- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Photocurrent in an organic solar cell is generated by a charge transfer reaction between electron donors and acceptors. Charge transfer is expected to proceed from thermalized
states, but this picture has been challenged by recent studies that have investigated the role of hot excitons. Here we show a direct link between excess excitation energy and photocarrier
mobility. Charge transfer from excited donor molecules generates hot photocarriers with excess energy coming from the offset between the lowest unoccupied molecular orbital of the donor and
that of the acceptor. Hot photocarriers manifest themselves through a short-lived spike in terahertz photoconductivity that decays on a picosecond timescale as carriers thermalize. Different
dynamics are observed when exciting the acceptor at its absorption edge to a thermalized state. Charge transfer in this case generates thermalized carriers described by terahertz
photoconductivity dynamics consisting of an instrument-limited rise to a long-lived signal. You have full access to this article via your institution. Download PDF SIMILAR CONTENT BEING
VIEWED BY OTHERS THE ROLE OF EXCITON LIFETIME FOR CHARGE GENERATION IN ORGANIC SOLAR CELLS AT NEGLIGIBLE ENERGY-LEVEL OFFSETS Article 31 August 2020 LONG-LIVED AND DISORDER-FREE CHARGE
TRANSFER STATES ENABLE ENDOTHERMIC CHARGE SEPARATION IN EFFICIENT NON-FULLERENE ORGANIC SOLAR CELLS Article Open access 05 November 2020 DECREASING EXCITON DISSOCIATION RATES FOR REDUCED
VOLTAGE LOSSES IN ORGANIC SOLAR CELLS Article Open access 27 March 2024 INTRODUCTION Organic solar cells show increasing promise for renewable energy1,2 with rapid progress over the past
decade leading to power conversion efficiencies >10% (refs 3, 4). The greatest advances have come from improved control over sample morphology5,6 and the development of novel electron
donors7,8. As the performance of organic solar cells approaches the limits envisaged with fullerene acceptors9, development of new donor/acceptor systems requires a more thorough theoretical
understanding of what drives the charge transfer reaction and experimental tools to probe these processes10. One pathway to new systems is to use excess excitation energy to drive the
charge transfer reaction. Several recent studies have concluded that hot charge transfer states boost the charge generation yield11,12,13. Bakulin _et al_.14 showed that delocalization could
be induced by excited state absorption, which in turn led to a rise in photocurrent. While these studies examine excited state dynamics, optical pump-probe measurements cannot address the
relationship between excess energy and carrier mobility. Time-resolved terahertz spectroscopy (TRTS) probes photoconductivity with the temporal resolution needed to access donor–acceptor
interactions in real time15. TRTS yields valuable insight, but it is an open question whether the initial terahertz photoconductivity dynamics in organic semiconductors are due to carrier
localization15,16 or recombination17,18,19. Here we address these issues by combining TRTS with transient absorption (TA) spectroscopy and through selective excitation of the electron donor
or acceptor. We show that the short-lived terahertz photoconductivity is the signature of a hot electron and its magnitude is sensitive to the interfacial volume between donors and
acceptors. Efficient charge transfer is found when exciting the electron acceptor at its absorption edge, demonstrating that the excess energy of a hot photocarrier is not necessary to
initiate the charge transfer reaction. RESULTS MODEL SYSTEM FOR CHARGE TRANSFER We selected three widely studied materials with technological relevance as model systems for charge transfer:
buckminsterfullerene (C60) is the electron acceptor and zinc phthalocyanine (ZnPc) and alpha-sexithiophene (α-6T) are the electron donors. Cnops _et al_.20 achieved organic solar cells with
a power conversion efficiency of 8.4% with using an α-6T donor and subphthalocyanine acceptors and the 12% record efficiency for organic solar cells was achieved in a sublimed tandem
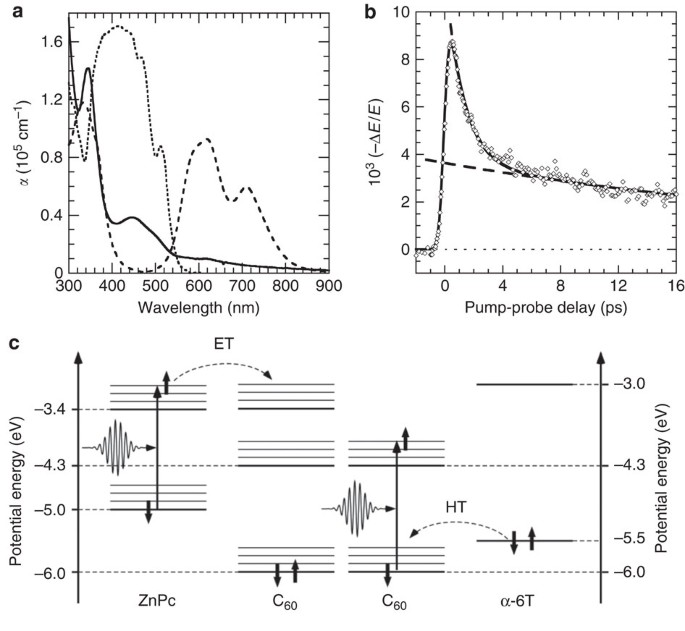
structure3. Figure 1a shows the absorption spectra of C60, ZnPc and α-6T. Figure 1b shows an example of terahertz photoconductivity dynamics, measured by scanning the delay between optical
pump and terahertz probe with the system set to sample the peak of the terahertz electric field _E_(_t_). The experimental data is fit to biexponential decay over the first 20 ps following
excitation: The slower decay constant _t_2 was determined by a linear regression where _t_/_t_1>>1 and then the residual was similarly fit. The rise in terahertz photoconductivity is
well described by an error function with a rise time (_t_0) of ∼0.3 ps. TA11,12,13,14 and broadband (∼10 THz) terahertz photoconductivity15 studies have shown that charge transfer occurs
within 0.1 ps, so we consider the rise to be instrument limited by the bandwidth of the spectrometer (∼3 THz). The decay dynamics of donor/acceptor films are complex, in part due to parallel
charge transfer processes. These include charge transfer from an excited donor to an unoccupied orbital of the acceptor (electron transfer), charge transfer from an occupied orbital of the
electron donor to an excited acceptor (hole transfer), as well as charge transfer between molecules of the same type (autoionization)21. We disentangle these processes, shown in Fig. 1c, by
varying the sample composition and through selective excitation of the electron donor or acceptor. We focus on multi-layered structures as these enable us to vary the interfacial volume
while maintaining the superior mobility of neat films. Alternate deposition of ultrathin layers yields a phase-segregated morphology with superior charge transport vis-à-vis co-sublimed
blends22. TERAHERTZ PHOTOCONDUCTIVITY OF FILMS WITH C60 AND ZNPC We first consider terahertz photoconductivity following electron transfer from an excited donor molecule. Figure 2a shows
pump scans of layered C60/ZnPc films excited at 615 nm and fits to the data. The fitting parameters of equation (1) are shown in Table 1. The extinction coefficient of ZnPc (92,500 cm−1) is
nine times that of C60 (10,200 cm−1) at 615 nm such that ZnPc is preferentially excited. The signal magnitude increases as the layer thickness decreases, but the initial decay rate is
independent of layer thickness. TA measurements of C60/ZnPc films show no significant recombination over the first 10 ps following charge transfer19. Hence, the early terahertz dynamics in
the layered system arise from a reduction in mobility as transferred carriers thermalize and become localized. This interpretation is consistent with Cooke _et al_.15, who showed that charge
carriers in polymer:fullerene blends localize within 1 ps. The initial terahertz photoconductivity dynamics observed for donor–acceptor systems, thus, arise from ultrafast charge transfer
following excitation of a donor molecule at a heterojunction, followed by decay due to decreasing mobility as the carrier localizes. Excitation of a donor molecule within a layer, on the
other hand, requires exciton migration to the interface before charge transfer. For the exciton diffusivity in organic semiconductors (10−6 m2 s−1)23, exciton diffusion to an interface
requires ∼10 ps. Charge transfer from molecules excited within a layer does not contribute to the initial transient, which in turn decreases with increasing layer thickness. Decay dynamics
are more complex when both the donor and acceptor are excited. Figure 2b shows pump scans of layered C60/ZnPc films for excitation at 400 nm. Weaker trends in the peak signal magnitude are
observed but the initial decay is sensitive to layer thickness. Using the same decay parameters as above, we obtained good fits for all dynamics for _t_>1 ps and a similar trend with
increasing layer thickness. There is an additional short-lived component to the signal that increases in magnitude with increasing layer thickness. This component has the same dynamics as
for excitation of neat C60 at 400 nm (Fig. 2c). Excitation of C60 molecules within a separate domain results in autoionization and recombination to a neutral exited state, contributing an
additional short-lived component to the terahertz response of the film. No significant terahertz photoconductivity was observed following excitation of C60 at 615 nm (Fig. 2c). The
absorption edge of solid C60 is at ∼750 nm and, thus, the excess energy (∼0.3 eV) is insufficient for autoionization. The presence of a neighbouring electron provides competing pathway to
autoionization and, thus, there is little difference in dynamics when exciting at different wavelengths for films with the thinnest layers. This is most evident when comparing the terahertz
photoconductivity dynamics of a 1:1 composite film, excited at 400 or 615 nm (Fig. 2d). The rise time is faster for excitation at 615 nm as with layered films, but the decay dynamics are not
affected by the excitation wavelength. Transfer of an electron follows excitation of ZnPc at 400 or 615 nm to an upper excited state of C60, generating a hot carrier. Charge transfer also
occurs following excitation of C60, a process analogous to hole transfer. In that case, C60 is excited to a higher lying Franck–Condon level and charge transfer precedes thermalization. The
excess excitation energy results in the same terahertz photoconductivity decay dynamics when the donor is excited. TERAHERTZ PHOTOCONDUCTIVITY OF FILMS WITH C60 AND Α-6T Measurements of
C60/α-6T films give further weight to our interpretation that the initial decay of terahertz photoconductivity arises from thermalization of a transferred photocarrier. The absorption
spectra of C60 and α-6T permit the selective excitation of either molecule as the extinction coefficient of α-6T (169,000 cm−1) is 4.8 times that of C60 at 400 nm (35,000 cm−1), but α-6T
does not absorb at 615 nm (Fig. 1a). Figure 3a shows the terahertz photoconductivity dynamics following excitation of the electron acceptor (C60 at 615 nm) and Fig. 3b shows the terahertz
photoconductivity dynamics following excitation of the electron donor (α-6T at 400 nm). When exciting the electron acceptor, there is a sharp rise (_t_0=0.3 ps) to an equilibrium signal with
only a weak overshoot (∼10% of the initial signal). The terahertz photoconductivity is stable out to a delay of 100 ps, shown in Fig. 3c. The terahertz photoconductivity dynamics arise from
electron transfer from α-6T to an excited C60 molecule. As the excitation is near the absorption edge, the resulting photocarrier is thermalized and hence there is no short-lived terahertz
photoconductivity. This result demonstrates unambiguously that efficient charge transfer occurs from thermalized states. Even though the absorbance of C60/α-6T films is relatively weak at
615 nm (∼50% of incident radiation is absorbed), the magnitude of the long-lived signal is comparable to that following charge transfer with excess energy. When exciting the electron donor
α-6T at 400 nm, however, the terahertz photoconductivity dynamics (Fig. 3b) are qualitatively similar to those measured following excitation of ZnPc—a short-lived transient, followed by a
long-lived component. The decay dynamics are significantly faster for C60/α-6T than for C60/ZnPc (see Table 1) with the equilibrium value being reached within 2 ps. The faster decay dynamics
may reflect differences in transfer dynamics as the hot carrier is transferred to C60 in both cases. As with excitation of C60/α-6T at 615 nm, the terahertz photoconductivity is stable up
to 100 ps delay. There is a remarkable agreement between the magnitude of the initial peak in terahertz photoconductivity when exciting the donor, shown in Fig. 3d. For both C60/ZnPc and
C60/α-6T films, the signal scales inversely with the square root of layer thickness (solid line). The absorbance of the two films at the pump wavelength is similar (>97%) and the same
photon flux was used. As the hole mobility of polycrystalline α-6T films (ca. 10−2 cm2 V−1 s)24 is two orders of magnitude larger than that of ZnPc (ca. 10−4 cm2 V−1 s)25, the initial
photoconductivity dynamics following electron transfer from an excited donor molecule reflect thermalization of the transferred carrier rather than relaxation of the electron donor. The same
trend is seen for excitation of C60/ZnPc films at 400 nm after neglecting the contribution from autoionization. The signal is higher, reflecting the additional contribution of hole transfer
following excitation of C60. Finally, the autoionization transient increases with layer thickness as an increasing fraction of excited C60 molecules lie within a separate domain. TRANSIENT
ABSORPTION OF FILMS WITH C60 AND Α-6T Regardless as to whether the donor or acceptor is excited, the terahertz photoconductivity of C60/α-6T films is stable from ∼5 to 100 ps pump-probe
delay. We performed TA measurements to correlate TRTS with excited state populations. Figure 4a shows the TA of C60, α-6T and layered films excited at 387.5 nm and probed at 470 nm. The
probe wavelength was selected where there are minimal contributions from α-6T and C60 (see Supplementary Fig. 1) and, thus, absorption bleaching dynamics can be tracked with minimal
interference. Neat films show a short-lived transient bleaching (decay constants 0.5 ps for C60; 1.1 ps for α-6T) with a crossover to TA at 6 ps for C60 and 2 ps for α-6T. The correlation
between the TA and THz decay dynamics (see Fig. 2) led us to assign the short-lived state in C60 to autoioinization. Lanzani _et al_.26 previously identified a short-lived intrachain singlet
state of α-6T that converts to a charge transfer state within several picoseconds. The layered films have a short-lived component similar to that of α-6T, but exhibit long-lived bleaching
of ground-state absorption that increases with decreasing layer thickness. The initial dynamics reflect a combination of transient bleaching and absorption of neutral as well as charged
states. We eliminated the contribution of neutral states to the transient transmission dynamics by subtracting off a scaled fraction of the neat film to eliminate the initial decay (Fig.
4b). The resulting dynamics match the terahertz photoconductivity and scale similarly with layer thickness. In summary, we find that charge transfer to a state with excess energy generates a
hot photocarrier that localizes within a picosecond. Photogeneration of a hot carrier manifests itself through short-lived terahertz photoconductivity that decays on a picosecond timescale.
The insensitivity of the photoconductivity magnitude to hole mobility suggests that C60 anions are the dominant contributor to terahertz photoconductivity in the films studied here.
Excitation of the electron acceptor at its optical gap generates localized photocarriers, demonstrating efficient charge transfer even in the absence of excess excitation energy. These
results illustrate the value of terahertz spectroscopy for probing charge transfer reactions at organic heterojunctions. METHODS SAMPLE PREPARATION C60, α-6T and ZnPc were purified by
consecutive vacuum train sublimation before use. We did not use a functionalized fullerene such as phenyl-C61-butyric acid methyl ester (PCBM), because such materials partially decompose
under sublimation. Films (∼300 nm thick) were prepared by sublimation under dynamic vacuum (3.0 × 10−7 Torr) from resistive heating furnaces onto fused silica substrates that were cleaned
with organic solvents and subjected to ozone treatment prior to use. Films with a layered nanostructure were prepared by alternate layer deposition and composite films were prepared by
co-sublimation. Quartz crystal microbalances were used to monitor the rate of deposition and determine the film composition and layer thickness. The absorption spectra of the films were
measured using a spectrophotometer (Lambda 1,050, Perkin Elmer). A custom spectrometer with an integrating sphere was used to measure absorption at low absorbance levels. TERAHERTZ
SPECTROSCOPY The terahertz spectrometer is driven by 60 fs pulses from a kilohertz Ti:Sapphire amplifier and interrogated with optically gated and synchronized terahertz probe pulses. The
output of the amplifier passed through a 50/50 beam splitter and the resulting pulses were used to generate the terahertz probe and optical pump pulses. Probe pulses were generated by a 2-mm
thick ZnTe[110] crystal and detected using a matched ZnTe crystal and free-space electro-optical sampling. The output of the Ti:sapphire amplifier was frequency doubled by a β-BaB2O4
crystal to 400 nm or used to excite a near infrared optical parametric amplifier and then frequency doubled to 615 nm. The pump intensity was 20 μJ per pulse at 615 nm or 30 μJ per pulse at
400 nm (6 × 1013 photons per cm2 per pulse). All measurements were performed in the linear regime for this spectrometer (see Supplementary Fig. 2). The temporal dynamics of the terahertz
photoconductivity were measured by scanning the delay between the pump and probe beam with the probe gated such that the peak of the terahertz pulse was sampled. There is a slight phase
shift between the peak of the terahertz pulse and the change in the terahertz transmission, although the amplitude does not change significantly (see Supplementary Fig. 3). The peak of the
pump-induced change in terahertz transmission was not affected by changing the pump-probe delay. TRANSIENT ABSORPTION TA experiments were based on a 1 kHz Ti:Sapphire amplifier (775 nm). A
portion of the output was frequency doubled by a β-BaB2O4 crystal for sample excitation. A small amount of power was focused into a sapphire plate to produce the white-light continuum probe.
The excited state spectra were resolved using a scanning monochromator. Ground-state bleach and excited state absorption dynamics were recorded by adjusting the delay between the pump and
probe pulses. The pump fluence was adjusted to ∼6 × 1013 photons per cm2 per pulse. A temporal resolution of ∼280 fs was measured through cross-correlation. ADDITIONAL INFORMATION HOW TO
CITE THIS ARTICLE: Lane, P. A. _et al_. Hot photocarrier dynamics in organic solar cells. _Nat. Commun._ 6:7558 doi: 10.1038/ncomms8558 (2015). REFERENCES * Mishra, A. & Bauerle, P.
Small molecule organic semiconductors on the move: promises for future solar energy technology. _Angew. Chem. Int. Ed. Engl._ 51, 2020–2067 (2012) . Article CAS Google Scholar * Scharber,
M. C. & Sariciftci, N. S. Efficiency of bulk-heterojunction organic solar cells. _Prog. Poly. Sci._ 38, 1929–1940 (2013) . Article CAS Google Scholar * Overton, G. Organic PV
efficiency reaches 12%: Heliatek breaks its prior record. _Laser Focus World._ http://www.laserfocusworld.com/articles/2013/01/heliatek-opv-12-percent.html (2013) . * Liu, Y. et al.
Solution-processed small-molecule solar cells: breaking the 10% power conversion efficiency. _Sci. Rep._ 3, 3356 (2013) . Article Google Scholar * Walker, B. et al. Nanoscale phase
separation and high photovoltaic efficiency in solution-processed, small-molecule bulk heterojunction solar cells. _Adv. Func. Mater._ 19, 3063–3069 (2009) . Article CAS Google Scholar *
Fitzner, R. et al. Correlation of pi-conjugated oligomer structure with film morphology and organic solar cell performance. _J. Am. Chem. Soc._ 134, 11064–11067 (2012) . Article CAS Google
Scholar * Helgesen, M., Sondergaard, R. & Krebs, F. C. Advanced materials and processes for polymer solar cell devices. _J. Mater. Chem._ 20, 36–60 (2010) . Article CAS Google
Scholar * Lin, Y. Z., Li, Y. F & Zhan, X. W. Small molecule semiconductors for high-efficiency organic photovoltaics. _Chem. Soc. Rev._ 41, 4245–4272 (2012) . Article CAS Google
Scholar * Dennler, G. et al. Design rules for donors in bulk-heterojunction tandem solar cells-towards 15% energy-conversion efficiency. _Adv. Mater._ 20, 579–583 (2008) . Article CAS
Google Scholar * Koster, L. J. A., Shaheen, S. E. & Hummelen, J. C. Pathways to a new efficiency regime for organic solar cells. _Adv. Energy Mater._ 2, 1246–1253 (2012) . Article CAS
Google Scholar * Cowan, S. R., Banerji, N., Leong, W. L. & Heeger, A. J. Charge formation, recombination, and sweep-out dynamics in organic solar cells. _Adv. Func. Mater._ 22,
1116–1128 (2012) . Article CAS Google Scholar * Jailaubekov, A. E. et al. Hot charge-transfer excitons set the time limit for charge separation at donor/acceptor interfaces in organic
photovoltaics. _Nat. Mater._ 12, 66–73 (2013) . Article ADS CAS Google Scholar * Grancini, G. et al. Hot exciton dissociation in polymer solar cells. _Nat. Mater._ 12, 29–33 (2013) .
Article ADS CAS Google Scholar * Bakulin, A. A. et al. The role of driving energy and delocalized states for charge separation in organic semiconductors. _Science_ 335, 1340–1344 (2012)
. Article ADS CAS Google Scholar * Cooke, D. G., Krebs, F. C. & Jepsen, P. U. Direct observation of sub-100 fs mobile charge generation in a polymer-fullerene film. _Phys. Rev.
Lett._ 108, 056603 (2012) . Article ADS CAS Google Scholar * Lane, P. A. et al. Photoexcitation dynamics in films of C60 and Zn phthalocyanine with a layered nanostructure. _Phys. Rev.
Lett._ 108, 077402 (2012) . Article ADS Google Scholar * Hegmann, F. A., Tykwinski, R. R., Lui, K. P. H., Bullock, J. E. & Anthony, J. E. Picosecond transient photoconductivity in
functionalized pentacene molecular crystals probed by terahertz pulse spectroscopy. _Phys. Rev. Lett._ 89, 227403 (2002) . Article ADS CAS Google Scholar * Hendry, E., Schins, J. M.,
Candeias, L. P., Siebbeles, L. D. A. & Bonn, M. Efficiency of exciton and charge carrier photogeneration in a semiconducting polymer. _Phys. Rev. Lett._ 92, 196601 (2004) . Article ADS
CAS Google Scholar * Ponseca, C. S. et al. Ultrafast terahertz photoconductivity of bulk heterojunction materials reveals high carrier mobility up to nanosecond time scale. _J. Am. Chem.
Soc._ 134, 11836–11839 (2012) . Article CAS Google Scholar * Cnops, K. et al. 8.4% efficient fullerene-free organic solar cells exploiting long-range exciton energy transfer. _Nat.
Commun._ 5, 3406 (2014) . Article Google Scholar * Wojcik, M., Michalak, P. & Tachiya, M. Geminate electron-hole recombination in organic solids in the presence of a donor-acceptor
heterojunction. _Appl. Phys. Lett._ 96, 162102 (2010) . Article ADS Google Scholar * Hong, Z. R. et al. Improved efficiency of zinc phthalocyanine/C60 based photovoltaic cells via
nanoscale interface modification. _Appl. Phys. Lett._ 90, 203505 (2007) . Article ADS Google Scholar * Liu, F., Ruden, P. P., Campbell, I. H. & Smith, D. L. Device model for
electronic processes at organic/organic interfaces. _J. Appl. Phys._ 111, 094507 (2012) . Article ADS Google Scholar * Horowitz, G., Hajlaoui, M. E. & Hajlaoui, R. Temperature and
gate voltage dependence of hole mobility in polycrystalline oligothiophene thin film transistors. _J. Appl. Phys._ 87, 4456–4463 (2000) . Article ADS CAS Google Scholar * Maennig, B. et
al. Controlled p-type doping of polycrystalline and amorphous organic layers: Self-consistent description of conductivity and field-effect mobility by a microscopic percolation model. _Phys.
Rev. B_ 64, 195208 (2001) . Article ADS Google Scholar * Lanzani, G. et al. Transient spectroscopy of frenkel and charge transfer excitons in α-sexithienyl films. _Phys. Rev. Lett._ 79,
3066–3069 (1997) . Article ADS CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank the Office of Naval Research and National Institute of Standards and Technology for
funding. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Optical Sciences Division, Naval Research Laboratory, Washington, 20375, DC, USA P. A. Lane * Electronics Division, Naval Research
Laboratory, Washington DC 20375, USA P. D. Cunningham & J. S. Melinger * Radiation Physics Division, National Institute of Standards and Technology, Gaithersburg, Maryland 20899, USA O.
Esenturk & E. J. Heilweil Authors * P. A. Lane View author publications You can also search for this author inPubMed Google Scholar * P. D. Cunningham View author publications You can
also search for this author inPubMed Google Scholar * J. S. Melinger View author publications You can also search for this author inPubMed Google Scholar * O. Esenturk View author
publications You can also search for this author inPubMed Google Scholar * E. J. Heilweil View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
The authors jointly conceived the study and designed the experiments. P.A.L. prepared the samples and measured the absorption spectra. P.A.L., O.K. and E.J.H. performed the terahertz
photoconductivity measurements and analysed the data. P.D.C. performed the transient absorption experiments and P.A.L. and P.D.C. jointly analysed the data. P.A.L. drafted the manuscript and
all authors jointly edited the manuscript. CORRESPONDING AUTHOR Correspondence to P. A. Lane. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing financial interests.
SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION Supplementary Figures 1-3 (PDF 145 kb) RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Lane, P.,
Cunningham, P., Melinger, J. _et al._ Hot photocarrier dynamics in organic solar cells. _Nat Commun_ 6, 7558 (2015). https://doi.org/10.1038/ncomms8558 Download citation * Received: 12
September 2014 * Accepted: 19 May 2015 * Published: 16 July 2015 * DOI: https://doi.org/10.1038/ncomms8558 SHARE THIS ARTICLE Anyone you share the following link with will be able to read
this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative



