information. RNA EXTRACTION, CDNA SYNTHESIS AND REVERSE TRANSCRIPTION–PCR Total RNA was isolated from cells
with TRIZOL lysis solution (Invitrogen, Life Technologies Ltd, Paisley, UK). One microgram of total RNA was treated with 5 U/μl of DNase I recombinant RNase free (Roche) and it was subjected
to reverse transcription with SuperScript III reverse transcriptase (Invitrogen). The cDNA obtained was subjected to PCR using specific primers for PBGD, B2M and ABL as control of cDNA
quality. CDNA MICROARRAY AND ANALYSIS Total RNA was extracted using the TRIZOL reagent (Invitrogen) and purified using the RNeasy total RNA Isolation Kit (Qiagen, Hilden, Germany). RNA
integrity was verified by means of a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). Hybridization was performed on HumanWG-12v4 BeadChips (Illumina, San Diego, CA, USA),
using biological triplicates for each condition. cDNA and biotinylated cRNA were generated by Illumina TotalPrep RNA Amplification Kit (Ambion, Life Technologies Ltd, Paisley, UK). Data were
processed with the Illumina GenomeStudio software using the following thresholds for significant differential expression: ‘Illumina Custom’ test _P_-value <0.001, fold-change >2. Gene
expression data were clustered and visualized with the GEDAS software.29 REAL-TIME QUANTITATIVE PCR cDNA was transcribed using SuperSCRIPT III following the manufacturer’s instructions
(Invitrogen). Quantitative reverse transcription–PCR was performed in triplicate on ICycler iQ Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA) with SYBR green dye.
IMMUNOFLUORESCENCE AND IMMUNOHISTOCHEMISTRY Cells were grown for 12 h on glass coverlips, fixed in phosphate-buffered saline containing 4% paraformaldehyde at room temperature (10 min), and
then permeabilized with phosphate-buffered saline containing 0.3% Triton X-100 for 5 min. Anti-EGFR (1:100, Cell Signaling Technology, Danvers, MA, USA), ERBB2 (1:300, Dako) and ERBB3
(1:100, LabVision, ThermoScientific, Kalamazoo, MI, USA) were incubated O/N, and heterodimer formation was detected using DUOLINK II assay (Olink Bioscience, Uppsala, Sweden). Images were
acquired by Imager.Z1 fluorescent microscope (Zeiss) and analysed with the Isis and Metaphor software. Immunohistochemistry was performed on a semi-automated immunostainer as described
previously.30 The following antibodies were used: anti-ALK (clone D5F3; Cell Signaling Technology) and anti-ERBB2 (clone CB11; Novocastra, Leica, Milano, Italy). MICE AND _IN VIVO_
BIOLUMINESCENCE EXPERIMENTS CB/17 scid and NOD-SCID mice (Charles River Laboratories Italia S.p.A., Wilmington, DE, USA) were inoculated subcutaneously in both flanks (single-cell suspension
1 × 107 in 0.2 ml phosphate-buffered saline). Mice were treated twice a day with 100 mg/kg of Comp-2. Tumor growth was measured over time. For imaging studies, luciferase-positive cell
xenografts were analyzed _in vivo_ by IVIS100 (Xenogen, Grantham, Lincolnshire, UK) instrument after intraperitoneal injection with D-luciferin (10 μl/g body weight, 15 mg/ml). Mice were
handled and treated in accordance with the European Community guidelines. REFERENCES * Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D . Global cancer statistics. _CA Cancer J Clin_
2011; 61: 69–90. Article PubMed Google Scholar * Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D _et al_. Oncogenic pathway signatures in human cancers as a guide to targeted
therapies. _Nature_ 2006; 439: 353–357. Article CAS PubMed Google Scholar * Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G _et al_. Patterns of somatic mutation in
human cancer genomes. _Nature_ 2007; 446: 153–158. Article CAS PubMed PubMed Central Google Scholar * Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R _et al_. Characterizing the
cancer genome in lung adenocarcinoma. _Nature_ 2007; 450: 893–898. Article CAS PubMed PubMed Central Google Scholar * Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H _et al_. Global
survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. _Cell_ 2007; 131: 1190–1203. Article CAS PubMed Google Scholar * Kohno T, Ichikawa H, Totoki Y, Yasuda
K, Hiramoto M, Nammo T _et al_. KIF5B-RET fusions in lung adenocarcinoma. _Nat Med_ 2012; 18: 375–377. Article CAS PubMed PubMed Central Google Scholar * Lipson D, Capelletti M,
Yelensky R, Otto G, Parker A, Jarosz M _et al_. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. _Nat Med_ 2012; 18: 382–384. Article CAS PubMed
PubMed Central Google Scholar * Hynes NE, Lane HA . ERBB receptors and cancer: the complexity of targeted inhibitors. _Nat Rev Cancer_ 2005; 5: 341–354. Article CAS PubMed Google
Scholar * Jackman DM, Chirieac LR, Janne PA . Bronchioloalveolar carcinoma: a review of the epidemiology, pathology, and treatment. _Semin Respir Crit Care Med_ 2005; 26: 342–352. Article
PubMed Google Scholar * Sekido Y, Fong KM, Minna JD . Molecular genetics of lung cancer. _Annu Rev Med_ 2003; 54: 73–87. Article CAS PubMed Google Scholar * Takeuchi K, Soda M, Togashi
Y, Suzuki R, Sakata S, Hatano S _et al_. RET, ROS1 and ALK fusions in lung cancer. _Nat Med_ 2012; 18: 378–381. Article CAS PubMed Google Scholar * Pao W, Girard N . New driver
mutations in non-small-cell lung cancer. _Lancet Oncol_ 2011; 12: 175–180. Article CAS PubMed Google Scholar * Haber DA, Settleman J . Cancer: drivers and passengers. _Nature_ 2007; 446:
145–146. Article CAS PubMed Google Scholar * Pao W, Kris MG, Iafrate AJ, Ladanyi M, Janne PA, Wistuba II _et al_. Integration of molecular profiling into the lung cancer clinic. _Clin
Cancer Res_ 2009; 15: 5317–5322. Article PubMed Google Scholar * Sequist LV, Heist RS, Shaw AT, Fidias P, Rosovsky R, Temel JS _et al_. Implementing multiplexed genotyping of
non-small-cell lung cancers into routine clinical practice. _Ann Oncol_ 2011; 22: 2616–2624. Article CAS PubMed PubMed Central Google Scholar * Stommel JM, Kimmelman AC, Ying H,
Nabioullin R, Ponugoti AH, Wiedemeyer R _et al_. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. _Science_ 2007; 318: 287–290. Article
CAS PubMed Google Scholar * Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E _et al_. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC.
_Cancer Cell_ 2010; 17: 77–88. Article CAS PubMed PubMed Central Google Scholar * Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO _et al_. MET amplification leads to
gefitinib resistance in lung cancer by activating ERBB3 signaling. _Science_ 2007; 316: 1039–1043. Article CAS PubMed Google Scholar * Engelman JA, Janne PA . Mechanisms of acquired
resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. _Clin Cancer Res_ 2008; 14: 2895–2899. Article PubMed Google Scholar * Katayama R,
Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B _et al_. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. _Sci Transl Med_ 2012; 4: 120ra117. Article
Google Scholar * Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ _et al_. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell
lung cancer. _Clin Cancer Res_ 2012; 18: 1472–1482. Article CAS PubMed PubMed Central Google Scholar * Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T _et al_. EML4-ALK
mutations in lung cancer that confer resistance to ALK inhibitors. _N Engl J Med_ 2010; 363: 1734–1739. Article CAS PubMed Google Scholar * Soda M, Choi YL, Enomoto M, Takada S,
Yamashita Y, Ishikawa S _et al_. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. _Nature_ 2007; 448: 561–566. Article CAS PubMed Google Scholar *
Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G . The anaplastic lymphoma kinase in the pathogenesis of cancer. _Nat Rev Cancer_ 2008; 8: 11–23. Article CAS PubMed Google Scholar *
Piva R, Chiarle R, Manazza AD, Taulli R, Simmons W, Ambrogio C _et al_. Ablation of oncogenic ALK is a viable therapeutic approach for anaplastic large-cell lymphomas. _Blood_ 2006; 107:
689–697. Article CAS PubMed PubMed Central Google Scholar * Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA _et al_. Effect of crizotinib on overall survival in patients
with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. _Lancet Oncol_ 2011; 12: 1004–1012. Article CAS PubMed PubMed Central Google Scholar
* Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG _et al_. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. _N Engl J Med_ 2010; 363: 1693–1703. Article CAS
PubMed PubMed Central Google Scholar * Ott GR, Wells GJ, Thieu TV, Quail MR, Lisko JG, Mesaros EF _et al_. 2,7-disubstituted-pyrrolo[2,1-f][1,2,4]triazines: new variant of an old
template and application to the discovery of anaplastic lymphoma kinase (ALK) inhibitors with in vivo antitumor activity. _J Med Chem_ 2011; 54: 6328–6341. Article CAS PubMed Google
Scholar * Cheng M, Quail MR, Gingrich DE, Ott GR, Lu L, Wan W _et al_. CEP-28122, a highly potent and selective orally active inhibitor of anaplastic lymphoma kinase with antitumor activity
in experimental models of human cancers. _Mol Cancer Ther_ 2012; 11: 670–679. Article CAS PubMed Google Scholar * Voena C, Panizza E, D’Amico L, Ambrogio C, Martinengo C, Boccalatte FE
_et al_. EML4-ALK signaling is required for the maintenance of neoplastic phenotype of non-small cell lung cancer cells: novel strategy for lung cancer tailored therapies. _AACR 101ST Annual
Meeting 2010_. American Association of Cancer Research (AACR), Washington DC, USA, 2010. Google Scholar * Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS _et al_.
Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. _J Clin Oncol_ 2009; 27: 4247–4253. Article CAS PubMed PubMed Central Google Scholar *
Yamada T, Takeuchi S, Nakade J, Kita K, Nakagawa T, Nanjo S _et al_. Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor-resistance in
EML4-ALK lung cancer cells. _Clin Cancer Res_ 2012; 18: 3592–3602. Article CAS PubMed Google Scholar * Takezawa K, Okamoto I, Nishio K, Janne PA, Nakagawa K . Role of ERK-BIM and
STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. _Clin Cancer Res_ 2011; 17: 2140–2148. Article CAS PubMed Google Scholar * Yano S,
Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T _et al_. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating
mutations. _Cancer Res_ 2008; 68: 9479–9487. Article CAS PubMed Google Scholar * Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW _et al_. Combating trastuzumab resistance by targeting
SRC, a common node downstream of multiple resistance pathways. _Nat Med_ 2011; 17: 461–469. Article PubMed Google Scholar * Janku F, Stewart DJ, Kurzrock R . Targeted therapy in
non-small-cell lung cancer--is it becoming a reality? _Nat Rev Clin Oncol_ 2011; 7: 401–414. Article Google Scholar * Gerber DE, Minna JD . ALK inhibition for non-small cell lung cancer:
from discovery to therapy in record time. _Cancer Cell_ 2010; 18: 548–551. Article CAS PubMed PubMed Central Google Scholar * Sasaki T, Janne PA . New strategies for treatment of
ALK-rearranged non-small cell lung cancers. _Clin Cancer Res_ 2011; 17: 7213–7218. Article CAS PubMed PubMed Central Google Scholar * Katayama R, Khan TM, Benes C, Lifshits E, Ebi H,
Rivera VM _et al_. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. _Proc Natl Acad Sci USA_ 2011; 108:
7535–7540. Article CAS PubMed PubMed Central Google Scholar * Guo A, Villen J, Kornhauser J, Lee KA, Stokes MP, Rikova K _et al_. Signaling networks assembled by oncogenic EGFR and
c-Met. _Proc Natl Acad Sci USA_ 2008; 105: 692–697. Article CAS PubMed PubMed Central Google Scholar * Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W _et al_. A novel
ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. _Cancer Res_ 2011; 71: 6051–6060. Article CAS PubMed PubMed Central Google Scholar * McDermott U,
Pusapati RV, Christensen JG, Gray NS, Settleman J . Acquired resistance of non-small cell lung cancer cells to MET kinase inhibition is mediated by a switch to epidermal growth factor
receptor dependency. _Cancer Res_ 2010; 70: 1625–1634. Article CAS PubMed PubMed Central Google Scholar * Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D _et al_.
Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. _Nature_ 2012; 483: 100–103. Article CAS PubMed Google Scholar * Sauer L, Gitenay D, Vo C,
Baron VT . Mutant p53 initiates a feedback loop that involves Egr-1/EGF receptor/ERK in prostate cancer cells. _Oncogene_ 2010; 29: 2628–2637. Article CAS PubMed PubMed Central Google
Scholar * Thiel G, Cibelli G . Regulation of life and death by the zinc finger transcription factor Egr-1. _J Cell Physiol_ 2002; 193: 287–292. Article CAS PubMed Google Scholar *
Uramoto H, Shimokawa H, Hanagiri T, Kuwano M, Ono M . Expression of selected gene for acquired drug resistance to EGFR-TKI in lung adenocarcinoma. _Lung Cancer_ 2011; 73: 361–365. Article
PubMed Google Scholar * Park K, Chung YJ, So H, Kim K, Park J, Oh M _et al_. AGR2, a mucinous ovarian cancer marker, promotes cell proliferation and migration. _Exp Mol Med_ 2011; 43:
91–100. Article CAS PubMed PubMed Central Google Scholar * Kosriwong K, Menheniott TR, Giraud AS, Jearanaikoon P, Sripa B, Limpaiboon T . Trefoil factors: tumor progression markers and
mitogens via EGFR/MAPK activation in cholangiocarcinoma. _World J Gastroenterol_ 2011; 17: 1631–1641. Article CAS PubMed PubMed Central Google Scholar Download references
ACKNOWLEDGEMENTS We thank Dr Paola Bernabei for the Flow Cytometry studies and Cell Sorting, Dr Flavio Cristofani for the mouse housing and care, and Dr Daniela Cantarella for
microarray-based gene expression profiling. We thank Dr Klingelhutz J for the immortalized airway epithelial cells (NuLi-1). This work was supported by the Associazione Italiana per la
Ricerca sul Cancro (AIRC No. IG 9196, Milan, Italy), Ministero dell’Università e Ricerca Scientifica and Regione Piemonte and Compagnia di San Paolo, Torino (Progetto Oncologia). AUTHOR
INFORMATION Author notes * F Di Giacomo and E Panizza: These authors contributed equally to this work. AUTHORS AND AFFILIATIONS * Department of Biomedical Sciences and Human Oncology, Center
for Experimental Research and Medical Studies (CERMS), University of Torino, Torino, Italy C Voena, F Di Giacomo, E Panizza, L D'Amico, F E Boccalatte, E Pellegrino, M Todaro, C
Ambrogio, C Martinengo, L Bonello, R Chiarle & G Inghirami * Department of Molecular Biotechnology and Health Sciences, University of Torino, Torino, Italy C Voena, F Di Giacomo, E
Panizza, L D'Amico, F E Boccalatte, E Pellegrino, M Todaro, D Recupero, F Tabbò, C Ambrogio, C Martinengo, L Bonello, R Chiarle & G Inghirami * Department of Molecular Biotechnology
and Health Science, Molecular Biotechnology Center, University of Torino, Torino, Italy R Pulito & J Hamm * Discovery Research, Cephalon Inc., West Chester, PA, USA M Cheng & B
Ruggeri * Laboratory of Oncogenomics, Institute for Cancer Research at Candiolo, Candiolo, Italy E Medico * Department of Oncological Sciences, University of Torino, Torino, Italy E Medico *
Department of Pathology and NYU Cancer Center, New York University School of Medicine, New York, NY, USA G Inghirami Authors * C Voena View author publications You can also search for this
author inPubMed Google Scholar * F Di Giacomo View author publications You can also search for this author inPubMed Google Scholar * E Panizza View author publications You can also search
for this author inPubMed Google Scholar * L D'Amico View author publications You can also search for this author inPubMed Google Scholar * F E Boccalatte View author publications You
can also search for this author inPubMed Google Scholar * E Pellegrino View author publications You can also search for this author inPubMed Google Scholar * M Todaro View author
publications You can also search for this author inPubMed Google Scholar * D Recupero View author publications You can also search for this author inPubMed Google Scholar * F Tabbò View
author publications You can also search for this author inPubMed Google Scholar * C Ambrogio View author publications You can also search for this author inPubMed Google Scholar * C
Martinengo View author publications You can also search for this author inPubMed Google Scholar * L Bonello View author publications You can also search for this author inPubMed Google
Scholar * R Pulito View author publications You can also search for this author inPubMed Google Scholar * J Hamm View author publications You can also search for this author inPubMed Google
Scholar * R Chiarle View author publications You can also search for this author inPubMed Google Scholar * M Cheng View author publications You can also search for this author inPubMed
Google Scholar * B Ruggeri View author publications You can also search for this author inPubMed Google Scholar * E Medico View author publications You can also search for this author
inPubMed Google Scholar * G Inghirami View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHORS Correspondence to C Voena or G Inghirami.
ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no conflict of interest. ADDITIONAL INFORMATION Supplementary Information accompanies this paper on the Oncogenesis website .
SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURES (PDF 1339 KB) SUPPLEMENTARY FIGURE LEGENDS (DOC 35 KB) RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons
Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/ Reprints and permissions ABOUT THIS ARTICLE
CITE THIS ARTICLE Voena, C., Di Giacomo, F., Panizza, E. _et al._ The EGFR family members sustain the neoplastic phenotype of ALK+ lung adenocarcinoma via EGR1. _Oncogenesis_ 2, e43 (2013).
https://doi.org/10.1038/oncsis.2013.7 Download citation * Received: 18 February 2013 * Accepted: 28 February 2013 * Published: 08 April 2013 * Issue Date: April 2013 * DOI:
https://doi.org/10.1038/oncsis.2013.7 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * lung cancer * ALK tyrosine kinase * HER family * EGR1