- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The association between tissue damage, chronic inflammation and cancer is well known. However, the underlying mechanisms are unclear. Here we characterize a mouse model in which
constitutive epidermal extracellular-signal-regulated kinase-MAP-kinase signalling results in epidermal inflammation, and skin wounding induces tumours. We show that tumour incidence
correlates with wound size and inflammatory infiltrate. Ablation of tumour necrosis factor receptor (TNFR)-1/-2, Myeloid Differentiation primary response gene 88 or Toll-like receptor
(TLR)-5, the bacterial flagellin receptor, but not other innate immune sensors, in radiosensitive leukocytes protects against tumour formation. Antibiotic treatment inhibits, whereas
injection of flagellin induces, tumours in a TLR-5-dependent manner. TLR-5 is also involved in chemical-induced skin carcinogenesis in wild-type mice. Leukocytic TLR-5 signalling mediates
upregulation of the alarmin HMGB1 (High Mobility Group Box 1) in wound-induced papillomas. HMGB1 is elevated in tumours of patients with Recessive Dystrophic Epidermolysis Bullosa, a disease
characterized by chronic skin damage. We conclude that in our experimental model the combination of bacteria, chronic inflammation and wounding cooperate to trigger skin cancer. SIMILAR
CONTENT BEING VIEWED BY OTHERS THE COMMENSAL SKIN MICROBIOTA TRIGGERS TYPE I IFN–DEPENDENT INNATE REPAIR RESPONSES IN INJURED SKIN Article 13 July 2020 IL-36 RECEPTOR ANTAGONIST DEFICIENCY
RESULTED IN DELAYED WOUND HEALING DUE TO EXCESSIVE RECRUITMENT OF IMMUNE CELLS Article Open access 08 September 2020 NK CELLS IN HYPOXIC SKIN MEDIATE A TRADE-OFF BETWEEN WOUND HEALING AND
ANTIBACTERIAL DEFENCE Article Open access 04 August 2021 INTRODUCTION The association between skin wounding, inflammation and cancer is well established1,2. For example, Marjolin’s ulcers
are aggressive squamous cell carcinomas (SCCs) that specifically develop on areas of previous skin trauma3. Keloid scarring is a consequence of aberrant wound healing and is also described
as benign fibrotic tumour formation4. In addition, malignancies at wound sites are often overlooked in chronic ulcers in diabetic and elderly patients5. Another context in which the
association between skin wounding and cancer is well established is Recessive Dystrophic Epidermolysis Bullosa (RDEB). This inherited skin blistering disease is characterized by repetitive
cycles of wounding and repair and is linked with a high incidence of SCC formation. Almost 100% of RDEB patients will develop at least one SCC6. However, despite the clear link between skin
damage and cancer, little is known about the underlying mechanisms. We previously described a mouse model of wound-induced skin cancer that mimics key features of human hyperproliferative
skin conditions. Wounded human skin and psoriatic lesions are characterized by misexpression of β1 integrin extracellular matrix receptors in the differentiating epidermal cell layers, and
consequent upregulation of extracellular signal-regulated kinase-MAP-kinase signalling7. When this is modelled in transgenic mice by expression of constitutively active MAP-kinase kinase 1
under the control of the involucrin promoter (InvEE mice), there is chronic skin inflammation and epidermal hyperproliferation, and mice develop benign tumours (papillomas) on wounding8,9.
We previously identified a pro-tumorigenic role for macrophages and peripheral γδT cells in this model8. Here we set out to identify the molecular signalling events that underlie the link
between chronic inflammation, tissue damage and skin cancer. We have found a previously unknown role for Toll-like receptor (TLR)-5, the receptor for bacterial flagellin, in skin tumour
formation, both in the InvEE mouse model and in wild-type (WT) mice treated with chemical carcinogens. We further show that TLR-5 plays a role in upregulation of the alarmin HMGB1 (High
Mobility Group Box 1) in wound-induced mouse tumours and demonstrate that HMGB1 is also elevated in tumours of RDEB patients. RESULTS EFFECT OF WOUND SIZE AND IMMUNE INFILTRATE ON
TUMORIGENESIS To test whether wound size, wound closure rate or inflammatory response to wounding influenced tumour incidence, full-thickness skin wounds of different sizes (2, 4, 5, 6 and 8
mm2) were made on back skin of WT and InvEE (Inv) mice, and papilloma formation at the wound site was monitored. Wounds in InvEE and WT littermates healed at the same rate but only InvEE
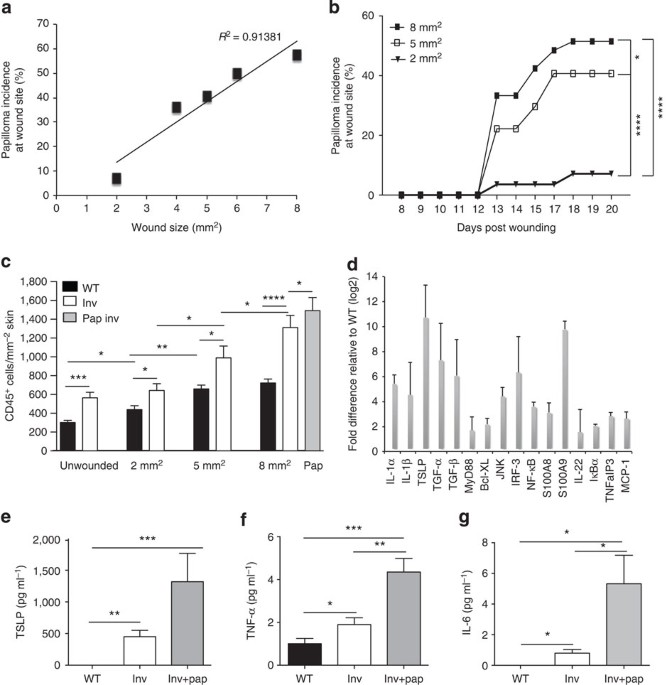
mice developed tumours (Supplementary Fig. 1a,b). Although onset of tumour formation was independent of wound size, there was a linear correlation between wound size and tumour incidence
(_R_2=0.91381; Fig. 1a,b). Wound size and total immune cell infiltrate (CD45+ cells) were correlated in both WT and InvEE skin, but CD45+ cells were significantly more abundant in InvEE skin
both before wounding9 and at the time of wound closure (Fig. 1c and Supplementary Fig. 1c,d). There were even more CD45+ cells in the tumour stroma than in newly closed 8 mm2 InvEE wounds
(Fig. 1c). These results indicate that the degree of inflammation remaining once the acute response to injury has resolved correlates with the extent of the primary insult and subsequent
tumour incidence. As nuclear factor-κB (NF-κB) is an important mediator of inflammation-associated cancer10, we analysed expression of NF-κB target genes in InvEE and control epidermis. All
16 of the genes examined were significantly upregulated in InvEE epidermis relative to WT epidermis (_P_<0.0001 for each individual gene product; Fig. 1d). The effects were systemic, as
levels of thymic stromal lymphopoietin (TSLP), tumour-necrosis factor (TNF)-α and interleukin (IL)-6 were elevated in serum of tumour-free InvEE mice and increased further in tumour-bearing
animals (Fig. 1e–g). TUMOUR FORMATION REQUIRES HAEMATOPOIETIC TNFR SIGNALLING TNF-α is well known for its context-dependent pro- and anti-tumorigenic roles11 and downstream TNF-α signalling
is mediated by TNFR-1 and TNFR-2. Mice deficient in both receptors are resistant to skin cancer induced by chemical carcinogens12. To examine whether wound-induced tumorigenesis was
dependent on TNFR signalling specifically in leukocytes, sub-lethally irradiated InvEE mice were reconstituted with TNFR-1/-2−/− (TNFR−/−) bone marrow (BM) and subsequently wounded.
Successful engraftment was verified by Y chromosome-fluorescence _in situ_ hybridization (Y-FISH) in spleens of reconstituted mice as previously described8 (Supplementary Fig. 2a). TNFR−/−
chimeric mice were highly resistant to wound-induced tumour formation (Fig. 2a). Only 8.3% of TNFR−/− chimeric mice developed papillomas, compared with 50% of control chimeras, and time of
tumour onset was delayed in TNFR−/− chimeric mice (Fig. 2a). Although wounds closed more rapidly in TNFR−/− chimeras (Supplementary Fig. 2b), in agreement with observations on TNFR-1−/−
mice13, the epidermis remained thickened and hyperproliferative, consistent with the ability of MEK1 to stimulate keratinocyte proliferation in the absence of other cell types7 (Fig. 2b).
Serum levels of TNF-α were markedly reduced in tumour-free but not tumour-bearing TNFR−/− chimeras (Supplementary Fig. 2c), consistent with MEK1 activation in epidermal tumour cells driving
NF-κB activation14. The tumour-protective effect of TNFR ablation in radiosensitive leukocytes correlated with changes in the skin immune cell infiltrate. CD4+ T cells were markedly reduced
in wounds and papillomas of TNFR−/− BM chimeras (Fig. 2c,d). When irradiated InvEE mice were reconstituted with BM from mice expressing enhanced green fluorescent protein under the control
of the β-actin cytomegalovirus (CMV) promoter and subsequently wounded, both the wounds and wound-induced tumours were heavily infiltrated with F4/80+ macrophages (Supplementary Fig. 2d).
Macrophage (F4/80+ CD11b+) and mast cell numbers were similar in healed wounds of TNFR−/− and control chimeras but significantly reduced in tumour stroma of TNFR−/− chimeras (Fig. 2e–g).
Epidermal γδ T cells infiltrated wounds of both TNFR−/− and control chimeras to the same extent. They were never present within the tumour epithelium, but did accumulate in adjacent
epidermis (Supplementary Fig. 2e), suggesting that the previously observed reduction in tumours on γδ T-cell ablation is an indirect effect of reduced macrophage recruitment8. TNFR ablation
in the BM did not affect numbers of dendritic cells (CD207+ CD11c+), NK or NKT cells infiltrating wounds or tumours (Supplementary Fig. 2f–h). B cells (CD19+) were not detectable in
unwounded skin or healed wound beds15 and there was no difference in the stromal B-cell content of TNFR−/− and control chimeric tumours (Supplementary Fig. 2i). We conclude that TNFR
ablation in leukocytes protected mice from developing tumours. It also led to a selective reduction in CD4+ T cells in wounded skin and a reduction in several immune cell subsets in tumour
stroma. MYD88 AND TLR-5 SIGNALLING MEDIATE TUMOUR FORMATION MyD88 (Myeloid Differentiation primary response gene 88) is a master regulator of innate signalling events as it is the key
adaptor for most TLRs, IL-1R1 and IL-18R16,17,18. Loss of MyD88 prevents tumour formation in various tissues19,20,21,22. Given that MyD88 controls TNF-α production, we analysed the effect of
reconstituting InvEE mice with MyD88−/− radiosensitive leukocytes. BM chimeras lacking MyD88 in the haematopoietic compartment exhibited a striking protection against wound-induced tumour
formation (Fig. 3a). Although InvEE keratinocytes express elevated levels of IL-1α and administration of the IL-1 receptor antagonist Kineret decreases tumour formation8,9, no differences in
wound-induced tumour formation were observed between IL-1R1−/− BM and control chimeras (Fig. 3b). We therefore examined the effects of deleting TLRs. Replacement of the radiosensitive
haematopoietic compartment with TLR-2/-4−/− or TLR-9−/− cells did not affect tumour formation (Fig. 3c), in contrast to the role of TLR-4 on haematopoietic and non-haematopoietic cells in
chemically induced skin carcinogenesis23. InvEE mice reconstituted with TLR-7/-8−/− BM exhibited accelerated wound closure (Supplementary Fig. 3a) but no difference in tumour incidence was
observed (Fig. 3d). TLR-3 and TLR-4 can signal via TIR-domain-containing adapter-inducing interferon-β (TRIF), instead of MyD88. However, reconstitution with TRIF−/− BM cells had no effect
on papilloma formation (Fig. 3e). Ablation of TLR-5 in radiosensitive leukocytes markedly reduced the number of tumours that developed on wounding (Fig. 3d). Wound closure rates were similar
in TLR-5−/− and control BM chimeras, suggesting that the dynamics of wound closure does not affect wound-induced tumour formation (Fig. 3f). These findings reveal the significance of an
innate MyD88-TLR-5-sensing axis specifically in BM-derived leukocytes that drives wound-induced tumour initiation. BACTERIAL PRODUCTS MEDIATE TUMOUR INITIATION IN INVEE MICE As flagellin
(Fla), the sole known TLR-5 ligand24, is the main protein constituent of bacterial flagella, we analysed whether lowering the microbial content of the skin would affect wound-induced tumour
incidence. When mice were treated with the broad-spectrum antibiotic enrofloxacin (enr), either by administration in drinking water or topical application, the skin bacterial load was
decreased (Supplementary Fig. 3c, d) and wound-induced tumour formation was greatly reduced (Fig. 4a,c). Tumour size in antibiotic-treated mice was greatly reduced (Fig. 4b), an effect that
was previously observed in intestinal tumours25. No reduction in tumour initiation was observed when mice were topically treated with methicillin (met; Fig. 4c), a narrow-spectrum antibiotic
that targets Gram-positive bacteria that are highly abundant on the skin, including the non-flagellated species _Staphylococcus aureus_ and _Staphylococcus epidermidis_. In mice topically
treated with antibiotics, there was no significant reduction in faecal bacterial load, excluding involvement of the gut microbiome in wound-induced skin carcinogenesis (Supplementary Fig.
3e). Broad-spectrum antibiotic treatment resulted in a transient increase in wound closure in WT but not InvEE mice (Supplementary Fig. 3f). In contrast to the tumour-suppressive effects of
TLR-5 ablation and antibiotic treatment, topical application of flagellin to InvEE wounds increased tumour incidence in a dose-dependent manner (Fig. 4d) and delayed wound closure (Fig. 4e
and Supplementary Fig. 3b). Strikingly, intradermal injection of flagellin was sufficient to induce small tumours in InvEE mice in the absence of wounding (Fig. 4f). WT mice never developed
tumours after administration of flagellin to wounds or intradermal injection (data not shown). Flagellin did not induce tumours when injected into TLR-5−/− BM chimeras. When flagellin was
administered to wounds of TLR-5−/− BM chimeras, tumour formation was greatly diminished compared with control chimeras treated with flagellin (Fig. 4g). Flagellated bacterial strains are
commensals on murine skin26. When we labelled unwounded InvEE skin with an antibody to the flagellated _Escherichia coli_ (_E. coli_) strain K12, we observed strong immunoreactivity in hair
follicles, sebaceous glands and cornified skin layers (Fig. 4h), in agreement with a previous report26. As expected, _E. coli_ labelling was markedly reduced in antibiotic-treated skin. All
epidermal layers stained positive for K12 _E. coli_ in InvEE healed wound beds and papillomas (Fig. 4h). Taken together, these data demonstrate that exposure to bacterial flagellin sensed by
TLR-5 on radiosensitive leukocytes promotes tumour formation in InvEE mice. ROLE OF TLR-5 SIGNALLING IN CARCINOGEN-INDUCED TUMOURS To validate our observations in a second experimental
setting, we induced tumours in WT mice via the classic two-stage DMBA/TPA (7,12-dimethylbenz(a)anthracene and 12-_O_-tetradecanoylphorbol-13-acetate) chemical carcinogenesis protocol in
which DMBA induces H-Ras mutations and TPA causes chronic inflammation, promoting tumour development27. Irradiated WT mice were reconstituted with WT (control) or TLR-5−/− BM and topically
treated with DMBA. Mice subsequently received repeated applications of TPA with or without prior wounding (Fig. 5a). Wound closure was accelerated in TLR-5−/− BM chimeras treated with TPA
(Supplementary Fig. 4a). Mice treated with DMBA, but not TPA, and subsequently wounded, did not develop tumours (data not shown). Control chimeras that were wounded before TPA treatment
developed tumours after 2 weeks of promotion, which is significantly faster than mice treated with TPA only (Fig. 5). There was a substantial delay in the development of DMBA/TPA-induced
tumours in wounded TLR-5−/− BM chimeras relative to wounded WT chimeras (Fig. 5b). The tumour-protective effect of TLR-5−/− BM was also apparent in non-wounded DMBA/TPA-treated mice, albeit
less marked (Fig. 5c). TLR-5−/− BM reconstitution did not reduce the final number of papillomas that formed (Supplementary Fig. 4b,c) but decreased tumour size considerably (Fig. 5d,e). We
conclude that TLR-5-mediated signalling is involved in tumour initiation in two different skin cancer models. UPREGULATION OF HMGB1 IN WOUND-INDUCED TUMOURS To investigate the relevance of
mouse wound-induced tumour formation to human skin cancer, we analysed SCCs from RDEB patients. RDEB is a rare skin blistering condition in which repetitive cycles of wounding and repair
predispose the skin to the development of SCCs6. HMGB1 is a nuclear danger-associated molecular pattern that is passively released from necrotic cells and actively secreted by inflammatory
cells28,29. HMGB1 is upregulated in RDEB patients30,31 and HMGB1 serum levels correlate with RDEB disease severity30. HMGB1 is also upregulated in a mouse RDEB model and mediates recruitment
of BM-derived cells in injured tissue31. Furthermore, HMGB1 is induced in epithelial cells upon exposure to flagellin32. We therefore investigated HMGB1 as a candidate biomarker linking
human and mouse wound-associated skin cancer. In lesional skin from RDEB patients, HMGB1 was highly upregulated compared with normal human skin and there was strong immunoreactivity for
HMGB1 in epidermis and dermis (Fig. 6a; _n_=6 patients per group). There was an even greater increase in HMGB1 immunoreactivity in RDEB SCCs (Fig. 6a; _n_=6 patients). Although HMGB1 was
mainly nuclear in normal human skin, we observed cytoplasmic HMGB1 in lesional skin and SCCs from RDEB patients (Fig. 6a), which is indicative of HMGB1 secretion in these inflammatory
conditions29. Consistent with these findings, HMGB1 was elevated in unwounded InvEE skin relative to WT (Fig. 6b,c) and further increased on wounding and in wound-induced papillomas (_n_=8;
Fig. 6b,c). HMGB1 expression was significantly downregulated in skin of unwounded TLR-5−/−/Inv relative to Inv/Inv BM chimeras and the absence of TLR-5 prevented HMGB1 upregulation on
wounding (Fig. 6c). The reduced immunolabelling in skin correlated with a reduction in serum HMGB1 levels in wounded mice (Fig. 6d). We conclude that HMGB1 is upregulated in wound-associated
mouse and human skin tumours and that HMGB1 levels are regulated by leukocytic TLR-5 signalling. DISCUSSION Host–microbe interactions are crucial for tissue homeostasis and evidence showing
cancer-promoting effects of the commensal microbiome in various organs is accumulating33,34,35. Our studies establish, for the first time, a role for innate sensing of flagellated bacteria
in wound-induced skin cancer. We demonstrate that TLR-5 ablation in BM-derived leukocytes reduced tumour incidence in mice and injection of the TLR-5 ligand flagellin induced papillomas in a
TLR-5-dependent manner. Our analysis of RDEB tumours points to the relevance of our findings to human skin cancer. HMGB1, which is induced in epithelial cells in response to flagellin32,
was strongly upregulated in wound-associated tumours in mice and RDEB patients. Ablation of TLR-5 in BM cells led to significantly reduced murine HMGB1 serum levels. The skin microbiome
composition is different across body surfaces36 and directs local immune responses26,37. The human microbiome project reference genome database reveals that intact skin contains flagellated
bacterial species (based on the presence of _FliC_ and _FliA_ genes; http://www.hmpdacc.org/catalog/38). Consistent with this, a flagellated _E. coli_ strain is present on InvEE skin26.
Although most bacteria are kept at bay by the skin barrier, in a wound situation the moist and metabolite-rich environment can promote growth of opportunistic bacteria, including
energetically expensive flagellated species39. Profiling the microbial configurations in chronic diabetic wounds and leg ulcers has indicated an increased representation of _E. coli_,
_Pseuodomonas aeruginosa_, _Shigella_ and other species comprising flagellated strains, whereas Staphylococci, a genus that does not comprise flagellates, are negatively correlated with
ulcer duration40,41. Consistent with the observations in human skin, topical application of a broad-spectrum antibiotic was tumour protective in InvEE mice, whereas an antibiotic that
targets Staphylocci was not. These observations lead us to propose that a key, and previously unrecognized, driver of wound-induced skin cancer is an increased exposure of leukocytes to
flagellated bacteria. Both of the mouse models in which we demonstrated a role for leukocytic TLR-5 sensing in wound-induced skin cancer are characterized by chronic inflammation. In InvEE
mice, the inflammation results from epidermal expression of constitutively active MEK1 and in WT mice inflammation is induced by TPA treatment. TLR-5 ablation in BM does not abolish the
inflammatory infiltrate in InvEE skin or TPA-treated WT skin. Our studies indicate that in the context of chronic inflammation, TLR-5 signalling in leukocytes can tip the balance between
normal wound repair and tumour formation. The tumour-inhibitory effect of ablating TNFR or MyD88 in radiosensitive leukocytes in InvEE mice was greater than that of TLR-5 ablation (Figs 2a
and 3a,d; 0.01<_P_<0.05; one-way analysis of variance), suggesting that additional components of the immune system that converge on NF-κB may contribute (Fig. 1d). Our findings raise
the possibility that the incidence of SCCs in non-healing ulcers and skin from RDEB patients could be reduced by systemic antibiotics treatment and the use of flagellin-specific targeting
strategies in wound-induced malignancies might present an interesting clinical avenue. METHODS MICE InvEE mice9 were maintained on an F1 genetic background (CBA × C57Bl/6) and kept
heterogeneous for the MEK1 transgene. Transgene negative littermates were used as WT controls and in DMBA/TPA experiments. All donor mice in the BM transplantation experiments were on a
C57/Bl6 genetic background. TLR-5−/− and TNFR-1/-2−/− mice were purchased from Jackson Laboratories. TLR-2/-4−/− mice were obtained from Simon Clare, IL-1R1−/− mice from Nancy Rothwell,
TRIF−/− mice from Frederic Geissmann, TLR-7/-8−/− mice from Lena Alexapoulou, MyD88−/− mice from Caetano Reis e Sousa and TLR-9−/− mice from Kinya Otsu. Animal procedures were subject to
local ethical approval and performed under a UK Government Home Office Licence. Sample sizes were determined on the basis of prior power calculations. No mice died as a direct result of
wounding or tumour formation. Mice that died as a result of myeloablation were excluded from analysis. WOUNDING AND BM RECONSTITUTION Full-thickness wounds were made on the back skin by
using 2, 4, 5, 6 or 8 mm2 punch biopsy needles (Stiefel Instruments) under analgesia and general anaesthesia in 8- to 16-week-old age- and sex-matched InvEE and control littermates.
Statistical power was calculated using the resource equation and animals were randomly assigned to treatment groups. No animals were excluded from any experiment. Tumour formation and size
were measured by two independent researchers, who were blinded to group allocations. In BM transplantation experiments8, 8- to 16-week-old female recipient mice were treated with acidified
water at least 10 days before irradiation. Allogenic BM transplants were performed 24 h after myeloablative total body irradiation42 (two times 5 Gy, separated by 3 h) of InvEE mice. Donor
BM was isolated from the tibia and femur of male mice. BM reconstitution was performed by intravenous injection of 5 × 106 BM cells in 200 μl of PBS 24 h after irradiation. Chimerism was
confirmed using Y-chromosome _in situ_ hybridization (Cytocell, probe AMPOYR) on spleens of reconstituted mice8. Mice were wounded 4 to 6 weeks post BM reconstitution and papilloma formation
was monitored for 50 to 60 days post wounding. Each InvEE BM reconstitution experiment was repeated two to three times. TREATMENT WITH ANTIBIOTICS AND FLAGELLIN Mice were treated with the
broad-spectrum antibiotic enrofloxacin by oral administration (5 ml Baytril per liter drinking water) starting 8 days before wounding and continuing until the end of the experiment. For
topical applications, shaved back skin of mice was treated daily with 200 μl of vehicle (acetone) or antibiotics in acetone starting 8 days before wounding. Antibiotics used were 0.25%
enrofloxacin or 0.2 mg ml−1 methicillin. Skin swabs were taken on day 10 post wounding and plated on lysogeny broth (LB) and blood agar plates to verify bacterial depletion. 16S rRNA
quantification was performed on skin biopsies taken at the time of wounding. 1 or 4 μg flagellin (high-purity flagellin, EnzoLifeSciences) was injected once intradermally in back skin in 100
μl PBS; control mice were injected with 100 μl PBS. Flagellin was applied once topically to 8 mm2 full-thickness skin wounds at time of wounding. QUANTITATIVE PCR Epidermis was separated
from the underlying dermis by scraping skin after incubation at 60 °C for 7 s in RNase-free water. Isolated epidermis was homogenized in lysis buffer using ceramic beads. RNA extraction was
performed according to the manufacturer’s instructions using a QIAGEN RNeasy Kit (Qiagen). Complementary DNA was generated using reverse transcriptase III (Invitrogen). Quantitative
real-time PCR reactions were set up using gene-specific primer sets (see Supplementary Table 1) and reactions were performed on a 7900HT real-time PCR machine (Applied Biosystems) on
biological triplicates. MICROBIOTA QUANTIFICATION Bacterial load was quantified in skin biopsy samples (8 mm2) and in faeces collected from mice 8 days after antibiotic treatment. Samples
were digested with Ready-Lyse Lysozyme Solution (Epidentre Biotechnologies) in lysis buffer (20 mM Tris, pH 8.0, 2 mM EDTA, 1.2% Triton X-100, DNA-free water). Samples were homogenized using
5 mm Stainless Steel Beads (Qiagen) in a Precellys homogenizer twice at 6,000 r.p.m. for 50 s. DNA was extracted using a Purelink Genomic DNA kit (Invitrogen). For 16S rRNA quantification,
the following primers were used: 5′-AGAGTTTGATCCTGGCTCAG-3′ and 5′-CTGCTGCCTCCCGTAGGAGT-3′. Data for skin bacterial load were normalized against the relative mouse genomic DNA amplicon
quantification. Primers used were 5′-TTAGCAGTTTGGCACAGCTAGG-3′ and 5′-CTAGGTTGGCAAGGAATTGTGG-3′. ELISA Blood was collected by cardiac puncture and left to clot for 2 h. Serum was collected
after centrifugation for 20 min at 4 °C. TNF-α, IL-6 and TSLP levels were quantified using Quantikine ELISA kits (R&D systems). HMGB1 serum levels were measured by ELISA (Shino-test
Corporation). DMBA/TPA TWO-STAGE CARCINOGENESIS Chemical carcinogenesis experiments were performed as previously described27. In brief, back skin of mice was shaved and treated once with 100
nmol (25 μg) DMBA in 200 μl acetone 5 weeks after BM reconstitution. Seven days later, mice were either wounded with an 8 mm2 full-thickness skin biopsy punch or treated with 6 nmol (3.7
μg) TPA. After 2 days, all mice were treated three times weekly with TPA for 15 weeks. Tumour incidence and burden were assessed once a week by two independent researchers who were blinded
to the group allocations. IMMUNOFLUORESCENCE STAINING Frozen sections were fixed in acetone/methanol (1:1) or 4% paraformaldehyde/PBS pH 7.4 and blocked with 2% BSA, 0.02% fish skin gelatin
and 10% goat serum for 1 h in PBS at room temperature. Paraffin sections were subjected to heat-mediated antigen retrieval (citrate buffer, pH6). When fluorochrome-conjugated primary
antibodies were used, sections were incubated overnight at 4 °C in antibody solution containing 4',6-diamidino-2-phenylindole, washed in PBS and then mounted using the DAKO mounting
reagent (DAKO). In the case of the unconjugated primary antibodies (HMGB1, Abcam, ab79823; anti-_Escherichia coli_ antibody, DAKO, B0357), incubation was overnight at 4 °C followed by 2 h
incubation at room temperature in secondary antibody (AlexaFluor 555 goat-anti-rabbit) containing 4,6-diamidino-2-phenylindole. Alexafluor 647-, 660- or 488-conjugated antibodies to CD3
(BioLegend, clone 17.A2), CD4 (eBioscience, clone RM4-5), F4/80 (eBioscience, clone BM8), CD19 (eBioscience, eFluor 660), CD11b (eBioscience, clone M1/70), CD11c (Cambridge Bioscience), γδ
TCR (BD Pharmingen, clone GL3), NK1.1 (BioLegend, clone PK136) and CD207 (eBioscience, eFluor 660) were used. IMMUNOHISTOCHEMISTRY Dewaxed paraffin sections were subjected to heat-mediated
antigen retrieval (citrate buffer, pH6). Mast cells were quantified by counting toluidine blue-stained cells on paraffin skin sections. CD45 (BD Pharmingen, clone 30-F11) antibody was used
on paraffin sections at 1:100 dilution. Quantification of immune cell infiltration was performed within the wound and adjacent to the wound (maximum distance from wound/pap: 4 mm) or in
tumour stroma. HUMAN SAMPLES All human samples were collected after informed, written consent and in accordance with Helsinki guidelines, in compliance with the UK Human Tissue Act. The
study protocol was approved by the Universities of Monterrey and New South Wales. STATISTICS Statistical analyses were performed using GraphPad Prism v6. Specific tests included Fisher’s
exact tests, one or two-way analysis of variance, unpaired _t_-tests and Mann–Whitney tests. *0.01<_P_<0.05, **0.01<_P_<0.001, ***0.0001<_P_<0.001, ****_P_<0.0001.
ADDITIONAL INFORMATION HOW TO CITE THIS ARTICLE: Hoste, E. _et al._ Innate sensing of microbial products promotes wound-induced skin cancer. _Nat. Commun._ 6:5932 doi: 10.1038/ncomms6932
(2015). REFERENCES * Arwert, E. N., Hoste, E. & Watt, F. M. Epithelial stem cells, wound healing and cancer. _Nat. Rev. Cancer_ 12, 170–180 (2012). Article CAS Google Scholar *
Dunham, L. J. Cancer in man at site of prior benign lesion of skin or mucous membrane: a review. _Cancer Res._ 32, 1359–1374 (1972). CAS PubMed Google Scholar * Fleming, M. D., Hunt, J.
L., Purdue, G. F. & Sandstad, J. Marjolin’s ulcer: a review and reevaluation of a difficult problem. _J. Burn Care Rehabil._ 11, 460–469 (1990). Article CAS Google Scholar * Seifert,
O. & Mrowietz, U. Keloid scarring: bench and bedside. _Arch. Dermatol. Res._ 301, 259–272 (2009). Article Google Scholar * Margolis, D. J., Bilker, W., Santanna, J. & Baumgarten,
M. Venous leg ulcer: incidence and prevalence in the elderly. _J. Am. Acad. Dermatol._ 46, 381–386 (2002). Article Google Scholar * Fine, J. D., Johnson, L. B., Weiner, M., Li, K. P. &
Suchindran, C. Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986-2006. _J. Am. Acad. Dermatol._ 60, 203–211 (2009). Article Google
Scholar * Haase, I., Hobbs, R. M., Romero, M. R., Broad, S. & Watt, F. M. A role for mitogen-activated protein kinase activation by integrins in the pathogenesis of psoriasis. _J. Clin.
Invest._ 108, 527–536 (2001). Article CAS Google Scholar * Arwert, E. N. et al. Tumor formation initiated by nondividing epidermal cells via an inflammatory infiltrate. _Proc. Natl Acad.
Sci. USA_ 107, 19903–19908 (2010). Article ADS CAS Google Scholar * Hobbs, R. M., Silva-Vargas, V., Groves, R. & Watt, F. M. Expression of activated MEK1 in differentiating
epidermal cells is sufficient to generate hyperproliferative and inflammatory skin lesions. _J. Invest. Dermatol._ 123, 503–515 (2004). Article CAS Google Scholar * Li, N., Grivennikov,
S. I. & Karin, M. The unholy trinity: inflammation, cytokines, and STAT3 shape the cancer microenvironment. _Cancer cell_ 19, 429–431 (2011). Article CAS Google Scholar * Balkwill, F.
Tumour necrosis factor and cancer. _Nat. Rev. Cancer_ 9, 361–371 (2009). Article CAS Google Scholar * Arnott, C. H. et al. Expression of both TNF-alpha receptor subtypes is essential for
optimal skin tumour development. _Oncogene_ 23, 1902–1910 (2004). Article CAS Google Scholar * Mori, R., Kondo, T., Ohshima, T., Ishida, Y. & Mukaida, N. Accelerated wound healing in
tumor necrosis factor receptor p55-deficient mice with reduced leukocyte infiltration. _FASEB J._ 16, 963–974 (2002). Article CAS Google Scholar * Hobbs, R. M. & Watt, F. M.
Regulation of interleukin-1alpha expression by integrins and epidermal growth factor receptor in keratinocytes from a mouse model of inflammatory skin disease. _J. Biol. Chem._ 278,
19798–19807 (2003). Article CAS Google Scholar * Andreu, P. et al. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. _Cancer cell_ 17, 121–134 (2010). Article
CAS Google Scholar * Kawai, T., Adachi, O., Ogawa, T., Takeda, K. & Akira, S. Unresponsiveness of MyD88-deficient mice to endotoxin. _Immunity_ 11, 115–122 (1999). Article CAS
Google Scholar * Medzhitov, R. et al. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. _Mol. Cell_ 2, 253–258 (1998). Article CAS Google Scholar * Muzio,
M., Ni, J., Feng, P. & Dixit, V. M. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. _Science_ 278, 1612–1615 (1997). Article ADS CAS Google
Scholar * Rakoff-Nahoum, S. & Medzhitov, R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. _Science_ 317, 124–127 (2007). Article ADS CAS
Google Scholar * Naugler, W. E. et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. _Science_ 317, 121–124 (2007). Article ADS CAS Google
Scholar * Ochi, A. et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. _J. Exp. Med._ 209, 1671–1687 (2012). Article CAS Google
Scholar * Cataisson, C. et al. IL-1R-MyD88 signaling in keratinocyte transformation and carcinogenesis. _J. Exp. Med._ 209, 1689–1702 (2012). Article CAS Google Scholar * Mittal, D. et
al. TLR4-mediated skin carcinogenesis is dependent on immune and radioresistant cells. _EMBO J._ 29, 2242–2252 (2010). Article CAS Google Scholar * Hayashi, F. et al. The innate immune
response to bacterial flagellin is mediated by Toll-like receptor 5. _Nature_ 410, 1099–1103 (2001). Article ADS CAS Google Scholar * Grivennikov, S. I. et al. Adenoma-linked barrier
defects and microbial products drive IL-23/IL-17-mediated tumour growth. _Nature_ 491, 254–258 (2012). Article ADS CAS Google Scholar * Naik, S. et al. Compartmentalized control of skin
immunity by resident commensals. _Science_ 337, 1115–1119 (2012). Article ADS CAS Google Scholar * Abel, E. L., Angel, J. M., Kiguchi, K. & DiGiovanni, J. Multi-stage chemical
carcinogenesis in mouse skin: fundamentals and applications. _Nat. Protoc._ 4, 1350–1362 (2009). Article CAS Google Scholar * Scaffidi, P., Misteli, T. & Bianchi, M. E. Release of
chromatin protein HMGB1 by necrotic cells triggers inflammation. _Nature_ 418, 191–195 (2002). Article ADS CAS Google Scholar * Bonaldi, T. et al. Monocytic cells hyperacetylate
chromatin protein HMGB1 to redirect it towards secretion. _EMBO J._ 22, 5551–5560 (2003). Article CAS Google Scholar * Petrof, G. et al. Serum levels of high mobility group box 1
correlate with disease severity in recessive dystrophic epidermolysis bullosa. _Exp. Dermatol._ 22, 433–435 (2013). Article CAS Google Scholar * Tamai, K. et al. PDGFRalpha-positive cells
in bone marrow are mobilized by high mobility group box 1 (HMGB1) to regenerate injured epithelia. _Proc. Natl Acad. Sci. USA_ 108, 6609–6614 (2011). Article ADS CAS Google Scholar *
Liu, S. et al. HMGB1 is secreted by immunostimulated enterocytes and contributes to cytomix-induced hyperpermeability of Caco-2 monolayers. _Am. J. Physiol. Cell Physiol._ 290, C990–C999
(2006). Article ADS CAS Google Scholar * Schwabe, R. F. & Jobin, C. The microbiome and cancer. _Nat. Rev. Cancer_ 13, 800–812 (2013). Article CAS Google Scholar * Gagliani, N.,
Hu, B., Huber, S., Elinav, E. & Flavell, R. A. The Fire Within: Microbes Inflame Tumors. _Cell_ 157, 776–783 (2014). Article CAS Google Scholar * Belkaid, Y. & Hand, T. W. Role of
the microbiota in immunity and inflammation. _Cell_ 157, 121–141 (2014). Article CAS Google Scholar * Grice, E. A. et al. Topographical and temporal diversity of the human skin
microbiome. _Science_ 324, 1190–1192 (2009). Article ADS CAS Google Scholar * Shen, W. et al. Adaptive immunity to murine skin commensals. _Proc. Natl Acad. Sci. USA_ 111, E2977–E2986
(2014). Article CAS Google Scholar * The Human Microbiome Project consortium. Structure, function and diversity of the healthy human microbiome. _Nature_ 486, 207–214 (2012). * Grice, E.
A. & Segre, J. A. Interaction of the microbiome with the innate immune response in chronic wounds. _Adv. Exp. Med. Biol._ 946, 55–68 (2012). Article CAS Google Scholar * Gontcharova,
V., Youn, E., Sun, Y., Wolcott, R. D. & Dowd, S. E. A comparison of bacterial composition in diabetic ulcers and contralateral intact skin. _Open Microbiol. J._ 4, 8–19 (2010). Article
Google Scholar * Gardner, S. E., Hillis, S. L., Heilmann, K., Segre, J. A. & Grice, E. A. The neuropathic diabetic foot ulcer microbiome is associated with clinical factors. _Diabetes_
62, 923–930 (2013). Article CAS Google Scholar * Lanzkron, S. M., Collector, M. I. & Sharkis, S. J. Hematopoietic stem cell tracking in vivo: a comparison of short-term and long-term
repopulating cells. _Blood_ 93, 1916–1921 (1999). CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We thank Watt lab members for input. We thank Ludmila Sabolcikova, Matthew
Clayton, Jennifer Arthur, Claire Pearce, Ellie and Mark Pryor and the CRUK Cambridge Research Institute Histopathology Lab for technical assistance. We thank Lena Alexopoulou, Caetano Reis
e Sousa, Simon Clare, Frederic Geissmann, Kinya Otsu and Nancy Rothwell for supplying mutant mice or bone marrow. We acknowledge financial support from the MRC, Wellcome Trust, Cancer
Research UK and European Union (HEALING EU-FP7). The research was funded/supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St
Thomas' NHS Foundation Trust and King's College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Cancer Research UK Cambridge Research institute, Li Ka Shing Centre, Robinson Way, Cambridge, CB2 0RE, UK Esther Hoste, Esther N. Arwert &
Giacomo Donati * Centre for Stem Cells and Regenerative Medicine, King’s College London, 28th Floor, Tower Wing, Guy’s Campus, London, SE1 9RT, UK Esther Hoste, Giacomo Donati & Fiona M.
Watt * Cancer Clinical Academic Group, Guy's and St Thomas' NHS Trust, Bermondsey Wing, Guy's Hospital, Great Maze Pond, London, SE1 9RT, UK Rohit Lal * Division of Cancer
Research, Ninewells Hospital and Medical School, University of Dundee, Dundee DD1 9SY, UK Andrew P. South * Department of Dermatology and Cutaneous Biology, Thomas Jefferson University,
Philadelphia, Pennsylvania 19107, USA Andrew P. South * Basic Science Department, Medicine School, University of Monterrey, Mexico, Nuevo Leon 64849 Julio C. Salas-Alanis * Department of
Dermatology, St George Hospital, University of New South Wales, Sydney, New South Wales 2217, Australia, Dedee F. Murrell Authors * Esther Hoste View author publications You can also search
for this author inPubMed Google Scholar * Esther N. Arwert View author publications You can also search for this author inPubMed Google Scholar * Rohit Lal View author publications You can
also search for this author inPubMed Google Scholar * Andrew P. South View author publications You can also search for this author inPubMed Google Scholar * Julio C. Salas-Alanis View author
publications You can also search for this author inPubMed Google Scholar * Dedee F. Murrell View author publications You can also search for this author inPubMed Google Scholar * Giacomo
Donati View author publications You can also search for this author inPubMed Google Scholar * Fiona M. Watt View author publications You can also search for this author inPubMed Google
Scholar CONTRIBUTIONS E.H. conception and design, acquisition of data, analysis and interpretation of data, drafting and revising the article; E.N.A., R.L., G.D. acquisition of data,
analysis of data; A.P.S., J.C.S.-A., D.F.M. acquisition of data; F.M.W. conception and design, analysis and interpretation of data, drafting and revising the article. CORRESPONDING AUTHOR
Correspondence to Fiona M. Watt. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing financial interests. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION
Supplementary Figures 1-3 and Supplementary Table 1 (PDF 1136 kb) RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0 International License. The images or
other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the
Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Hoste, E., Arwert, E., Lal, R. _et al._ Innate sensing of microbial products
promotes wound-induced skin cancer. _Nat Commun_ 6, 5932 (2015). https://doi.org/10.1038/ncomms6932 Download citation * Received: 28 October 2014 * Accepted: 21 November 2014 * Published: 09
January 2015 * DOI: https://doi.org/10.1038/ncomms6932 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable
link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative