- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The nuclear lamina is thought to be the primary mechanical defence of the nucleus. However, the lamina is integrated within a network of lipids, proteins and chromatin; the
interdependence of this network poses a challenge to defining the individual mechanical contributions of these components. Here, we isolate the role of chromatin in nuclear mechanics by
using a system lacking lamins. Using novel imaging analyses, we observe that untethering chromatin from the inner nuclear membrane results in highly deformable nuclei _in vivo_, particularly
in response to cytoskeletal forces. Using optical tweezers, we find that isolated nuclei lacking inner nuclear membrane tethers are less stiff than wild-type nuclei and exhibit increased
chromatin flow, particularly in frequency ranges that recapitulate the kinetics of cytoskeletal dynamics. We suggest that modulating chromatin flow can define both transient and long-lived
changes in nuclear shape that are biologically important and may be altered in disease. SIMILAR CONTENT BEING VIEWED BY OTHERS MECHANICS AND FUNCTIONAL CONSEQUENCES OF NUCLEAR DEFORMATIONS
Article 05 May 2022 NONLINEAR MECHANICS OF LAMIN FILAMENTS AND THE MESHWORK TOPOLOGY BUILD AN EMERGENT NUCLEAR LAMINA Article Open access 04 December 2020 CORRELATIVE SINGLE MOLECULE LATTICE
LIGHT SHEET IMAGING REVEALS THE DYNAMIC RELATIONSHIP BETWEEN NUCLEOSOMES AND THE LOCAL CHROMATIN ENVIRONMENT Article Open access 16 May 2024 INTRODUCTION Cells are subjected to mechanical
stress, both from extracellular sources (such as forces exerted on the skin or sheer flow across the endothelia of the vasculature) and internal sources (such as forces exerted by the
cytoskeleton). Thus, mechanisms to adapt to and dissipate mechanical stress are necessary for cell survival. It is well established that extracellular mechanical forces can be transmitted to
the nucleus1,2. Further, disruption of the mechanical properties of the nucleus during normal cellular processes like migration compromises cell survival3,4 and can sometimes lead to
disease5. Defining the factors that underlie the mechanical properties of the nucleus and how these factors can be remodelled is an essential challenge in the field. The nuclear lamina is
considered to be the primary mechanical defense of the mammalian nucleus6,7. Strong evidence suggests that A-type lamins provide a mechanical buffer to cellular forces in differentiated
cells8,9, particularly those that reside in stiff tissues, whereas the B-type lamins are essential for organogenesis10. Mutations in A-type lamins are associated with numerous diseases,
including Hutchinson–Gilford progeria syndrome in which nuclei are stiff and fragile11,12 and Emery–Dreifuss muscular dystrophy in which nuclei are unusually soft13. Further, increasing the
concentration of lamin A augments the viscoelasticity and rigidity of the nucleus, which has been shown to contribute to mechanical scaling between nuclei and the tissue in which they
reside14,15,16. In addition to the ability of lamins to oligomerize into a polymeric assembly that can directly contribute to the stiffness of the nucleus6,7,17, the lamina is also
associated with peripheral heterochromatin18,19. Indeed, lamins associate with chromatin at the nuclear envelope either through association with integral inner nuclear membrane (INM)
proteins such as LAP1 and proteins of the LEM domain family (LAP2, emerin, Man1) or a host of soluble nuclear factors20. Mutations in INM proteins, such as emerin and LAP1, underlie a
broader set of genetic diseases termed ‘nuclear envelopathies’ that in some cases phenocopy lamin mutations21. Likely, it is this integrated network of proteins and polymers18,22 that define
the ensemble mechanical properties of the nucleus. This integrated network creates a challenge for dissecting the relative contributions of lamins, INM proteins and the associated chromatin
to nuclear mechanics, particularly because of the co-dependence of their localization23,24. Unlike lamins, LEM domain proteins were present in the last eukaryotic common ancestor25,
suggesting that the association of chromatin with the INM is an ancient aspect of nuclear organization. Interestingly, numerous organisms that are largely protected from external forces by a
cell wall lack genes homologous to the lamins, suggesting a lamin-independent mechanism capable of buffering internal forces, such as those delivered by the cytoskeleton onto nuclei. Both
_Saccharomyces cerevisiae_ and _Schizosaccharomyces pombe_ express two LEM domain proteins, Heh1 and Heh2 (refs 26, 27, 28). Heh1 and Heh2 are orthologues of the mammalian LEM domain
proteins Man1, LEMD2 and emerin26,28. _S. pombe_ has an additional, conserved INM protein, Ima1, which also associates with chromatin29 and is homologous to mammalian Net5/Samp1 (refs 29,
30). Importantly, loss of INM proteins leads to apparent defects in nuclear structure in fission yeast28,29,31. In this study, we investigate how association of chromatin with the nuclear
periphery influences the ability of the nucleus to withstand mechanical forces in the model organism, _S. pombe. S. pombe_ lacks lamins25, allowing us to isolate the contribution of
chromatin to nuclear mechanics. In _S. pombe_, the spindle pole body (SPB), the centrosome equivalent in yeast, is tethered to the nuclear envelope (NE) through NE-spanning LINC (Linker of
Nucleoskeleton and Cytoskeleton) complexes32,33,34. Microtubules (MTs) emanating from the SPB grow stochastically towards the cell tips35; once the growing MT reaches the cell cortex,
continued MT polymerization applies a rear-ward force onto the nucleus. This mechanism maintains the nucleus in the middle of the cell to support symmetric cell division36. Interestingly,
_S. pombe_ chromosomes are organized in the ‘Rabl’ conformation37, with the centromeres clustered at the SPB interface38. Thus, centromeric heterochromatin is associated with the region of
the NE that is physically linked to dynamic MTs in the cytoplasm. Here, we use new quantitative approaches to investigate _S. pombe_ nuclear mechanics _in vivo_ and _in vitro_. Our work
demonstrates that the association of chromatin with the INM contributes substantially to nuclear stiffness. Further, increased mobility of chromatin in the absence of INM tethers allows for
chromatin flow in response to MT-induced fluctuations, leading to longer-lived defects in nuclear shape. This work suggests that the rate of chromatin flow can be tuned by modulating
chromatin tethering to the nuclear periphery. RESULTS MICROTUBULES DRIVE NUCLEAR ENVELOPE FLUCTUATIONS _IN VIVO_ To assess the nuclear response to intracellular forces, we first sought to
develop an assay to measure NE fluctuations _in vivo_. To visualize the NE, we used strains expressing Cut11-GFP (green fluorescent protein) from its endogenous locus; Cut11 is an integral
membrane protein of the nuclear pore complex39. Monitoring Cut11-GFP in living cells provided a means to monitor nuclear shape, which responds to thermal forces as well as MT forces _in
vivo_29,36,39. We specifically analysed cells in the G2 phase of the cell cycle, which were visually identified on the basis of the criteria that they were single cells (that is, they had
completed cytokinesis) and did not have Cut11-GFP associated with the spindle pole body (which occurs at mitotic entry39). Three-dimensional (3D) structural information of individual nuclei
was encoded in two-dimensional (2D) sequential microscope images collected at different focal depths (10 z-slices each separated by 400 nm) collected every 2.5 s for 5 min. To quantitatively
assess nuclear fluctuations at each time point, we reconstructed a 3D contour of the NE with sub-pixel resolution. We used a novel optimization scheme that seeks to maximize the contour
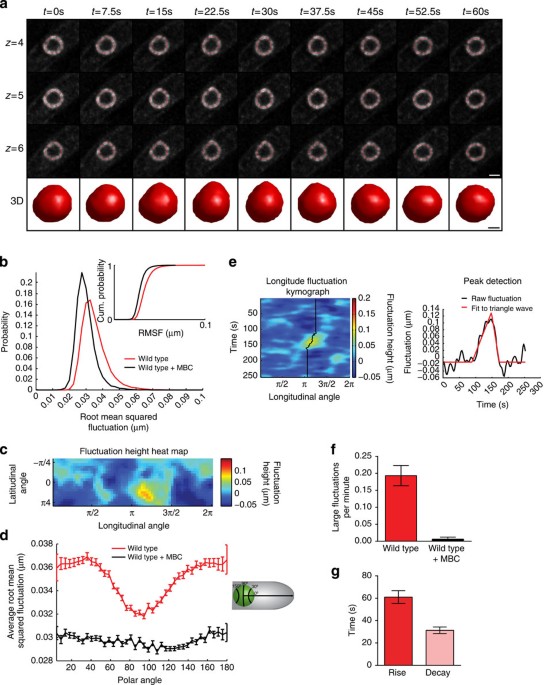
intensity at each image stack while simultaneously minimizing the bending of the contour. A representative reconstruction of a wild-type NE at various time points is shown in Fig. 1a as both
a 3D rendering and 2D cross-sections at three z-slices about the centre of the nucleus (see also Supplementary Movie 1). Using spherical coordinates centred on the nucleus at each time
point, each NE fluctuation was defined as the difference between the radial coordinate of the contour and its time-averaged radial value; this approach also allows us to correct for changes
in nuclear position over time. The frequency and magnitude of fluctuations, sampled at equally spaced spherical angles around the nucleus, were assessed by computing the root mean square
fluctuation (RMSF). The RMSF thus provides a quantitative metric for changes in nuclear shape over time. We compared the RMSF of cells imaged with and without carbendazim (MBC), which
depolymerizes MTs. The distribution of RMSF from a population of nuclei over time at each spherical angle clearly shows that untreated nuclei have larger fluctuations on average compared
with MBC-treated nuclei (Fig. 1b). Thus, our fluctuation analysis is sufficiently sensitive to capture the impact of active MT dynamics on nuclear shape. To test whether the actin
cytoskeleton also applies forces that can drive fluctuations of the NE, we analysed nuclear fluctuations in cells treated with latrunculin A (Lat A), which depolymerizes actin. Actin
depolymerization in _S. pombe_ causes a population of cells to arrest in G2 (ref. 40), increasing the size of the cells and, therefore, the size of the nuclei41. After applying a size filter
to account for this effect (Supplementary Fig. 1a), we found that actin depolymerization in cells caused a minor shift towards larger RMSF values compared with untreated cells
(Supplementary Fig. 1b). Thus, MT forces are the primary driver of large NE fluctuations in this system. To discriminate between fluctuations caused by MT dynamics from thermal fluctuations,
we generated a sequence of spatial maps of NE fluctuations (Fig. 1c,e). The landscape of each 3D nuclear contour was obtained by plotting fluctuation size (represented as a heat map) at
each latitudinal and longitudinal angle (Fig. 1c). We assessed the angle at which these fluctuations occur using a polar coordinate system based on the long axis of the cell (Fig. 1d). While
NE fluctuations in MBC-treated nuclei are small and are uniformly distributed along the NE (Fig. 1d, black line), untreated nuclei have a distinct pattern in which larger NE fluctuations
occur at 30 degrees and 150 degrees along the polar axis (Fig. 1d, red line) with a minimum at 90 degrees (orthogonal to the long axis of the cell). Given that MT polymerization is aligned
with the long axis of the cell, this result confirms that large NE fluctuations are primarily due to MT-derived forces. The NE fluctuations maps were next interrogated to spatially track the
fluctuation over time, allowing us to monitor the amplitude and duration of a fluctuation event. The one-dimensional angular trajectory of a fluctuation event can be represented with a
kymograph (Fig. 1e). The resulting amplitude of each NE fluctuation time series was fit with a single peak asymmetric triangle waveform, allowing us to determine the rise and decay times
(Fig. 1e). Comparing untreated versus MBC-treated cells, we established criteria to identify MT-induced fluctuations. These criteria require that a fluctuation maintains a minimum height of
50 nm for a duration longer than 25 s (Supplementary Fig. 1c). This approach was validated by comparing the frequency of fluctuations that meet these criteria in untreated versus MBC-treated
cells (Fig. 1f). The duration of large NE fluctuations corresponds very well within the known duration of MT forces on the nucleus (∼90 s) in _S. pombe_36 (Fig. 1g). Interestingly,
MT-dependent fluctuations decay faster than they form; since MTs in _S. pombe_ undergo a rapid catastrophe, the rate at which the fluctuations resolve is likely dependent on the innate
physical properties of the nucleus. UNTETHERING CHROMATIN ALTERS NE FLUCTUATIONS _IN VIVO_ Our previous work suggests that association of heterochromatin with the INM plays an important role
in supporting nuclear mechanics29. To quantitatively investigate this model, we focused on three conserved integral INM proteins: Heh1 (also called Lem2), Heh2 (also called Man1) and Ima1,
which contribute to chromatin tethering to the nuclear periphery28,42,43,44. On the basis of proteomic studies, there are ∼1,000 copies of Heh1, 500 copies of Heh2 and 200 copies of Ima1 per
cell45. In _S. pombe_, a fraction of Heh1-GFP and GFP-Ima1 co-localize with the _S. pombe_ SUN domain protein, Sad1-mCherry, which resides at the centromere–spindle pole body interface of
the NE29 (Fig. 2a). By contrast, Heh2-GFP is dispersed throughout the NE where it is found in faint foci (Fig. 2a). Importantly, Heh1, Heh2 and Ima1 target independently to the NE and loss
of these chromatin tethers does not substantially alter the focal nature of heterochromatin within the nucleus as assessed by visualization of heterochromatin-binding proteins46,47
(Supplementary Fig. 2b–d). Further, while there are subtle alterations in the total nuclear intensity of heterochromatic foci in _heh1Δ_ and _heh2Δ_ cells, these differences do not correlate
with the observed effects on nuclear mechanics (see below and Supplementary Fig. 2e). Qualitatively, cells lacking INM proteins have apparent defects in nuclear shape (Fig. 2b and
Supplementary Movies 2–8). To quantitatively assess these defects, we compared the RMSF distribution in the absence of individual INM proteins as in Fig. 1. While _ima1_Δ cells display a
modest increase in RMSF, both _heh1Δ_ and _heh2Δ_ cells exhibit a prominent shift towards larger RMSF, with a substantial tail towards very large RMSF values that are rarely observed in
wild-type cells (Fig. 2b,c, solid lines). The increased tail of the RMSF distributions seen in _heh1Δ_ and _heh2Δ_ cells indicates a higher occurrence of very large NE fluctuations. We next
investigated the different combinations of INM protein knockouts. In general, increasing release of chromatin from the INM shifts the distribution to larger RMSF values and further increases
the tail of the distribution (Fig. 2b,c, dashed lines). The increase in unusually large NE fluctuations is more clearly revealed when displayed as a semi-log plot (Fig. 2c, inset). Here, it
can be seen that all genetic backgrounds that lack Heh1 display a marked increase in very large fluctuations. Importantly, depolymerization of MTs leads to a loss of all large fluctuations
for every genotype (Fig. 1b and Supplementary Fig. 3a). Interestingly, the RSMF distributions correlate with the impact of the alleles on growth, with enhancing effects of _heh1_ and _ima1_
and suppressive effects of _heh2_ (Supplementary Fig. 3b). Indeed, _heh1Δheh2Δ_ cells show improved growth and smaller RMSF compared with _heh1Δ_ cells (Supplementary Fig. 3a,b). We next
investigated whether decoupling chromatin from the nuclear periphery impacted the timescale of NE fluctuations using the same approach as in Fig. 1g. The mean rise time for the deformations
was not statistically different between wild-type and cells lacking INM proteins (Fig. 2d), consistent with the expectation that the MT polymerization rate dictates rise time. Interestingly,
the decay time of fluctuations was longer in all of the mutant cells compared with wild type. This property appears distinct from the impact on NE fluctuation size. Thus, not only are NE
fluctuations generally larger when tethering of chromatin to the nuclear periphery is compromised, but these NE fluctuations also persist longer. Together, these two defects likely underlie
the overall changes in nuclear shape that are seen in cells lacking INM proteins (Fig. 2b). We next compared the effect of releasing chromatin from the NE due to loss of chromatin tethers
with the effective ‘dilution’ of chromatin tethers at the nuclear periphery by increasing nuclear volume. To achieve this, we took advantage of the temperature-sensitive _cdc25-22_ strain,
which arrests in G2 due to insufficient Cdc2 (Cdk1) activity48. During this G2 arrest, the cells continue to grow, leading to a concomitant growth in the nuclear volume41 without changes in
the amount of chromatin. The _cdc25-22_ nuclei have on average an ∼50% increase in their nuclear volume compared with wild-type nuclei (Fig. 3a). Consistent with a model in which chromatin
plays a mechanical role at the NE, we find that _cdc25-22_ nuclei have both larger RMSF values (although this effect is milder than all but the _ima1Δ_ cells) and a slightly longer decay
time than wild-type nuclei (Fig. 3b–d and Supplementary Movie 9). This suggests that effective ‘dilution’ of chromatin tethers impacts nuclear mechanics, although to a lesser extent than
decoupling chromatin from the NE through loss of tethering factors. Last, we evaluated whether the chromatin state impacts nuclear fluctuation size by treating the cells with trichostatin A
(TSA), a histone deacetylase inhibitor. In this condition, we observed abundant tubulation of the NE in both wild-type and INM protein knockout backgrounds (Supplementary Fig. 3c). These
tubules disappeared once the cells were treated with MBC, however, MBC-treated nuclei never fully recovered their spherical shape (Supplementary Fig. 3c). While we are cautious to
over-interpret these findings given the pleiotropic nature of this perturbation (which leads to extensive changes in gene transcription), these effects suggest that changes in the chromatin
state can have a mechanical impact. CHARACTERIZATION OF WILD-TYPE NUCLEAR MECHANICS _IN VITRO_ Although 3D contour fluctuation analysis allows for robust characterization of the nuclear
response to MT forces _in vivo_, a full biophysical characterization of nuclear mechanics requires that the nuclear response be measured with calibrated forces across a wide range of
timescales _in vitro_. To achieve this, we isolated nuclei from the same strains that were characterized in Fig. 2 by an adaptation of protocols for purification of intact budding yeast
nuclei49 (see Methods). These nuclei appear intact by scanning electron microscopy (SEM, Fig. 4a) and are impermeable to 70 kDa dextran (Supplementary Fig. 4a). Importantly, these nuclei
appear to have the same heterochromatin organization as in intact cells by visualization of GFP-Swi6, the HP1 orthologue (Supplementary Fig. 4b). Nuclei prepared in this manner are
sufficiently pure for further experimentation given our ability to select individual nuclei for study (see below). We developed a novel force spectroscopy assay that allows us to directly
measure the stiffness of isolated nuclei under a variety of experimental conditions (Fig. 4b,c). In a flow cell designed with two input ports and one output port (Supplementary Fig. 4c), we
nonspecifically adhere large (5.2 μm) poly-ornithine-coated silica beads to the coverslip (Fig. 4b). _S. pombe_ nuclei are flowed into the flow cell and an individual nucleus is trapped
using the optical tweezers. We select individual nuclei that are between 1.5 and 3 μm, spherical in shape (as assessed by Cut11-GFP fluorescence) and lack any residual tubules of endoplasmic
reticulum (visible by transmitted light microscopy, Fig. 4c). Once trapped, each nucleus is associated with the side of one of the large, poly-ornithine-coated silica beads, which elevates
the nucleus off of the coverslip and provides a hard wall against which forces can be applied. After the desired nucleus is immobilized, the flow cell design allows the remainder of the
nuclear preparation to be washed away by flowing buffer through one of the two input ports. Subsequently, small (1.2 μm) poly-ornithine-coated latex beads are flowed into the flow cell
through the second input port, trapped and manoeuvred next to the nucleus. By gently placing the small bead in contact with the nucleus, we are able to induce nonspecific electrostatic
interactions between the nucleus and the poly-ornithine-coated surface. Movement of the coverslip (and thereby the large bead and nucleus) while holding the small bead in the optical trap
allows us to apply rounds of tension and compression at a set amplitude and frequency, as well as a means to measure the mechanical response of the nucleus (Fig. 4b). To assess nuclear
mechanics over a broad range of timescales, we begin by sinusoidally driving the stage with slow oscillations (0.01–0.5 Hz) that recapitulate the timescale of MT polymerization _in vivo_36
and then incrementally increase the frequency of the oscillations on a single nucleus to timescales much faster than MT polymerization rates (1–2 Hz). Importantly, we expended great efforts
to improve drift control methods that allow us to obtain reliable data at these biologically relevant timescales, which are often inaccessible by force spectroscopy50. We first chose to test
oscillation amplitudes that drive 50–60 nm nuclear deformations, which approximately recapitulate the scale of MT-dependent NE fluctuations _in vivo_ (Supplementary Fig. 1c). The mechanical
response of wild-type nuclei appeared elastic (that is, the force versus extension relationship was linear) with ∼3–4 pN of force at maximal extension at all frequencies (Fig. 4d). Given
this linear behaviour, we fit the data to _F_=_k_nucleusΔ_x_nucleus where _F_ is the force applied, _k_nucleus is the effective stiffness of the nucleus and Δ_x_nucleus is the displacement
of the NE from equilibrium51. We then compared _k_nucleus for each individual oscillation (Fig. 4e). The nuclei appear to have a very modest frequency-dependent stiffening behaviour, with a
slightly softer apparent stiffness between the slowest timescale (0.01 Hz) and all faster timescales (0.025–2 Hz; Fig. 4e). These observations are in line with several mechanical studies of
wild-type mammalian nuclei, which also demonstrate time-dependent stiffening behaviour52,53,54. In addition, we tested whether the presence of adenosine triphosphate (ATP) or an ATP
regeneration system impacts the mechanical response of the nucleus in this experimental regime, but observed no substantial difference when an individual nucleus was tested sequentially
under both conditions (Supplementary Fig. 4d). We chose to carry out the rest of the described experiments in the presence of 2 mM ATP. The hint of frequency dependence observed for small
amplitude oscillations between 0.01 and 0.025 Hz (Fig. 4e) suggests the possibility that wild-type nuclei have a minor viscous component that is not apparent in the individual force versus
extension plots (Fig. 4d). To better investigate whether there is such a viscous component to the mechanical response of wild-type nuclei, we applied large oscillatory deformations (∼170
nm), which represent the length scales of rare _in vivo_ fluctuations, followed by a series of small oscillatory deformations (∼60 nm), and then another series of large oscillations. At the
large oscillations, the force versus extension curves exhibit a reproducible hysteresis, indicating that the nuclei have a minor viscous component (Fig. 4f and Supplementary Fig. 4e).
Interestingly, when the compression force reaches ∼4–5 pN, the response of the nucleus begins to plateau. During the intervening small oscillations, we observe the same linear (elastic)
response as before (Fig. 4e). As a further test, we also performed force clamp experiments in which we apply a sudden pull on a nucleus with a constant force and monitor the nuclear
extension over time (Fig. 4g and Supplementary Fig. 4f). From these experiments, we can access the characteristic timescale of this creep behaviour, which we found is reproducibly in the
range of ∼10 s. Interestingly, the characteristic timescale of this viscous component is similar to that described previously for mammalian nuclei55. In these studies, it was suggested that
the flow of chromatin within the volume of the nucleus underlies a viscous component of nuclear mechanics; such chromatin flow likely explains the mild decrease in apparent stiffness at slow
timescales observed here for _S. pombe_ nuclei. UNTETHERING CHROMATIN ALTERS NUCLEAR MECHANICS _IN VITRO_ Next, we isolated nuclei lacking individual INM proteins and assessed their
mechanical properties using the force spectroscopy assay. Similar to wild-type nuclei, _heh1Δ_, _heh2Δ_ and _ima1_Δ nuclei display time-dependent stiffening behaviour at small deformations,
which is particularly prominent for _heh2Δ_ nuclei (Fig. 5a). This time-dependent effect manifests as an increase in compliance at slow oscillation frequencies (0.01–0.1 Hz). Because this
timescale recapitulates the _in vivo_ lifetime of NE fluctuations, this suggests that nuclei lacking INM proteins are softest at the frequency range that is most relevant for buffering MT
forces _in vivo_. Decreasing the chromatin to nuclear volume ratio also led to a modest decrease in nuclear stiffness at all frequencies compared with wild type (_cdc25-22_, Fig. 5a),
consistent with the _in vivo_ measurements (Fig. 3). By fitting the time-dependent stiffening behaviour to a simple viscoelastic model (Supplementary Fig. 4g and Methods), we can clearly see
that each INM protein contributes to the observed stiffness of the nucleus, as _heh1Δ_, _ima1_Δ and particularly _heh2Δ_ nuclei are all softer than wild type (Fig. 5a,b). Further, wild-type
nuclei exhibit higher viscosity than nuclei lacking INM proteins, suggesting that tethering of chromatin to the NE attenuates chromatin flow (Fig. 5a,c). The characteristic timescale _τ_,
at which the stiffness plateaus for each type of nucleus, indicates the timescale required for chromatin to respond to external forces. Nuclei lacking INM proteins have shorter _τ_ values
than wild-type nuclei, further supporting a greater propensity for chromatin to flow in the absence of NE tethers (Fig. 5a,d). By contrast, lowering the chromatin to nuclear volume ratio led
to only a slight decrease in stiffness and intermediate value for viscosity and _τ_, despite the fact that _cdc25-22_ nuclei having 50% greater volume than wild-type nuclei (Fig. 5c,d). As
a control, we applied similar oscillatory forces on vesicles that mimic nuclei in their size, lipid composition and internal viscosity (see Methods); these vesicles are highly deformable
relative to all nuclei (Fig. 4a–d). To further evaluate whether tethering of chromatin to the NE underlies the observed viscous component of nuclear mechanics, we took advantage of the fact
that _S. pombe_ undergo a closed mitosis in which the NE remains intact but chromatin is globally released from the INM to facilitate chromosome segregation56. We allowed cells to accumulate
in mitosis by culturing them in the presence of the spindle poison, MBC, followed by isolation of the nuclei. Consistent with our expectation, mitotic nuclei are softer than any of the
nuclei lacking individual INM proteins (Fig. 5a,b). Interestingly, they also show clear time-dependent stiffening behaviour (Fig. 5a). By fitting the data to the viscoelastic model, we see
that the mitotic nuclei are the most compliant and exhibit the lowest viscosity and shortest characteristic time (_τ_; Fig. 5a–d). This supports a model in which untethering of chromatin
from the nuclear periphery decreases both nuclear stiffness and viscosity. DISCUSSION Taken together, these data support a model in which tethering of chromatin to the nuclear periphery
supports two critical functions: it imparts stiffness to nuclei and attenuates the flow of chromatin within the nucleus. When stress is delivered onto isolated chromosomes, they respond
elastically57,58. However, within the confines of the nucleus, chromatin can respond to force by either flowing with or bearing the applied stress. Nuclei with a normal extent of chromatin
tethers predominantly respond elastically to external forces below 4 pN, suggesting that tethering of chromatin to the NE largely restrains chromatin flow and favours the bearing of stress
by chromatin. In this way, the nucleus takes on the mechanical properties of its contents—the chromatin. Untethering of chromatin allows it to flow unabated; this property leads to
compromised nuclear stiffness that is particularly weak when nuclei are deformed slowly. While in wild-type nuclei, the characteristic timescale of chromatin flow is sufficiently slow to
largely prevent migration of chromatin into MT-dependent deformations (Fig. 6a), the absence of NE chromatin tethers allows chromatin to respond to a deformation by flowing to a new (lower
energy) configuration (Fig. 6b). Consequently, residual memory of the deformation persists even after the force is removed, as seen in the slower decay of MT-dependent fluctuations _in vivo_
(Fig. 2d). Thus, microtubule bundles impart, on average, deformations smaller than those that impart hysteretic behaviour (Fig. 4f and Supplementary Fig. 4d). This work also provides new
insights into how _in vivo_ measurements of NE fluctuation size and timescales relate to nuclear mechanics, as nuclear stiffness correlates well with fluctuation size whereas the viscosity
correlates well with fluctuation decay time. Further, delineating this relationship may allow insights into nuclear mechanics to be drawn from live-cell imaging rather than requiring complex
experimental methods such as force spectroscopy. The model put forth here suggests that a balance between the biological timescales of nuclear deformations and the rate of chromatin flow
defines whether the nucleus retains ‘memory’ of previous force. While in wild-type fission yeast such ‘plastic’ change appears to be avoided, in other contexts, for example in migrating
mammalian cells, physical ‘memory’ could facilitate maintaining an altered nuclear shape that supports efficient migration. While it has been observed that A-type lamins impart a
viscoelastic property to nuclei in some14,15, but not all9, studies, the association of A-type lamins with chromatin raises the possibility that at least a component of this behaviour is due
to the ability of lamins to serve as chromatin tethers, particularly, as we observe viscous behaviour in a system lacking nuclear lamins with decay times on the same order of magnitude as
these previous studies53,54,59. Thus, in addition to the clear contribution of the A-type lamin polymer to nuclear stiffness, the ability of the lamina composition to modulate chromatin flow
could also play a critical role in establishing the lifetime of changes in nuclear shape. Thus, changes in nuclear shape that are observed in ‘nuclear envelopathies’ could arise not just
from changes in nuclear stiffness, but also from changes in the kinetics of chromatin flow. While tethering of chromatin to the NE through INM proteins appears sufficient to support stable
nuclear mechanics in response to intracellular forces in _S. pombe_, we expect that these tethering proteins and their associated chromatin are insufficient to provide the stiffness needed
for nuclei to remain intact in the face of large forces exerted on tissues (such as the skin) and in those cells that reside within stiff mechanical environments (like bone). However, at
different stages of development, the stiffness of mammalian nuclei varies drastically. While stem cell nuclei are soft, increased nuclear stiffness occurs as cells differentiate in
conjunction with increasing lamin levels55. Thus, tethering of chromatin to the NE may contribute more substantially to the stiffness of stem cells or other cell types that are under low
levels of external mechanical stress7,60. Further studies will be required to investigate how lamins and INM proteins cooperate to define nuclear stiffness and the kinetics of chromatin
flow. Taking into account the measured _in vitro_ stiffness of wild-type nuclei (∼0.06 pN nm−1) and the average-sized MT-dependent fluctuation from the _in vivo_ measurements (∼50–60 nm,
Supplementary Fig. 1c), we expect that the microtubule bundle delivers ∼3–4 pN of force on the nucleus, which is within the range possible for MT polymerization from a small number of
individual MTs61. Interestingly, the yield point at which the elastic response begins to manifest a viscous component, irrespective of the timescales of the applied stress, is about 5 pN
(Fig. 4f), which likely exceeds physiological forces; thus, the mechanics of wild-type nuclei support an elastic response _in vivo_. Within this context, the organization of chromatin
appears critical. _S. pombe_ clusters its centromeres precisely at the region of the nucleus that is subjected to the most powerful MT forces. Accordingly, our findings show that
specifically untethering chromatin from the SPB interface (as in _heh1Δima1Δ_ cells) leads to the most pronounced mechanical defects _in vivo_. By contrast, loss of Heh2, which is broadly
distributed throughout the NE, appears to mildly suppress increased fluctuation size in this context. Such a differential distribution of Heh1 and Heh2 tethers could allow them to act
antagonistically, with Heh1 more likely to promote chromatin migration into MT-dependent deformations and Heh2 more likely to restrain this chromatin flow. In our current _in vitro_ system,
we do not deliver forces specifically onto the centromeric heterochromatin via the SPB and therefore we cannot directly assess this possibility; testing this model will require further assay
development. Interestingly, yeasts are not the only organisms that cluster large blocks of heterochromatin at MT interfaces. Plants, which also do not encode orthologues of the lamin genes,
organize centromeres inside the nucleus at sites of MT interaction62. In mammalian cells, heterochromatin association with the lamina may, likewise, serve a mechanical role. Such a model
suggests new avenues to consider how nuclear mechanics can be modulated during development and in disease. METHODS CELL CULTURE AND STRAIN GENERATION The strains used in this study are
listed in Supplementary Table 1. _S. pombe_ cells were grown and maintained in standard cell culture conditions63. All the strains were grown at 30 °C. Gene replacements were made by
replacement of the open reading frames with the kanMX6 (ref. 64), hphMX6 or natMX6 (ref. 65) cassettes. C-terminal GFP tagging was performed with pFa6a-GFP-kanMX6 (ref. 64). The
pFa6a-mCherry-kanMX6 cassette was used as a template for C-terminal mCherry tagging66. N-terminal GFP tagging was performed as established using pFa6a-kanMX6-nmt41-GFP64. All strains
generated by cassette integration were confirmed by PCR. Strains made through genetic crosses were confirmed by the segregation of markers and/or by the presence of the appropriate
fluorescently tagged protein. GROWTH CURVES For growth assays, 5 ml cultures were grown in YE5S at 30 °C overnight to saturation. Cultures were diluted to an OD600 ∼0.05 in 50 ml of YE5S and
recovered at 30 °C for 2–3 h. OD600 measurements were taken every hour for 8 h. Doubling time was found using _T_=log(2_t_2_−t_1)/log(_y_/_x_), where _T_=generation time, _y_=cells per ml
at _t_2 and _x_=cells per ml at _t_1. Cells per ml were calculated using the following equation: _x_=(2 × 106)/(0.1 × OD600). MICROSCOPY _S. pombe_ strains were grown in YE5S plus 250 mg l−1
adenine to log phase (OD600 0.5–0.8). Cells were mounted on agarose pads (1.4% agarose in Edinburgh minimal media) and sealed with VALAP (1:1:1, vaseline:lanolin:paraffin). Cells treated
with MBC were incubated on agar pads with 50 μg ml−1 MBC for 10 min before imaging. Cells treated with TSA were grown in YE5S media plus 30 μg ml−1 TSA overnight at 30 °C and placed on agar
pads with 30 μg ml−1 TSA for imaging. Cells treated with Lat A were grown to log phase and treated with 100 μM Lat A 30 min before imaging and placed on agar pads with 100 μM Lat A for
imaging. Live-cell images were acquired on a Deltavision Widefield Deconvolution Microscope (Applied Precision/GE Healthcare) with a CoolSnap HQ2 CCD camera (Photometrics) or an Evolve 512
EMCCD camera (Photometrics). For nuclear contour imaging, 10 z-slices with 400 nm spacing were taken every 2.5 s for 10 min with the EMCCD camera. For INM protein localization imaging, 10
z-slices with 200 nm spacing were taken with the CCD camera. A representative z-slice from the image stack was chosen for figures. For intensity measurements of Chp1-GFP, we used a 3D
Gaussian function to fit each focus. The values of the number of foci and integrated intensity from each cell so-obtained are presented in Supplementary Fig. 2e. CONTOUR FITTING We developed
an optimization procedure that robustly reconstructs the shape of the nucleus in three spatial dimensions with sub-pixel resolution based on a stack of 10 z-plane 2D images of the NE
separated by 400 nm. Our algorithm seeks to find the contour that maximizes the sum of two terms, namely a term corresponding to the total intensity of the NE across the z-stack,
_I_(_r_,_θ_,_ϕ_), and a term corresponding to the negative of the total curvature of the NE, given according to: where the sum is over equally spaced angular vertices, _κ_ is the
computational bending constant, and _J__ij_ is equal to 1 for nearest-neighbour vertices and 0 otherwise. The bending constant of the NE was set automatically via an algorithm that uses the
measured intensities to impose a smoothness constraint on the NE, thus eliminating spurious fluctuations. Hence, bright pixels provide a better definition of the contour while noise, caused
by a low signal-to-noise ratio, is properly interpolated in dim membrane regions. Although our procedure provides a closed surface of the nuclear contour, the reconstructed cap regions of
the nucleus suffer from larger noise for several reasons: the distance between each z-stack interval is larger than the image pixel size, the point spread function is wider in the _z_ axis,
the aberration of the microscope becomes progressively worse for z-slices away from the focus and the drift of images between z-slices. To minimize these noise contributions, we chose to
proceed with our fluctuation analysis by only including a band around the centre of the nucleus defined by 45 degrees above and below the centre stack. To decouple the motions of the nucleus
due to dynamic fluctuations from overall motions of the nucleus, we apply a low-pass Butterworth filter on the centroid position of the nucleus and set these positions as the origin of the
spherical coordinate system at each time point. The radius at equally spaced spatial angles from the centre of the nucleus define the positions of the NE at time _t_ according to
_r_(_θ_,_φ_,_t_). Code available upon request. CONTOUR ANALYSIS To quantify the changes in contour shape, we use the RMSF calculated according to: where _T_ is the total number of frames,
_r_(_θ_,_ϕ_,_t_) is the radial distance from the centre of the contour at vertex angle (_θ_,_ϕ_) at frame _t_ and 〈_r_(_θ_,_ϕ_,_t_)〉_t_ is the time-averaged radial distance at vertex angle
(_θ_,_ϕ_). FLUCTUATION TIME ANALYSIS We identified all the large fluctuation events using a 2D single particle tracking technique. We first projected the fluctuations onto a 2D spatial map,
where the _x_ axis corresponds to a different longitudinal angle (0, 2π) and the _y_ axis corresponds to a different longitudinal angle and the _y_ axis corresponds to a different
latitudinal angle (−π/4, π/4). In this representation, a fluctuation can be approximated as a 2D Gaussian profile. We identified large fluctuations that have fitted amplitudes >50 nm in
each fluctuation map over time. We next applied a single particle tracking algorithm to connect the locations of the large fluctuations across sequential frames67. We only analyse
trajectories that last longer than 10 frames (25 s) to filter out short-lived fluctuations, which we ascribe to spontaneous, thermal fluctuations of the NE (validated by the analysis in
Supplementary Fig. 1c). The magnitude of each fluctuation trajectory is then fit to a single asymmetric triangle waveform to determine the rise time, decay time and peak height. NUCLEAR
ISOLATION Nuclei were isolated by modification of an established protocol described for _S. cerevisiae_49. Strains were grown to log phase at 30 °C in YE5S and diluted to an OD600 of 0.01 in
1 liter YE5S. After growth overnight, cells were collected at an OD600 between 0.8 and 1.0. To prepare for spheroplasting, cells were incubated in 100 mM Tris pH 9.4 and 10 mM DTT for 10
min at 33 °C. Cells were spheroplasted in 0.4 mg ml−1 zymolyase (MP Biomedicals), 0.6 mg ml−1 lysing enzymes (Sigma), 350 μl of beta-glucuronidase (MP Biomedicals) and 5 mM DTT in 1.1 M
sorbitol. Spheroplasting was allowed to proceed at 33 °C for 2–3 h. Spheroplasts were isolated from intact cells by collection through a cushion of 7.5% Ficoll in 1.1 M sorbitol spun in a
SW28 rotor (Beckman) at 10,000_g_ for 15 min at 4 °C. Cells were lysed by gentle homogenization in 0.25% Surfact-Amps X-100 (Thermo), 5 mM DTT, 200 μl protease inhibitor cocktail (Sigma),
0.2 mg ml−1 PMSF, 4 μg ml−1 Pepstatin A (American Bio) in 8% polyvinylpyrrolidone (Sigma). Three density-step sucrose gradients were prepared at the following concentrations: 1.875, 2.30 and
2.50 M. To purify nuclei, lysates were applied to the sucrose gradient and spun in a SW28 rotor (Beckman) at 14,118_g_ for 90 min at 4 °C. Isolated nuclei sedimented at the interface
between the 1.875 and 2.3 M sucrose layers. 100 μl aliquots were flash frozen in liquid N2 and stored at −80 °C. For use, isolated nuclei aliquots were dialysed in 20,000 MWCO Slide-A-Lyzer
MINI Dialysis Units (Thermo) overnight into 500 ml dialysis buffer (80 mM PIPES, 5% DMSO, 2 mM MgCl2, 1 mM EGTA, 500 mM sucrose). Nuclear integrity was confirmed by incubating the nuclei in
10 μg ml−1 FITC 70 kD Dextran (Sigma) and testing for nuclear exclusion. SEM SAMPLE PREPARATION AND IMAGING SEM samples were prepared on the basis of previously published methods68. Silicon
chips (5 × 5 mm2) were prepared by incubating in 1 mg ml−1 poly-L-lysine at room temperature for 30 min. Silicon chips were rinsed with Fixative-1 (4% paraformaldehyde, 20 mM potassium
phosphate pH 6.5, 0.5 mM MgCl2, 0.2 M sucrose) and placed into an inverted microcentrifuge tube lid separated from the top of the tube. The lid with silicon chip was then placed at the base
of a microcentrifuge tube with its bottom cut off. The chip was then overlayed with 20 μl of Fixative-1. Dialyzed, isolated nuclei (8 μl) were placed on top of the fixative in the
microcentrifuge chamber. To bind the nuclei to the silicon chip, the microcentrifuge chamber was spun at 1,000_g_ for 3 min at 4 °C. The silicon chips were then removed from the chamber and
rinsed with Fixative-1 without sucrose and incubated in Fixative-2 (2% glutaraldehyde, 0.2% tannic acid in 20 mM potassium phosphate, 0.5 MgCl2, pH 7.4) for 10 min at room temperature.
Silicon chips were rinsed with dH2O and fixed in 1% osmium tetroxide for 10 min. Samples were dehydrated by incubating for 2 min in each 30%, 50%, 70%, 95% (twice) and 100% (three times)
ethanol in series. After final dehydration in 100% ethanol, the samples were dried using a Polaron critical point dryer with liquid carbon dioxide as transitional fluid. The silicon chips
were glued to aluminium stubs and sputter coated with 15 nm gold (Electron Microscopy Science). The samples were viewed and digital images acquired in an FEI ESEM between 10 kV at a working
distance of 10 mm. VESICLE PREPARATION Vesicles were prepared on the basis of methods previously described69 and all lipids were purchased from Avanti. A 20:10:70 ratio of lipids DOPE
(1,2-dioleoyl-_sn_-glycero-3-phosphoethanolamine), DOPS (1,2-dioleoyl-_sn-_glycero-3-phospho-L-serine) and DOPC (1,2-dioleoyl-_sn_-glycero-3-phosphocholine), respectively were dissolved in
chloroform and dried to a film under nitrogen gas. The mixed lipids were resuspended in vesicle buffer (80 mM PIPES pH 6.8, 1 mM MgCl2, 1 mM EGTA, 170 mM sucrose, 1 mg ml−1 70 kD FITC
dextran) to 10–30 mM and subjected to seven freeze–thaw cycles. Resuspended lipids were extruded through two polycarbonate membranes (Whatman) with 3.0 μm pore size using the LipSoFast-Basic
extruder (Avestin) at room temperature and stored at −80 °C. For use, vesicles were diluted in nuclei dialysis buffer minus sucrose and spun at 10,000_g_ in a TLA 100.3 (Beckman) rotor for
30 min at 4 °C. Vesicles were resuspended in 500 μl dialysis buffer with sucrose for experiments. OPTICAL TWEEZERS SAMPLE PREPARATION Flow cells were constructed by cutting double-sided
sticky tape to create a flow channel between a microscope coverslip and a microscope slide that contains holes to allow for two inlet ports and one outlet port. Dry, 5.20 μm silica-NH2
microspheres (Bangs Laboratories) were resuspended in 100% EtOH. A concentrated slurry of microspheres was added to 170 μl of 0.01% poly-L-ornithine (Sigma), loaded into the flow cell and
incubated for a minimum of 2 h. Flow cells were then rinsed with dialysis buffer. Aliphatic amine latex beads (2% w/v 1.2 μm, Invitrogen) were pre-incubated for a minimum of 2 h in
poly-l-ornithine, then rinsed with dialysis buffer. Nuclei were loaded onto flow cell immediately before experiments. Latex beads were diluted 1:300 into dialysis buffer+2 mM ATP. The
dialysis buffer+2 mM ATP and the latex bead mixture were loaded into separate syringes and connected to the two inlet ports. OPTICAL TWEEZERS SETUP We use a single-beam gradient optical trap
for both trapping and back focal plane interferometry position detection. The trapping beam, produced by a 3 W 1,064 nm ND:YAG laser (Laser Quantum, Ventus IR), passes through an
acousto-optic deflector (IntraAction) to control the position and intensity of the laser beam and also acts as an optical isolator. The beam is then expanded by a factor of 3 to slightly
overfill the back aperture of the objective (Nikon, CFI × 100, 1.2 NA). The focused laser is used to trap a bead inside of a flow cell, which rests on a custom-built sample stage holder
mounted on a Thorlabs nanomax positioner, consisting of coarse stage motors with micron scale accuracy and a piezo stage with nanometre resolution. The forward scattered light is collected
with a condenser and is directed onto an infrared-enhanced quadrant photodiode (First Sensor) located in a conjugate plane to the back focal plane of the condenser. To visualize the nucleus
via fluorescence, an epi-fluorescence microscope is coupled into the optical tweezers setup. The excitation source, produced by a 470 nm blue light emitting diode (Luxeon Star), is reflected
with a long pass dichroic mirror (Semrock) to the back of the objective. The backscattered fluorescent light is collected with the same objective and is transmitted through a dichroic
mirror and two bandpass filters (Semrock) placed in series to attenuate non-fluorescence light, including backscattered light from the trapping laser. The fluorescence signal then travels
through a tube lens onto an Orca R2 CCD camera (Hamamatsu). All instrument controls and data acquisition is accomplished with custom-made virtual instruments programmed in Labview (National
Instruments). OPTICAL TWEEZERS OSCILLATION ASSAY Isolated nuclei were flowed into the flow cell and an individual nucleus was trapped using the optical tweezers and manipulated onto the side
of a surface-immobilized 5.20 μm silica-NH2 microsphere. The nuclei associate with the microspheres via electrostatic interactions. Excess nuclei were rinsed away by flowing in dialysis
buffer+2 mM ATP. Poly-ornithine-coated latex beads (1.20 μm) were loaded into the flow cell and a single bead was trapped with the optical tweezers. The trapped bead was brought close to the
nucleus opposite of the large bead-nucleus interface (see diagram in Fig. 4). As the trapped bead was brought into contact with the nucleus, we monitored its displacement within the optical
trap, which allowed us to determine the equilibrium position of the nucleus. Upon contact, electrostatic interactions adhere the latex bead with the nucleus. Excess beads were rinsed away
with dialysis buffer +2 mM ATP. The stage was driven sinusoidally with an amplitude of ∼60–70 nm or ∼150–170 nm to apply rounds of compression and tension forces on the nucleus at different
frequencies, as indicated. DRIFT CONTROL To enable high-precision measurements for extended durations, we have implemented a drift correction procedure50. Specifically, two template images
are acquired, both consisting of a window about an isolated surface-attached bead. One image is in focus. The other image is out of focus, displaced from the in-focus condition by a known
distance along the beam direction (_z_). To correct for in-plane (_xy_ plane) drift, the _x_ and _y_ axes of the piezo stage are adjusted to maintain the maximum cross-correlation between
the in-focus template image and the corresponding measured image at subsequent times. Thus, we are able to stabilize the position within the _xy_ plane to within 1 nm. To correct for drift
along _z_, we exploit the observation that the maximum cross-correlation between the out-of-focus template image and the measured image varies linearly with their _z_ displacement in the
relevant range. Therefore, we correct _z_-drift by holding fixed the value of the maximum cross-correlation between the out-of-focus template image and the measured image via a PID
controller implemented in LabView. In this way, we stabilize the _z_-position to within 2.5 nm. Key to the success of our approach is the use of partially coherent illumination from an LED
such that fringes are a prominent feature of the bead images and depend sensitively on the _z_-position. OPTICAL TWEEZERS ANALYSIS The effective stiffness of the nucleus, _k_nucleus, is
determined by applying a linear fit to _F_=_k_nucleusΔ_x_nucleus, where Δ_x_nucleus is the extension of the nucleus from equilibrium. By exploiting the fact that the force on the nucleus is
equal and opposite to the force on the trapped bead, the force on the nucleus is determined by _F=k_trapΔ_x_trap, where Δ_x_trap is the displacement of the bead from the centre of the trap,
and _k_trap is the optical trap stiffness. The stiffness of the optical trap is found by fitting the power spectral density of a trapped 1.2 μm latex bead to a Lorentzian function to
determine the corner frequency70. The stiffness is then determined as the ratio of the corner frequency to the bead’s friction coefficient, corrected using Faxen’s Law to account for the
proximity of the glass coverslip. Δ_x_trap is determined by a quadrant photodiode, which is calibrated by scanning the trapping laser over a surface-immobilized bead70. The extension of the
nucleus from equilibrium is determined as the difference between the displacement of the piezo stage from equilibrium and the displacement of the bead from the centre of the trap, according
to Δ_x_nucleus=Δ_x_piezostage−Δ_x_trap. VISCOELASTIC MODEL We fit the frequency dependence of the effective stiffness of the nucleus with a Maxwell viscoelastic model in which a spring and
dashpot in series captures the elastic component, _K_, and viscous component, _η_, respectively. The corresponding fitting function for the effective stiffness is given according to: and the
characteristic time is given according to ADDITIONAL INFORMATION HOW TO CITE THIS ARTICLE: Schreiner, S.M. _et al._ The tethering of chromatin to the nuclear envelope supports nuclear
mechanics. _Nat. Commun._ 6:7159 doi: 10.1038/ncomms8159 (2015). REFERENCES * Maniotis, A. J., Chen, C. S. & Ingber, D. E. Demonstration of mechanical connections between integrins,
cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. _Proc. Natl Acad. Sci. USA_ 94, 849–854 (1997). Article CAS ADS Google Scholar * Graumann, K., Irons, S. L.,
Runions, J. & Evans, D. E. Retention and mobility of the mammalian lamin B receptor in the plant nuclear envelope. _Biol. Cell_ 99, 553–562 (2007). Article CAS Google Scholar * Rowat,
A. C. et al. Nuclear envelope composition determines the ability of neutrophil-type cells to passage through micron-scale constrictions. _J. Biol. Chem._ 288, 8610–8618 (2013). Article CAS
Google Scholar * Harada, T. et al. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. _J. Cell Biol._ 204, 669–682 (2014). Article CAS Google Scholar
* Isermann, P. & Lammerding, J. Nuclear mechanics and mechanotransduction in health and disease. _Curr. Biol._ 23, R1113–R1121 (2013). Article CAS Google Scholar * Dahl, K.,
Discher, D. & Wilson, K. The nuclear envelope lamina network has elasticity and a compressibility limit suggestive of a "molecular shock absorber". _Mol. Biol. Cell_ 15,
119A–120A (2004). Google Scholar * Broers, J. L. et al. Decreased mechanical stiffness in LMNA-/- cells is caused by defective nucleo-cytoskeletal integrity: implications for the
development of laminopathies. _Hum. Mol. Genet._ 13, 2567–2580 (2004). Article CAS Google Scholar * Lammerding, J. et al. Lamins A and C but not lamin B1 regulate nuclear mechanics. _J.
Biol. Chem._ 281, 25768–25780 (2006). Article CAS Google Scholar * Schäpe, J., Prauße, S., Radmacher, M. & Stick, R. Influence of lamin A on the mechanical properties of amphibian
oocyte nuclei measured by atomic force microscopy. _Biophys. J._ 96, 4319–4325 (2009). Article ADS Google Scholar * Kim, Y. et al. Mouse B-type lamins are required for proper
organogenesis but not by embryonic stem cells. _Science_ 334, 1706–1710 (2011). Article CAS ADS Google Scholar * Dahl, K. et al. Distinct structural and mechanical properties of the
nuclear lamina in Hutchinson-Gilford progeria syndrome. _Proc. Natl Acad. Sci. USA_ 103, 10271–10276 (2006). Article CAS ADS Google Scholar * Verstraeten, V. L., Ji, J. Y., Cummings, K.
S., Lee, R. T. & Lammerding, J. Increased mechanosensitivity and nuclear stiffness in Hutchinson-Gilford progeria cells: effects of farnesyltransferase inhibitors. _Aging Cell_ 7,
383–393 (2008). Article CAS Google Scholar * Sullivan, T. et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. _J. Cell Biol._ 147,
913–920 (1999). Article CAS Google Scholar * Swift, J. et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. _Science_ 341, 1240104 (2013).
Article Google Scholar * Banerjee, A. et al. Viscoelastic behavior of human lamin A proteins in the context of dilated cardiomyopathy. _PLoS ONE_ 8, e83410 (2013). Article ADS Google
Scholar * Swift, J. & Discher, D. E. The nuclear lamina is mechano-responsive to ECM elasticity in mature tissue. _J. Cell Sci._ 127, 3005–3015 (2014). Article CAS Google Scholar *
Lammerding, J. et al. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. _J. Clin. Invest._ 113, 370–378 (2004). Article CAS Google Scholar * Guelen, L. et
al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. _Nature_ 453, 948–951 (2008). Article CAS ADS Google Scholar * Pickersgill, H. et al.
Characterization of the Drosophila melanogaster genome at the nuclear lamina. _Nat. Genet._ 38, 1005–1014 (2006). Article CAS Google Scholar * Gruenbaum, Y., Margalit, A., Goldman, R. D.,
Shumaker, D. K. & Wilson, K. L. The nuclear lamina comes of age. _Nat. Rev. Mol. Cell Biol._ 6, 21–31 (2005). Article CAS Google Scholar * Dauer, W. T. & Worman, H. J. The
nuclear envelope as a signaling node in development and disease. _Dev. Cell_ 17, 626–638 (2009). Article CAS Google Scholar * Zuleger, N. et al. Specific nuclear envelope transmembrane
proteins can promote the location of chromosomes to and from the nuclear periphery. _Genome Biol._ 14, R14 (2013). Article Google Scholar * Malik, P. et al. Cell-specific and
lamin-dependent targeting of novel transmembrane proteins in the nuclear envelope. _Cell Mol. Life Sci._ 67, 1353–1369 (2010). Article CAS Google Scholar * Guo, Y., Kim, Y., Shimi, T.,
Goldman, R. D. & Zheng, Y. Concentration-dependent lamin assembly and its roles in the localization of other nuclear proteins. _Mol. Biol. Cell_ 25, 1287–1297 (2014). Article Google
Scholar * Mans, B. J., Anantharaman, V., Aravind, L. & Koonin, E. V. Comparative genomics, evolution and origins of the nuclear envelope and nuclear pore complex. _Cell Cycle_ 3,
1612–1637 (2004). Article CAS Google Scholar * King, M. C., Lusk, C. P. & Blobel, G. Karyopherin-mediated import of integral inner nuclear membrane proteins. _Nature_ 442, 1003–1007
(2006). Article CAS ADS Google Scholar * Yewdell, W. T., Colombi, P., Makhnevych, T. & Lusk, C. P. Lumenal interactions in nuclear pore complex assembly and stability. _Mol. Biol.
Cell_ 22, 1375–1388 (2011). Article CAS Google Scholar * Gonzalez, Y., Saito, A. & Sazer, S. Fission yeast Lem2 and Man1 perform fundamental functions of the animal cell nuclear
lamina. _Nucleus_ 3, 60–76 (2012). Article Google Scholar * King, M., Drivas, T., Blobel, G. & Blobel, G. A network of nuclear envelope membrane proteins linking centromeres to
microtubules. _Cell_ 134, 427–438 (2008). Article CAS Google Scholar * Buch, C. et al. An integral protein of the inner nuclear membrane localizes to the mitotic spindle in mammalian
cells. _J. Cell Sci._ 122, 2100–2107 (2009). Article CAS Google Scholar * Hiraoka, Y. et al. Inner nuclear membrane protein Ima1 is dispensable for intranuclear positioning of
centromeres. _Genes Cells_ 16, 1000–1011 (2011). Article CAS Google Scholar * Ding, R. et al. The spindle pole body of Schizosaccharomyces pombe enters and leaves the nuclear envelope as
the cell cycle proceeds. _Mol. Biol. Cell_ 8, 1461–1479 (1997). Article CAS Google Scholar * Shimanuki, M. et al. A novel fission yeast gene, kms1, is required for the formation of
meiotic prophase-specific nuclear architecture. _Mol. Gen. Genet._ 254, 238–249 (1997). Article CAS Google Scholar * Wälde, S. & King, M. C. The KASH protein Kms2 coordinates mitotic
remodeling of the spindle pole body. _J Cell Sci._ 127, 3625 (2014). Article Google Scholar * Sawin, K. & Tran, P. T. Cytoplasmic microtubule organization in fission yeast. _Yeast_ 23,
1001–1014 (2006). Article CAS Google Scholar * Tran, P. T. et al. A mechanism for nuclear positioning in fission yeast based on microtubule pushing. _J. Cell Biol._ 153, 397–411 (2001).
Article CAS Google Scholar * Goto, B., Okazaki, K. & Niwa, O. Cytoplasmic microtubular system implicated in _de novo_ formation of a Rabl-like orientation of chromosomes in fission
yeast. _J. Cell Sci._ 114, 2427–2435 (2001). CAS PubMed Google Scholar * Funabiki, H., Hagan, I., Uzawa, S. & Yanagida, M. Cell cycle-dependent specific positioning and clustering of
centromeres and telomeres in fission yeast. _J. Cell Biol._ 121, 961–976 (1993). Article CAS Google Scholar * West, R. R., Vaisberg, E. V., Ding, R., Nurse, P. & McIntosh, J. R.
cut11(+): a gene required for cell cycle-dependent spindle pole body anchoring in the nuclear envelope and bipolar spindle formation in Schizosaccharomyces pombe. _Mol. Biol. Cell_ 9,
2839–2855 (1998). Article CAS Google Scholar * Rupes, I., Webb, B. A., Mak, A. & Young, P. G. G2/M arrest caused by actin disruption is a manifestation of the cell size checkpoint in
fission yeast. _Mol. Biol. Cell_ 12, 3892–3903 (2001). Article CAS Google Scholar * Neumann, F. & Nurse, P. Nuclear size control in fission yeast. _J. Cell Biol._ 179, 593–600 (2007).
Article CAS Google Scholar * Grund, S. E. et al. The inner nuclear membrane protein Src1 associates with subtelomeric genes and alters their regulated gene expression. _J. Cell Biol._
182, 897–910 (2008). Article CAS Google Scholar * Mekhail, K., Seebacher, J., Gygi, S. P. & Moazed, D. Role for perinuclear chromosome tethering in maintenance of genome stability.
_Nature_ 456, 667–670 (2008). Article CAS ADS Google Scholar * Steglich, B., Filion, G. J., van Steensel, B. & Ekwall, K. The inner nuclear membrane proteins Man1 and Ima1 link to
two different types of chromatin at the nuclear periphery in S. pombe. _Nucleus_ 3, 77–87 (2012). Article Google Scholar * Marguerat, S. et al. Quantitative analysis of fission yeast
transcriptomes and proteomes in proliferating and quiescent cells. _Cell_ 151, 671–683 (2012). Article CAS Google Scholar * Ishida, M. et al. Intrinsic nucleic acid-binding activity of
Chp1 chromodomain is required for heterochromatic gene silencing. _Mol. Cell_ 47, 228–241 (2012). Article CAS Google Scholar * Sadaie, M., Iida, T., Urano, T. & Nakayama, J. A
chromodomain protein, Chp1, is required for the establishment of heterochromatin in fission yeast. _EMBO J._ 23, 3825–3835 (2004). Article CAS Google Scholar * Russell, P. & Nurse, P.
Cdc25+ functions as an inducer in the mitotic control of fission yeast. _Cell_ 45, 145–153 (1986). Article CAS Google Scholar * Rout, M. P. & Kilmartin, J. V. Components of the yeast
spindle and spindle pole body. _J. Cell Biol._ 111, 1913–1927 (1990). Article CAS Google Scholar * Koo, P. K., Setru, S. U. & Mochrie, S. G. Active drift stabilization in three
dimensions via image cross-correlation. _Rev. Sci. Instrum._ 84, 103705 (2013). Article CAS ADS Google Scholar * Guo, M. et al. Probing the stochastic, motor-driven properties of the
cytoplasm using force spectrum microscopy. _Cell_ 158, 822–832 (2014). Article CAS Google Scholar * Guilluy, C. et al. Isolated nuclei adapt to force and reveal a mechanotransduction
pathway in the nucleus. _Nat. Cell Biol._ 16, 376–381 (2014). Article CAS Google Scholar * Dahl, K. et al. Power-law rheology of isolated nuclei with deformation mapping of nuclear
substructures. _Biophys. J._ 89, 2855–2864 (2005). Article CAS ADS Google Scholar * Rowat, A. C., Foster, L. J., Nielsen, M. M., Weiss, M. & Ipsen, J. H. Characterization of the
elastic properties of the nuclear envelope. _J. R. Soc. Interface_ 2, 63–69 (2005). Article CAS Google Scholar * Pajerowski, J. D., Dahl, K., Zhong, F., Sammak, P. & Discher, D.
Physical plasticity of the nucleus in stem cell differentiation. _Proc. Natl Acad. Sci. USA_ 104, 15619–15624 (2007). Article CAS ADS Google Scholar * Fujita, I. et al. Telomere-nuclear
envelope dissociation promoted by Rap1 phosphorylation ensures faithful chromosome segregation. _Curr. Biol._ 22, 1932–1937 (2012). Article CAS Google Scholar * Poirier, M. G. &
Marko, J. F. Micromechanical studies of mitotic chromosomes. _Curr. Top. Dev. Biol._ 55, 75–141 (2003). Article CAS Google Scholar * Cui, Y. & Bustamante, C. Pulling a single
chromatin fiber reveals the forces that maintain its higher-order structure. _Proc. Natl Acad. Sci. USA_ 97, 127–132 (2000). Article CAS ADS Google Scholar * Rowat, A. C., Lammerding, J.
& Ipsen, J. H. Mechanical properties of the cell nucleus and the effect of emerin deficiency. _Biophys. J._ 91, 4649–4664 (2006). Article CAS ADS Google Scholar * Broers, J. L. et
al. A- and B-type lamins are differentially expressed in normal human tissues. _Histochem. Cell Biol._ 107, 505–517 (1997). Article CAS Google Scholar * Dogterom, M. & Yurke, B.
Measurement of the force-velocity relation for growing microtubules. _Science_ 278, 856–860 (1997). Article CAS ADS Google Scholar * Batzenschlager, M. et al. The GIP gamma-tubulin
complex-associated proteins are involved in nuclear architecture in _Arabidopsis thaliana_. _Front. Plant Sci._ 4, 480 (2013). Article Google Scholar * Moreno, S., Klar, A. & Nurse, P.
Molecular genetic analysis of fission yeast _Schizosaccharomyces pombe_. _Methods Enzymol._ 194, 795–823 (1991). Article CAS Google Scholar * Bahler, J. et al. Heterologous modules for
efficient and versatile PCR-based gene targeting in _Schizosaccharomyces pombe_. _Yeast_ 14, 943–951 (1998). Article CAS Google Scholar * Hentges, P., Van Driessche, B., Tafforeau, L.,
Vandenhaute, J. & Carr, A. M. Three novel antibiotic marker cassettes for gene disruption and marker switching in _Schizosaccharomyces pombe_. _Yeast_ 22, 1013–1019 (2005). Article CAS
Google Scholar * Snaith, H. A., Samejima, I. & Sawin, K. E. Multistep and multimode cortical anchoring of tea1p at cell tips in fission yeast. _EMBO J._ 24, 3690–3699 (2005). Article
CAS Google Scholar * Crocker, J. & Grier, D. Methods of digital video microscopy for colloidal studies. _J. Colloid Interface Sci._ 179, 298–310 (1996). Article CAS ADS Google
Scholar * Kiseleva, E. et al. A protocol for isolation and visualization of yeast nuclei by scanning electron microscopy (SEM). _Nat. Protoc._ 2, 1943–1953 (2007). Article CAS Google
Scholar * Jotwani, A., Richerson, D. N., Motta, I., Julca-Zevallos, O. & Melia, T. J. Approaches to the study of Atg8-mediated membrane dynamics in vitro. _Methods Cell Biol._ 108,
93–116 (2012). Article CAS Google Scholar * Mack, A. H. Practical axial optical trapping. _Rev. Sci. Instrum._ 83, 103106 (2012). Article CAS ADS Google Scholar Download references
ACKNOWLEDGEMENTS We thank the members of the King Lab and Dr Patrick Lusk for critical reading of the manuscript. We also thank Dr Cecile Mejean and Dr Eric Dufresne for help during the
initial development of the force spectroscopy assay, Elisa Rodriguez for help purifying the nuclei, Dr Thomas Melia for help with the vesicle preparation and Morven Graham for help with the
SEM imaging. This work was supported by NSF CMMI-1334406, an American Heart Association Founders Affiliate Predoctoral Fellowship 13PRE17070004 (S.M.S.), NIH T32-GM007499 (S.M.S.), NSF PHY
1019147 (P.K.K.), the Searle Scholars Program (M.C.K.), G. Harold & Leila Y. Mathers Charitable Foundation (M.C.K.) and a seed grant from the Raymond and Beverly Sackler Institute for
Physical and Engineering Biology. AUTHOR INFORMATION Author notes * Sarah M. Schreiner and Peter K. Koo: These authors contributed equally to this work AUTHORS AND AFFILIATIONS * Department
of Cell Biology, Yale School of Medicine, 333 Cedar Street, New Haven, 06520-8002, Connecticut, USA Sarah M. Schreiner & Megan C. King * Department of Physics, Yale University, 217
Prospect Street, New Haven, 06511, Connecticut, USA Peter K. Koo & Simon G. J. Mochrie * Department of Applied Physics, Yale University, 15 Prospect Street, New Haven, 06511,
Connecticut, USA Yao Zhao & Simon G. J. Mochrie Authors * Sarah M. Schreiner View author publications You can also search for this author inPubMed Google Scholar * Peter K. Koo View
author publications You can also search for this author inPubMed Google Scholar * Yao Zhao View author publications You can also search for this author inPubMed Google Scholar * Simon G. J.
Mochrie View author publications You can also search for this author inPubMed Google Scholar * Megan C. King View author publications You can also search for this author inPubMed Google
Scholar CONTRIBUTIONS All the authors contributed to the experimental design. S.M.S. performed all the biological experiments and all final optical tweezers experiments. P.K.K. constructed
the optical tweezers apparatus and performed initial optical tweezers experiments. S.M.S. and P.K.K. both analysed the optical tweezers data. Y.Z. and P.K.K. contributed equally to the
development of the nuclear contour algorithm and fluctuation analysis. All the authors wrote and edited the manuscript. CORRESPONDING AUTHORS Correspondence to Simon G. J. Mochrie or Megan
C. King. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing financial interests. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION Supplementary Figures 1-4 and
Supplementary Tables 1-2 (PDF 438 kb) SUPPLEMENTARY MOVIE 1 2D cross-sections at three z-slices about the center of the nucleus (right) and the corresponding 3D rendering (left) of the wild
type nucleus from Figure 1a. Cells were imaged every 2.5s for 5 minutes. Movies shown at 3 frames/second. Scale bar equals 1 μm in 2D cross-sections. Nuclear deformations are highlighted
using the heat map shown in the movie. (MOV 2867 kb) SUPPLEMENTARY MOVIE 2 2D cross-sections at three z-slices about the center of the nucleus (right) and the corresponding 3D rendering
(left) of the heh1Δ nucleus from Figure 2b. Cells were imaged every 2.5 seconds for 5 minutes. Movies shown at 3 frames per second. Scale bar equals 1 μm in 2D cross-sections. Nuclear
deformations are highlighted using the heat map shown in the movie. (MOV 2950 kb) SUPPLEMENTARY MOVIE 3 2D cross-sections at three z-slices about the center of the nucleus (right) and the
corresponding 3D rendering (left) of the heh2Δ nucleus from Figure 2b. Cells were imaged every 2.5 seconds for 5 minutes. Movies shown at 3 frames per second. Scale bar equals 1 μm in 2D
crosssections. Nuclear deformations are highlighted using the heat map shown in the movie. (MOV 2867 kb) SUPPLEMENTARY MOVIE 4 2D cross-sections at three z-slices about the center of the
nucleus (right) and the corresponding 3D rendering (left) of the ima1Δ nucleus from Figure 2b. Cells were imaged every 2.5 seconds for 5 minutes. Movies shown at 3 frames per second. Scale
bar equals 1 μm in 2D crosssections. Nuclear deformations are highlighted using the heat map shown in the movie. (MOV 2836 kb) SUPPLEMENTARY MOVIE 5 2D cross-sections at three z-slices about
the center of the nucleus (right) and the corresponding 3D rendering (left) of the heh1Δheh2Δ nucleus from Figure 2b. Cells were imaged every 2.5 seconds for 5 minutes. Movies shown at 3
frames per second. Scale bar equals 1 μm in 2D cross-sections. Nuclear deformations are highlighted using the heat map shown in the movie. (MOV 2827 kb) SUPPLEMENTARY MOVIE 6 2D
cross-sections at three z-slices about the center of the nucleus (right) and the corresponding 3D rendering (left) of the heh1Δima1Δ nucleus from Figure 2b. Cells were imaged every 2.5
seconds for 5 minutes. Movies shown at 3 frames per second. Scale bar equals 1 μm in 2D cross-sections. Nuclear deformations are highlighted using the heat map shown in the movie. (MOV 2782
kb) SUPPLEMENTARY MOVIE 7 2D cross-sections at three z-slices about the center of the nucleus (right) and the corresponding 3D rendering (left) of the heh2Δima1Δ nucleus from Figure 2b.
Cells were imaged every 2.5 seconds for 5 minutes. Movies shown at 3 frames per second. Scale bar equals 1 μm in 2D cross-sections. Nuclear deformations are highlighted using the heat map
shown in the movie. (MOV 2836 kb) SUPPLEMENTARY MOVIE 8 2D cross-sections at three z-slices about the center of the nucleus (right) and the corresponding 3D rendering (left) of the
heh1Δheh2Δima1Δ nucleus from Figure 2b. Cells were imaged every 2.5 second for 5 minutes. Movies shown at 3 frames per second. Scale bar equals 1 μm in 2D cross-sections. Nuclear
deformations are highlighted using the heat map shown in the movie. (MOV 2754 kb) SUPPLEMENTARY MOVIE 9 2D cross-sections at three z-slices about the center of the nucleus (right) and the
corresponding 3D rendering (left) of the cdc25-22 nucleus from Figure 3b. Cells were imaged every 2.5 second for 5 minutes. Movies shown at 3 frames per second. Scale bar equals 1 μm in 2D
crosssections. Nuclear deformations are highlighted using the heat map shown in the movie. (MOV 2914 kb) RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0
International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the
material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Schreiner, S., Koo, P., Zhao, Y. _et al._ The tethering of chromatin to the nuclear
envelope supports nuclear mechanics. _Nat Commun_ 6, 7159 (2015). https://doi.org/10.1038/ncomms8159 Download citation * Received: 31 October 2014 * Accepted: 10 April 2015 * Published: 15
June 2015 * DOI: https://doi.org/10.1038/ncomms8159 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link
is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative





