- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Activation of the NLRP3 inflammasome enables monocytes and macrophages to release high levels of interleukin-1β during inflammatory responses. Concentrations of extracellular
calcium can increase at sites of infection, inflammation or cell activation. Here we show that increased extracellular calcium activates the NLRP3 inflammasome via stimulation of G
protein-coupled calcium sensing receptors. Activation is mediated by signalling through the calcium-sensing receptor and GPRC6A via the phosphatidyl inositol/Ca2+ pathway. The resulting
increase in the intracellular calcium concentration triggers inflammasome assembly and Caspase-1 activation. We identified necrotic cells as one source for excess extracellular calcium
triggering this activation. _In vivo_, increased calcium concentrations can amplify the inflammatory response in the mouse model of carrageenan-induced footpad swelling, and this effect was
inhibited in GPRC6A−/− mice. Our results demonstrate that G-protein-coupled receptors can activate the inflammasome, and indicate that increased extracellular calcium has a role as a danger
signal and amplifier of inflammation. SIMILAR CONTENT BEING VIEWED BY OTHERS PYROPTOTIC CELL CORPSES ARE CROWNED WITH F-ACTIN-RICH FILOPODIA THAT ENGAGE CLEC9A SIGNALING IN INCOMING
DENDRITIC CELLS Article Open access 04 December 2024 CALCIUM-SENSING RECEPTOR-MEDIATED NLRP3 INFLAMMASOME RESPONSE TO CALCIPROTEIN PARTICLES DRIVES INFLAMMATION IN RHEUMATOID ARTHRITIS
Article Open access 25 August 2020 INDIRECT REGULATION OF HMGB1 RELEASE BY GASDERMIN D Article Open access 11 September 2020 INTRODUCTION The multi-protein complex of the NLRP3 inflammasome
is assembled in response to danger-associated stimuli during infectious1 or sterile2 inflammatory events, but the involved proximal signalling pathways are only partially characterized.
Extracellular calcium concentrations (ex[Ca2+]) are tightly regulated in the extracellular interstitial fluid and presumed to range from 0.9 to 1.2 mM3,4. The ex[Ca2+] concentrations are
known to be increased at sites of infections5,6 and in the interstitial fluid around activated cells7, but the resulting cellular consequences are not known. The presence of calcifications
at sites of chronic inflammation or ischaemic necrosis8,9 suggests, however, that ex[Ca2+] can increase not only in infectious but also in sterile inflammation. We hypothesized, therefore,
that increased ex[Ca2+] conveys a danger signal to surrounding cells and triggers inflammasome activation. Here we show that increased extracellular calcium concentrations result in an
amplification of inflammatory responses, which can be inhibited by antagonists of calcium-sensing receptor (CaSR), and which is significantly reduced in monocytes from GPRC6A−/− mice.
RESULTS EX[CA2+] INDUCES IL-1Β THROUGH THE NLRP3 INFLAMMASOME To test the hypothesis that extracellular calcium triggers inflammasome activation, monocytes were stimulated _in vitro_ with
increasing ex[Ca2+]. To raise the concentration of free calcium ions in serum-supplemented tissue culture media, CaCl2 had to be added in excess (for titration, see Supplementary Table 1).
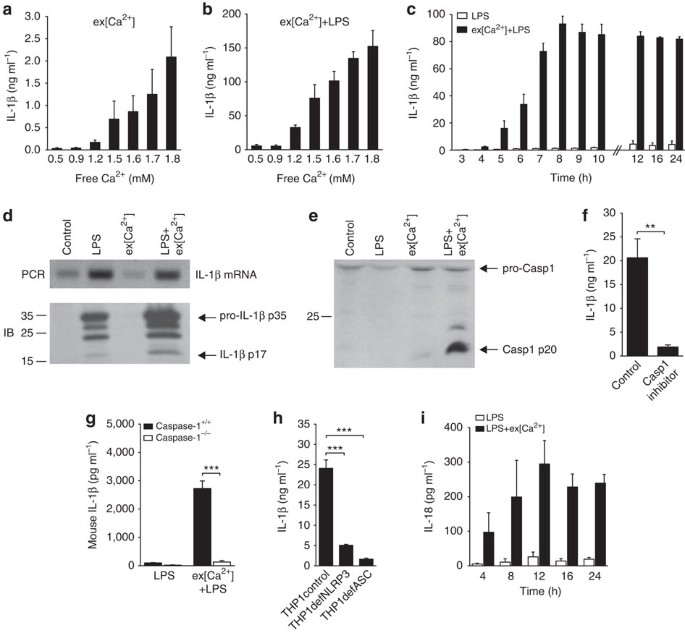
After 16 h, a concentration-dependent increase in ex[Ca2+]-induced IL-1β secretion was detected (Fig. 1a). For inflammasome-dependent IL-1β release, an initial signal via toll-like receptor
(TLR) stimulation is required to increase pro-IL-1β synthesis2. Accordingly, lipopolysaccharide (LPS) priming of monocytes prior to stimulation with ex[Ca2+] lead to a striking increase in
IL-1β production (Fig. 1b; for other TLR ligands, see Supplementary Fig. S1). IL-1β secretion started 4 h after stimulation with ex[Ca2+], peaked after 8 h and remained stable thereafter
(Fig. 1c), with no discernible influence of the duration of LPS priming or the ex[Ca2+] concentration during pre-incubation (Supplementary Fig. S2a,b). Increased ex[Ca2+] had no influence on
IL-1β mRNA expression, but was found to trigger proteolytic cleavage of pro-IL-1β protein into IL-1β p17 (Fig. 1d). Accordingly, activated Caspase-1 was detectable by western blot following
stimulation with ex[Ca2+] (Fig. 1e), whereas the Caspase-1 inhibitor Z-WEHD-FMK abrogated ex[Ca2+]-induced release of IL-1β (Fig. 1f). Most importantly, monocytic cells from Caspase-1−/−
mice did not produce IL-1β in response to increased ex[Ca2+], whereas wild-type (wt) Caspase-1+/+ mice (Fig. 1g) and IL-1R−/− mice (Supplementary Fig. S3) did. Evidence for inhibition of
ex[Ca2+]-induced IL-1β secretion by geldanamycin and high extracellular K+ concentration (Supplementary Fig. S4) suggested involvement of the inflammasome. This was confirmed in experiments
with THP-1 cells deficient for the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC), which were found not to respond to increased ex[Ca2+] (Fig. 1h).
Similarly, NLRP3-deficient THP-1 cells also showed a greatly reduced response (Fig. 1h), indicating that this inflammasome is required for the monocytic response to increased ex[Ca2+]. The
expression of NLRP3 mRNA in human monocytes was not influenced by stimulation with LPS+ex[Ca2+] (Supplementary Fig. S5). The inflammasome-dependent cytokine IL-1810 was found to be secreted
with similar kinetics as IL-1β (Fig. 1i). EX[CA2+] ACTIVATES THE INFLAMMASOME VIA CASR AND GPRC6A Cells sense ex[Ca2+] through two G-protein-coupled receptors (GPCRs), CaSR11 and
GPRC6A12,13. Both receptors are expressed in human monocytes as confirmed by western blot (Fig. 2a) and quantitative reverse transcriptase PCR (qRT–PCR) (data not shown). Two selective
inhibitors of CaSR and GPRC6A, Calhex231 and NPS2143 (ref. 14) were found to significantly decrease ex[Ca2+]-induced IL-1β production (Fig. 2b), but had no influence on adenosine
triphosphate (ATP)-induced inflammasome activation (Supplementary Fig. S6). In addition, Calhex231 inhibited the ex[Ca2+]+LPS-induced processing of active Caspase-1 (Fig. 2c). To obtain
definite proof for the involvement of calcium-sensing GPCRs in mediating ex[Ca2+]-induced inflammasome activation, experiments with GPRC6A−/− mice15 were performed. IL-1β secretion of
monocytic cells from GPRC6A−/− mice in response to ex[Ca2+] (Fig. 2d) was significantly reduced compared with those from wt C57BL/6 mice. _In vitro_ differentiated THP-1 cells express both
receptors and respond strongly to increased ex[Ca2+] (Supplementary Fig. S7). Transfecting those cells with GPRC6A- or CaSR-specific small interfering RNA (siRNA) resulted in a knockdown of
the receptors on the protein level, and reduced significantly the ex[Ca2+]-induced IL-1β production (Fig. 2e). Simultaneous knockdown of both receptors inhibited IL-1β secretion by >70%
(Fig. 2e), indicating that both receptors contribute equally towards ex[Ca2+]-induced IL-1β production. Two alternative agonists of those receptors, Gd3+ and Al3+, also stimulated monocytic
IL-1β release _in vitro_ (Fig. 2f and Supplementary Fig. S8a). The response to both ligands was reduced in GPRC6A−/− mice (Fig. 2g and Supplementary Fig. S8b), and was strictly inflammasome
dependent, because IL-1β production was abrogated in both ASC- and NLRP3-deficient THP-1 cells (Fig. 2h and Supplementary Fig. S8c). CaSR and GPRC6A are coupled to Gαi and Gαq
proteins12,13,16, implicating the cyclic adenosine monophosphate (cAMP) and phosphatidyl–inositol/Ca2+ signalling pathway in ex[Ca2+]-induced inflammasome activation. The
inflammasome-stimulating effect of Gαi/Gαq-coupled GPCR signalling was confirmed with two other ligands binding to Gαi/Gαq-coupled receptors, platelet-activating factor and thrombin, which
induced a concentration-dependent increase in IL-1β release in LPS-primed monocytes after 16 h, whereas C5a, Rantes and fMLP (all signalling only via Gαi-protein-coupled receptors) did not
(Fig. 2i). To test whether Gαq signalling via the phosphatidyl–inositol/Ca2+ pathway contributes to the inflammasome activation, U-73122, an inhibitor of the phospholipase C (PLC), was added
to the cultures and found to effectively abrogate ex[Ca2+]-induced IL-1β release (Fig. 2j). Accordingly, PLC activity in monocytes stimulated with ex[Ca2+] was increased, indicating
activation of this pathway (Fig. 2k). Measurement of cytosolic calcium showed an increase in [Ca2+]i concentration, which occurred immediately after stimulation with ex[Ca2+], and inhibition
of calcium-sensing GPCRs by NPS2143 diminished the increase in [Ca2+]i (Fig. 2l). Elevating [Ca2+]i using thapsigargin triggered IL-1β release without concomitant increase in IL-1β mRNA
expression (Supplementary Fig. S9). Pre-incubation with the cell-permeable Ca2+-chelator BAPTA-AM was found to dose-dependently inhibit inflammasome activation by ex[Ca2+] (Fig. 2m), thereby
corroborating a crucial role of intracellular calcium accumulation, which has also been shown to be required for other inflammasome stimuli17,18. cAMP induction by direct stimulation of the
adenylyl cyclases with forskolin did not induce IL-1β release, the adenylyl cyclase inhibitor SQ22536 had no effect, and no influence of stimulation with increased ex[Ca2+] on intracellular
cAMP levels was observed (Fig. 3a–c), indicating that Gαi signalling without concomitant Gαq has no role in inflammasome activation. A known stimulator of the NLRP3 inflammasome is
extracellular ATP, which acts as a danger signal when released from dying cells19. To exclude autocrine stimulation with ATP via the P2X7 receptor in ex[Ca2+]-induced IL-1β release, we
measured ATP levels, added pharmacological P2X7 inhibitors and depleted extracellular ATP using exogenous apyrase. No evidence for autocrine ATP effects was seen (Fig. 3d–f). Finally, the
ex[Ca2+]-induced IL-1β production of monocytic cells from P2X7−/− mice did not differ from wt mice (Fig. 3g), which all but excludes an influence of endogenously released ATP. Secretory
lysosome exocytosis is also not involved in Ca2+-induced augmentation of IL-1β release, because no concomitant increase in cathepsin B release was detected (Supplementary Fig. S10).
Substantial cell death was also not observed in the monocytes cultures, since the lactate dehydrogenase release was increased only minimally (1.5-fold) in relation to untreated monocytes.
EX[CA2+] INDUCES IL-1Α AND OTHER PROINFLAMMATORY CYTOKINES Intracellular calcium accumulation is involved in multiple signalling pathways and can result in diverse cellular outcomes, which
prompted us to investigate secretion of other cytokines following stimulation with ex[Ca2+]. Recently, IL-1α has been described to be induced by inflammasome activators17. Monocytes
stimulated with ex[Ca2+] secreted high concentrations of IL-1α (Fig. 4a). Induction of IL-1α was also mediated via CaSR and GPRC6A (Fig. 4b), and was absent in THP-1 cells deficient for ASC
or NLRP3 (Fig. 4d). In addition to IL-1β and IL-1α, monocytes stimulated with ex[Ca2+] were found to secrete IL-6 and TNF (Supplementary Fig. S11). Both cytokines were induced via GPRC6A and
CaSR signalling, because secretion was inhibited in GPRC6A−/− mice (Fig. 4e) and by Calhex231 and NPS2143 (Fig. 4f and Supplementary Fig. S11), and was dependent on BAPTA-AM-sensitive
intracellular calcium accumulation (Supplementary Fig. S11). The lipid mediator prostaglandin E2, which has been shown to be induced in macrophages following stimulation with silica
crystals20, was also detectable in ex[Ca2+]-stimulated monocytes (Fig. 4g). NECROTIC CELLS RELEASE CA2+ AND ACTIVATE THE INFLAMMASOME Necrotic cells are a potential source of excess
extracellular calcium. When necrosis was induced in monocytes with 7-BIO, increased extracellular calcium concentrations were detectable in the supernatant (Fig. 4h). Accordingly,
co-incubation of monocytes with necrotic cells was found to result in inflammasome activation, which could be inhibited by Calhex231 (Fig. 4i). Importantly, inflammasome activation by
necrotic cells was significantly inhibited in monocytes from GPRC6A−/− mice (Fig. 4j). EX[CA2+] INDUCES INFLAMMASOME ACTIVATION _IN VIVO_ To test the _in vivo_ relevance of increased
ex[Ca2+], the well-established model of carrageenan-induced footpad swelling was used. In C57BL/6 mice, co-injection of Ca2+ together with carrageenan resulted in significantly greater
footpad swelling compared with carrageenan alone after 1.5 and 4 h (Fig. 5a). Similarly, co-injection of Al3+ or Gd3+ significantly increased carrageenan-induced footpad swelling (Fig.
5b–d). The additional inflammatory effect of co-injected Ca2+ or Al3+ was dependent on inflammasome-mediated IL-1β release, because it was reduced both in Caspase-1−/− and in IL-1R−/− mice
(Fig. 5e–g). Final proof for the involvement of GPRC6A in this local inflammatory response was obtained by investigating the model in GPRC6A−/− mice. The results show that both the Ca2+- and
the Al3+-triggered increase in carrageenan-induced footpad swelling was inhibited in the absence of GPRC6A (Fig. 5h). Determination of the cytokine content of the injected paws revealed a
significant increase in IL-1β in response to Al3+, which is absent in paws from GPRC6A−/− mice (Fig. 5j). DISCUSSION In summary, we provide evidence that increased ex[Ca2+] can activate the
NLRP3 inflammasome and Caspase-1, thereby inducing high levels of bioactive IL-1β. Changes in ex[Ca2+] are sensed by monocytes through CaSR signalling7,11 or - as shown here - through GPRC6A
signalling, and activate the phosphatidyl–inositol/Ca2+ pathway, which in turn leads to inflammasome activation. Of note, this is the first report linking GPCR signalling to inflammasome
activation. _In vivo_, ex[Ca2+] has been reported to be increased at sites of infections5 and ischaemic necrosis8,9. We propose, therefore, that increased ex[Ca2+] might act as
danger-associated molecular pattern (DAMP) by inducing inflammasome activation. In addition, a rise in ex[Ca2+] can result from extrusion to the extracellular fluid during cytosolic calcium
signalling21,22, and the resulting calcium increase in the confined interstitial space can transmit intercellular signalling to neighbouring cells via CaSR7 or GPRC6A12,13. Our experiments
show that release of Ca2+ from necrotic cells can also trigger inflammasome activation in neighbouring cells, and that GPRC6A contributes to the transmission of this proinflammatory signal.
Consequently, increased ex[Ca2+] could provide a positive feedback loop at sites of inflammation, thereby amplifying secretion of IL-1β and aggravating tissue damage. Consequences of
excessive levels of bioactive IL-1β are seen in hereditary and autoimmune diseases, in which IL-1β blockade is clinically effective23. We propose that stimulation of monocytes by increased
ex[Ca2+] is likely to be of clinical relevance in those and other inflammasome-dependent inflammatory conditions, including atherosclerosis24 and type 2 diabetes25,26. While this manuscript
was undergoing final review, a related paper was published describing activation of the NLRP3 inflammasome by increased intracellular Ca2+ via CaSR and decreased cellular cyclic AMP27. The
study corroborates our finding of an ex[Ca2+]-induced increase in intracellular Ca2+, triggering inflammasome activation. In contrast to this study, however, no significant influence of cAMP
levels on inflammasone activation was detectable in our system. Most importantly, however, we identified GPRC6A as a second important calcium-sensitive receptor signalling towards
inflammasome activation, and also showed the _in vivo_ relevance of this system in an animal model. METHODS ANTIBODIES (ABS) AND REAGENTS Rabbit-polyclonal anti-Caspase-1 Ab was purchased
from Cell Signaling Technology. BAY 61-3606, Calhex231, NPS2143, rabbit-polyclonal anti-IL-1β, rabbit-polyclonal anti-CaSR and anti-GPRC6A Abs and peroxidase-conjugated goat-anti-rabbit
secondary Ab were obtained from Santa Cruz Biotechnology. LDH release assay, SQ22536, carrageenan, ATP, thrombin and apyrase were purchased from Sigma-Aldrich. Colchicine, U-73122, fMLP,
4-aminopyridine and forskolin were purchased from Merck. PAF, A-438079 and AZ-10606120 were from Tocris Bioscience. Thapsigargin was from MBL, Tenascin-C from Millipore and Z-WEHD-FMK, C5a
and Rantes from R&D Systems. The water-soluble geldanamycin analogue 17-DMAG was purchased from InvivoGen. HUMAN SUBJECTS Control subjects were recruited among healthy blood donors. The
local ethics committee approved all experiments with human materials, and informed consent was obtained from all subjects. ANIMAL EXPERIMENTS Homozygous genetically deficient
B6.129P2-P2rx7tm1Gab/J mice, P2X7−/− KO mice28, B6.129S7-Il1r1tm1Imx/J, NOD.129S2(B6)-Casp1tm1Sesh/LtJ and age-matched wt C57BL/6 and NOD/ShiLtJ controls were purchased from The Jackson
Laboratory. GPRC6A−/− mice on a C57BL/6 background were described previously15, were used here for _in vitro_ monocyte experiments and _in vivo_ carrageenan-induced footpad swelling and were
compared with C57BL/6 wt littermates. Wt or GPRC6A−/− C57BL/6 mice received a subplantar injection of 20 μl of carrageenan (1% w/v) in saline29. In some experiments, CaCl2 (50 mM),
Al-lactate (0.3 mg), GdCl3 (10 mM) or Calhex231 (100 μM) was co-injected. Footpad thickness was assessed by caliper measurement (DigiPlus; Vogel). Measurements were taken immediately before
administration of carrageenan and at the indicated time points. The assessment of paw swelling was performed in a double-blind fashion and by the same operator. The increase in paw swelling
was calculated by subtracting the initial paw thickness from the paw thickness measured at each time point. For some experiments, mice were euthanized with CO2 and their hind feet were
removed. Paws were put in 250 μl phosphate-buffered saline supplemented with 10% fetal calf serum (FCS) and centrifuged at 1,000 _g_ for 15 min30. Mouse IL-1β levels in paw exudates were
measured using enzyme-linked immunosorbent assay. Mice were bred and maintained at the animal facilities at the Medizinisch Experimentelles Zentrum, University of Leipzig, Germany. All
experiments were approved by and performed according to the institutional guidelines of the animal ethics committee at the University of Leipzig. CELL ISOLATION Human monocytes were isolated
as described previously31. Bone marrow was obtained by aspirating the femurs of the mice. CD11b+ mononuclear cells were isolated from bone marrow and peripheral blood by positive magnetic
separation (Miltenyi Biotech). Peritoneal macrophages were recovered by peritoneal lavage with 5 ml of cold phosphate-buffered saline and used without further separation. CELL CULTURE AND
CALCIUM TITRATION Monocytes (3 × 105 per 200 μl) and THP-1 cells were incubated in RPMI1640 supplemented with 10% heat-inactivated FCS, 2 mM L-glutamine, 100 U ml−1 penicillin and 100 μg
ml−1 streptomycin. NLRP3-deficient, ASC-deficient and control THP-1 cells were purchased from InvivoGen. LPS was used at a concentration of 100 ng ml−1. In the indicated experiments, other
TLR ligands (human TLR1-9 Agonist Kit; InvivoGen) were used at following concentrations: Pam3CSK4 (1 μg ml−1), heat-killed preparation of _Listeria monocytogenes_ (108 cells ml−1), Poly(I:C)
(10 μg ml−1), flagellin (1 μg ml−1), FSL1 (1 μg ml−1), Imiquimod (1 μg ml−1), ssRNA40 (1 μg ml−1) and ODN2006 (5 μM). To incubate monocytes in media containing increasing ionized calcium
concentration, CaCl2 was added to the cultures. Final Ca2+ concentrations in the media were measured after anaerobic sampling (sealed blood gas syringes were completely filled under an CO2
atmosphere and sealed with caps to avoid loss of CO2) with ABL 730 (Radiometer) by the potentiometric measuring principle, which uses an ion-selective Ca electrode (E733). The amount of
CaCl2 added into the culture medium and the resulting ionized calcium concentration are indicated in Supplementary Table 1. A final ionized calcium concentration of 1.7 mM was used in the
experiments, which was achieved by adding 2.5 mM CaCl2 to the culture medium. From a concentration of 1.9 mM Ca2+ in the media, crystals started to precipitate. The IL-1β response using
these high Ca2+ concentrations was not further increased (Supplementary Fig. S3c) Undifferentiated THP-1 cells do not express CaSR and GPRC6A on the protein level (Supplementary Fig. S1). To
achieve protein expression, THP-1 cells were differentiated for 2 days with phorbol-12-myristate-13-acetate (100 ng ml−1). Differentiation with vitamin D3 was not sufficient (Supplementary
Fig. S1). MEASUREMENT OF [CA2+]I IN HUMAN MONOCYTES For measurement of [Ca2+]i, human monocytes (1 × 106) were incubated for 30 min at 37 °C on coverslides with Fura-2/AM (4 μM; Invitrogen)
in HBS buffer with 0.2% BSA (Sigma) and 0.6 mM CaCl2. In some experiments, 5 μM NPS was added to the cells. Attached cells were rinsed with HBS (0.6 mM or 1.7 mM CaCl2) in a bath chamber and
viewed with an inverted epifluorescence microscope (Fluar 10 × /0.5; Axiovert 100 microscope; Carl Zeiss Jena). The fiber-coupled monochromator device (Polychrome V; Till-Photonics) excited
fluorescence at alternating wavelength of 340, 358 and 380 nm. Fluorescence emission was imaged with a camera (Sensicam HR; PCO) through a dichroic beam splitter (DCXR-510; Chroma) and a
515-nm long pass filter (OG 515; Schott). Changes of [Ca2+]i were analysed for 200 cells and three different blood donors. KNOCKDOWN OF CASR AND GPRC6A WITH SIRNA For inactivation of CaSR
and GPRC6A, Dharmacon Accell SMARTpool siRNA was applied to THP-1 cells for 4 days before differentiation, as recommended for protein knockdown. For this purpose, THP-1 cells were incubated
in RPMI1640 supplemented with 1% heat-inactivated FCS, 2 mM L-glutamine and incubated for 4 days in the presence of 1 μM non-targeting siRNA, CaSR-specific siRNA or GPRC6A-specific siRNA (in
each case, a mixture of four different siRNAs). For the final 48 h before detection, THP-1 cells were differentiated with phorbol-12-myristate-13-acetate (100 ng ml−1). After 2 days, cells
were lysed for western blotting or stimulated with LPS or LPS plus CaCl2. NECROTIC CELLS To induce necrosis, cells were exposed to shear stress by aspirating them repeatedly up and down a
syringe with a 26-needle gauge. For experiments with human cells, CD4+ T cells from the peripheral blood were used, and for experiment with mouse cells, spleen cells were used. CELL LYSIS,
GEL ELECTROPHORESIS AND WESTERN BLOTTING Caspase-1 was detected in cell culture supernatants of activated cells, whereas CaSR/GPRC6A was analysed in 3 × 106 cells lysed with RIPA buffer.
Protein concentrations were determined with a detergent-compatible protein assay (Bio-Rad). Aliquots of the supernatants were incubated in non-reducing Laemmli buffer for 30 min at room
temperature (CaSR/GPRC6A) or boiled in reducing Laemmli for 5 min (Caspase-1), and equal amounts of protein were resolved by SDS–PAGE. Gel electrophoresis and western blotting were performed
as described previously30. MEASUREMENT OF PHOSPHATIDYLCHOLINE-SPECIFIC PLC ACTIVITY The phosphatidylcholine-specific PLC activity in 1 h-stimulated monocytes was measured using the
AmplexRed phosphatidylcholine-specific PLC assay from Invitrogen following the manufacturer’s protocol. MEASUREMENT OF CYTOSOLIC CAMP CONCENTRATION The cytosolic cAMP concentration in
monocytes stimulated for 16 h was determined using the commercially available competition enzyme-linked immunoassay cAMP XP assay (Cell Signaling) following the manufacturer’s protocol.
Forskolin was used as a positive control. RNA EXTRACTION, CDNA SYNTHESIS AND QUANTITIVE AND SEMI-QUANTITIVE PCR Total RNA was prepared using the RNeasy Mini Kit (Qiagen) according to the
manufacturer’s protocol. cDNA was transcribed with Multiscribe Reverse Transcriptase (Applied Biosystems) using random hexamers. Quantitative PCR was performed using Taqman Gene Expression
Assays for CaSR, GPRC6A and control gene HPRT1 (Applied Biosystems) in the 7500 Real Time PCR System (Applied Biosystems). Results were analysed using 7500 System SDS Software (Applied
Biosystems). Semi-quantitative PCR was performed in an Eppendorf Master cycler gradient 5331 using Perkin Elmer PCR reagents. Nlrp3 was amplified using MEP_L_cop32pre-designed primers from
Santa Cruz Biotechnology with nested PCR. The first, larger fragment was amplified in 15 cycles and the second, smaller fragment with additional 20 cycles. A 193 bp-fragment of IL-1β was
amplified with following primers for 30 cycles (forward 5′-GCATCCAGCTACGAATCTCC-3′, reverse 5′-TCGTTATCCCATGTGTCGAA-3′). PCR fragments were resolved by elecrophoresis on a 2% agarose gel and
visualized by ethidium bromide staining. CYTOKINE MEASUREMENT Human and mouse IL-1β, TNF and IL-6, mouse IL-1α, (OptEIA; BD Bioscienes), PGE2, human IL-1α (R&D Systems) and human IL-18
(MBL) were measured by commercially available enzyme immunoassay following the manufacturer’s protocol. STATISTICAL ANALYSIS For statistical analysis, the software Sigma Stat was used.
Before all comparisons, a normality test was performed, which was uniformly passed. Therefore, statistical significance was assessed using the two-tailed Student’s _t_-test. ADDITIONAL
INFORMATION HOW TO CITE THIS ARTICLE: Rossol, M. _et al_. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. _Nat.
Commun._ 3:1329 doi: 10.1038/ncomms2339 (2012). REFERENCES * Martinon F., Burns K., Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and
processing of proIL-beta. _Mol. Cell._ 10, 417–426 (2002). Article CAS Google Scholar * Chen G. Y., Nuñez G. Sterile inflammation: sensing and reacting to damage. _Nat. Rev. Immunol._ 10,
826–837 (2010). Article CAS Google Scholar * Robertson W. G., Marshall R. W. Ionized calcium in body fluids. _Crit. Rev. Clin. Lab. Sci._ 15, 85–125 (1981). Article CAS Google Scholar
* Janle E. M., Sojka J. E. Use of ultrafiltration probes in sheep to collect interstitial fluid for measurement of calcium and magnesium. _Contemp. Top. Lab. Anim. Sci._ 39, 47–50 (2000).
CAS PubMed Google Scholar * Menkin In _Biochemical Mechanisms in Inflammation_ (ed. Thomas) Springfield, IL (1958). * Kaslick R. S. et al. Quantitative analysis of sodium, potassium and
calcium in gingival fluid from gingiva in varying degrees of inflammation. _J. Periodontol._ 41, 93–97 (1970). Article CAS Google Scholar * Hofer A. M., Curci S., Doble M. A., Brown E.
M., Soybel D. I. Intercellular communication mediated by the extracellular calcium-sensing receptor. _Nat. Cell. Biol._ 2, 392–398 (2000). Article CAS Google Scholar * Tzimas G. N. et al.
Correlation of cell necrosis and tissue calcification with ischemia/reperfusion injury after liver transplantation. _Transplant. Proc._ 36, 1766–1768 (2004). Article CAS Google Scholar *
Korff S. et al. Calcification of myocardial necrosis is common in mice. _Virchows Arch._ 448, 630–638 (2006). Article Google Scholar * Boraschi D., Dinarello C. A. IL-18 in autoimmunity:
review. _Eur. Cytokine. Netw._ 17, 224–252 (2006). CAS PubMed Google Scholar * Brown E. M. et al. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine
parathyroid. _Nature_ 366, 575–580 (1993). Article ADS CAS Google Scholar * Pi M. et al. Identification of a novel extracellular cation-sensing G-protein-coupled receptor. _J. Biol.
Chem._ 280, 40201–40209 (2005). Article CAS Google Scholar * Christiansen B., Hansen K. B., Wellendorph P., Bräuner-Osborne H. Pharmacological characterization of mouse GPRC6A, an
L-alpha-amino-acid receptor modulated by divalent cations. _Br. J. Pharmacol._ 150, 798–807 (2007). Article CAS Google Scholar * Faure H. et al. Molecular determinants of non-competitive
antagonist binding to the mouse GPRC6A receptor. _Cell Calcium_ 46, 323–332 (2009). Article CAS Google Scholar * Wellendorph P. et al. No evidence for a bone phenotype in GPRC6A knockout
mice under normal physiological conditions. _J. Mol. Endocrinol._ 42, 215–223 (2009). Article CAS Google Scholar * Khan M. A., Conigrave A. D. Mechanisms of multimodal sensing by
extracellular Ca(2+)-sensing receptors: a domain-based survey of requirements for binding and signalling. _Br. J. Pharmacol._ 159, 1039–1050 (2010). Article CAS Google Scholar * Gross O.
et al. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. _Immunity_ 36, 388–400 (2012).
Article CAS Google Scholar * Murakami T. et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. _Proc. Natl Acad. Sci. USA_ 109, 11282–11287 (2012).
Article ADS CAS Google Scholar * Piccini A. et al. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine
way. _Proc. Natl Acad. Sci. USA_ 105, 8067–8072 (2008). Article ADS CAS Google Scholar * Kuroda E. et al. Silica crystals and aluminum salts regulate the production of prostaglandin in
macrophages via NALP3 inflammasome-independent mechanisms. _Immunity_ 34, 514–526 (2011). Article CAS Google Scholar * Wolff T. et al. Evidence for agonist-induced export of intracellular
Ca2+ in epithelial cells. _Pflugers Arch._ 424, 423–430 (1993). Article CAS Google Scholar * Tepikin A. V., Voronina S. G., Gallacher D. V., Petersen O. H. Pulsatile Ca2+ extrusion from
single pancreatic acinar cells during receptor-activated cytosolic Ca2+ spiking. _J. Biol. Chem._ 267, 14073–14076 (1992). CAS PubMed Google Scholar * Gabay C., Lamacchia C., Palmer G.
IL-1 pathways in inflammation and human diseases. _Nat. Rev. Rheumatol._ 6, 232–241 (2010). Article CAS Google Scholar * Duewell P. et al. NLRP3 inflammasomes are required for
atherogenesis and activated by cholesterol crystals. _Nature_ 464, 1357–1361 (2010). Article ADS CAS Google Scholar * Larsen C. M. et al. Interleukin-1–receptor antagonist in type 2
diabetes mellitus. _N. Engl. J. Med._ 356, 1517–1526 (2007). Article CAS Google Scholar * Masters S. L. et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a
mechanism for enhanced IL-1β in type 2 diabetes. _Nat. Immunol._ 11, 897–904 (2010). Article CAS Google Scholar * Lee G. S. et al. The calcium-sensing receptor regulates the NLRP3
inflammasome through Ca(2+) and cAMP. _Nature_ 492, 123–128 (2012). Article ADS CAS Google Scholar * Solle M. et al. Altered cytokine production in mice lacking P2X(7) receptors. _J.
Biol. Chem._ 276, 125–132 (2001). Article CAS Google Scholar * Morris C. J. Carrageenan-induced paw edema in the rat and mouse. _Methods Mol. Biol._ 225, 115–121 (2003). PubMed Google
Scholar * Zhang Y., Shaffer A., Portanova J., Seibert K., Isakson P. C. Inhibition of cyclooxygenase-2 rapidly reverses inflammatory hyperalgesia and prostaglandin E2 production. _J.
Pharmacol. Exp. Ther._ 283, 1069–1075 (1997). CAS PubMed Google Scholar * Rossol M. et al. Interaction between transmembrane TNF and TNFR1/2 mediates the activation of monocytes by
contact with T cells. _J. Immunol._ 179, 4239–4248 (2007). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank Cornelia Arnold for technical assistance. This work was
supported by the Deutsche Forschungsgemeinschaft (RO 4037/1-1 and WA 2765/3-1) and the Federal Ministry of Education and Research (01EC1008A IMPAM TP1). Hans Bräuner-Osborne received
support for the breeding of the mice from The Novo Nordisk Foundation and the UNIK Center of Excellence for Food, Fitness and Pharma. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department
of Internal Medicine, Rheumatology Unit, University of Leipzig, Liebigstr. 20, D-04103 Leipzig, Germany, Manuela Rossol, Matthias Pierer, Nora Raulien, Dagmar Quandt, Undine Meusch, Kathrin
Rothe, Kristin Schubert, Christoph Baerwald & Ulf Wagner * Institute of Biochemistry, Medical Faculty, University of Leipzig, Johannisallee 30, D-04103 Leipzig, Germany, Torsten
Schöneberg * Rudolf-Boehm-Institute for Pharmacology and Toxicology, Medical Faculty, University of Leipzig, Härtelstr. 16-18, D-04107 Leipzig, Germany, Michael Schaefer & Ute Krügel *
Department of Drug Design and Pharmacology, Faculty of Health and Medical Sciences, University of Copenhagen, Fruebjergvej 3, 2100 Copenhagen, Denmark, Sanela Smajilovic & Hans
Bräuner-Osborne Authors * Manuela Rossol View author publications You can also search for this author inPubMed Google Scholar * Matthias Pierer View author publications You can also search
for this author inPubMed Google Scholar * Nora Raulien View author publications You can also search for this author inPubMed Google Scholar * Dagmar Quandt View author publications You can
also search for this author inPubMed Google Scholar * Undine Meusch View author publications You can also search for this author inPubMed Google Scholar * Kathrin Rothe View author
publications You can also search for this author inPubMed Google Scholar * Kristin Schubert View author publications You can also search for this author inPubMed Google Scholar * Torsten
Schöneberg View author publications You can also search for this author inPubMed Google Scholar * Michael Schaefer View author publications You can also search for this author inPubMed
Google Scholar * Ute Krügel View author publications You can also search for this author inPubMed Google Scholar * Sanela Smajilovic View author publications You can also search for this
author inPubMed Google Scholar * Hans Bräuner-Osborne View author publications You can also search for this author inPubMed Google Scholar * Christoph Baerwald View author publications You
can also search for this author inPubMed Google Scholar * Ulf Wagner View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS M.R. was involved in
the study design and the drafting of the manuscript, performed most of the experiments and performed data analysis. M.P. was responsible for performing the animal experiments and evaluating
the results. U.M. and K.S. performed western blot experiments and siRNA experiments. N.R., D.Q. and K.R. performed animal experiments, evaluated the results and performed data analysis. U.K.
and M.S. provided the P2X7 knockout model and performed part of the experiments with it. H.B-O. and S.S. provided the GPRC6A knockout model and were involved in drafting the manuscript.
C.B. and T.S. were involved in the study design and the drafting of the manuscript. U.W. conceived the project, was involved in data analysis and drafted the manuscript. CORRESPONDING AUTHOR
Correspondence to Ulf Wagner. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing financial interests. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION Supplementary
Figures S1-S11, Supplementary Table S1 (PDF 1555 kb) RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a
copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Rossol, M., Pierer, M., Raulien, N. _et al._
Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. _Nat Commun_ 3, 1329 (2012). https://doi.org/10.1038/ncomms2339
Download citation * Received: 05 September 2012 * Accepted: 27 November 2012 * Published: 27 December 2012 * DOI: https://doi.org/10.1038/ncomms2339 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative