- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Klotho is a membrane protein predominantly produced in the kidney that exerts some antiageing effects. Ageing is associated with an increased risk of heart failure; whether Klotho
is cardioprotective is unknown. Here we show that Klotho-deficient mice have no baseline cardiac abnormalities but develop exaggerated pathological cardiac hypertrophy and remodelling in
response to stress. Cardioprotection by Klotho in normal mice is mediated by downregulation of TRPC6 channels in the heart. We demonstrate that deletion of _Trpc6_ prevents stress-induced
exaggerated cardiac remodelling in Klotho-deficient mice. Furthermore, mice with heart-specific overexpression of TRPC6 develop spontaneous cardiac hypertrophy and remodelling. Klotho
overexpression ameliorates cardiac pathologies in these mice and improves their long-term survival. Soluble Klotho present in the systemic circulation inhibits TRPC6 currents in
cardiomyocytes by blocking phosphoinositide-3-kinase-dependent exocytosis of TRPC6 channels. These results provide a new perspective on the pathogenesis of cardiomyopathies and open new
avenues for treatment of the disease. You have full access to this article via your institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS SOLUBLE KLOTHO PROTECTS AGAINST
GLOMERULAR INJURY THROUGH REGULATION OF ER STRESS RESPONSE Article Open access 22 February 2023 KLF9 IS ESSENTIAL FOR CARDIAC MITOCHONDRIAL HOMEOSTASIS Article 08 November 2024 TDAG51
INDUCES RENAL INTERSTITIAL FIBROSIS THROUGH MODULATION OF TGF-Β RECEPTOR 1 IN CHRONIC KIDNEY DISEASE Article Open access 08 October 2021 INTRODUCTION Klotho is an antiageing protein
predominantly produced in the kidney and several other tissues including parathyroid glands and epithelial cells of the choroids plexus1. Mice homozygous for a hypomorphic _Klotho_ allele
(_kl_/_kl_) manifest multiple ageing-related phenotypes including skin and muscle atrophy, hyperphosphatemia, osteoporosis and vascular calcification, and die prematurely at around 2–3
months of age. The full-length Klotho protein is a type-1 membrane protein with a large extracellular domain of 952 amino acids in human, a membrane-spanning segment, and a short 11 amino
acids intracellular carboxyl terminus1. Membranous Klotho associates with fibroblast growth factor (FGF) receptors to form co-receptors for the ligand FGF23, a bone-derived circulating
hormone that lowers serum phosphate levels by increasing renal phosphate excretion, suppressing 1,25-dihyroxyvitamin D synthesis, and decreasing gastrointestinal phosphate absorption2,3,4,5.
Klotho-deficient mice have severe hyperphosphatemia due to defects in the Klotho-FGF23-vitamin D regulatory axis5,6,7. This phosphate retention is pivotal for growth retardation and
premature death of Klotho-deficient mice. Dietary phosphate restriction rescues growth defects and premature death of the mice5,6,7. The notion that FGF receptor and Klotho form obligatory
co-receptors for FGF23 is supported by the demonstration that systemic injection of bioactive FGF23 decreases serum levels of phosphate in wild–type (WT) mice, but not in Klotho-deficient
mice8. The extracellular domain of Klotho is composed of two internal repeats, KL1 and KL2, each sharing amino-acid sequence homology to family 1 glycosidases1. The extracellular domain of
Klotho is shed into the systemic circulation, urine and cerebrospinal fluid9. In urine, soluble Klotho regulates several ion transporters in the apical membrane of kidney tubules10,11,12.
The physiological function of soluble Klotho present in the systemic circulation is mostly unknown. The heart responds to injury and stress signals by pathological growth and remodelling
that often progresses to heart failure and sudden death13. One key regulatory step in the development of pathological cardiac growth and remodelling is activation of calmodulin-dependent
serine–threonine protein phosphatase calcineurin by abnormal calcium signalling14. Once activated by increases in intracellular calcium, calcineurin dephosphorylates and causes nuclear
translocation of nuclear factor of activated T cells (NFAT) transcription factors, which bind the regulator regions of cardiac genes and in conjunction with other transcription factors
induce gene expression and promote hypertrophic growth and remodelling. Extracellular stimuli increase intracellular Ca2+ levels by either promoting its release from intracellular organelles
or its entry across the plasma membrane. The TRPC family channels are Ca2+-permeable cation channels expressed in the plasma membrane of many tissues including the heart15. The TRPC family
includes seven members, and is divided into two groups based on structural and functional similarities: TRPC1/4/5, which are not sensitive to diacylglycerol (DAG), and TRPC3/6/7, which are
activated by DAG. TRPC2 is not expressed in humans. Evidence indicates that Ca2+ influx through cardiac TRPC channels—including TRPC1, 3, 4, 5 and 6—is important in calcineurin signalling
and hypertrophic growth of hearts16,17,18,19,20,21,22. The expression of TRPC1, 3, 4, 5 and/or 6 is increased in hypertrophic hearts stimulated by various types or forms of stresses and
their downregulation protects against cardiac hypertrophy. Some members of TRPC family channels, such as TRPC6, contain NFAT-responsive elements in their promoters, which have a pivotal role
in amplifying and sustaining gene expression through a feed-forward circuit16. Thus, TRPC6 is an important modulator of cardiac hypertrophy and a potential target for treatment. However,
physiological function of TRPC6 in the heart and its regulation remain poorly understood, limiting therapeutic strategies for targeting the pathway. Here we show that soluble Klotho inhibits
cardiac TRPC6 channels and protects the heart against stress-induced pathological hypertrophy and remodelling. RESULTS KLOTHO DEFICIENCY AGGRAVATES PATHOLOGICAL HEART GROWTH Klotho
expression is decreased in aging, a condition associated with increased risk for heart failure23,24. We examined the role of Klotho in protecting the heart using _Klotho_-hypomorphic mice
rescued by dietary phosphate restriction. To avoid potential variations caused by strain and gender differences, we studied male mice congenic for the 129/SvJ background by backcrossing for
>6 generations. As reported previously5,6,7, dietary phosphate restriction lowered serum phosphate levels and rescued growth defects and premature death of Klotho-deficient mice
(Supplementary Figs S1 and S2a). Serum levels of sodium, potassium, chloride, calcium, magnesium and urea nitrogen were not different between WT and _kl_/_kl_ mice on a phosphate-restricted
diet (Supplementary Table S1). Phosphate restriction did not affect the growth of WT mice (Supplementary Fig. S1). _Klotho_-hypomorphic mice on low-phosphate diet remained markedly
Klotho-deficient (Supplementary Fig. S2b). To investigate the potential cardioprotective effect of Klotho, we measured heart weight indices (heart weight normalized to body weight or tibia
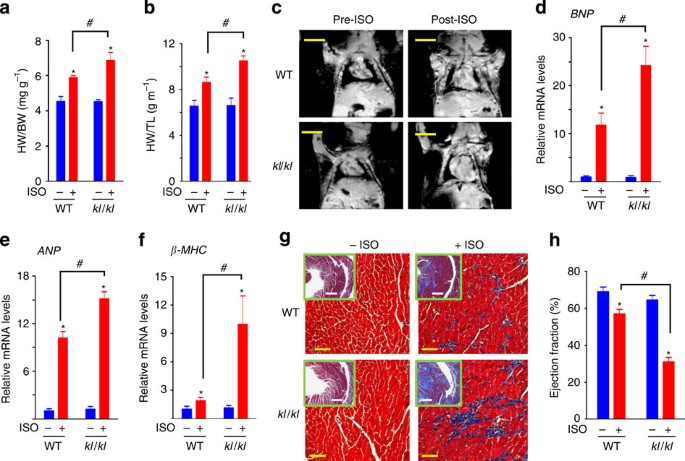
length) as well as the overall heart size in WT and Klotho-deficient mice. Heart weight indices (Fig. 1a) and the overall heart size measured using magnetic resonance imaging (MRI) (Fig. 1c)
were not different between WT and Klotho-deficient mice at baseline. Overstimulation by isoproterenol (ISO) induced pathological hypertrophy in WT mice as reflected by increase in heart
weight indices and the overall heart size, and these ISO-induced changes were aggravated in Klotho-deficient mice (Fig. 1a–c). ISO overstimulation is a well-accepted experimental model of
stress-induced cardiac hypertrophy25,26. Phosphate restriction itself did not alter cardiac responses to stress, as baseline and ISO-induced increases in heart mass were not different
between WT mice fed normal and phosphate-restricted diets (Supplementary Fig. S2c). Pathological cardiac hypertrophy and remodelling are also characterized by increased (re)expression of
fetal genes that are normally quiescent in adult hearts, including brain natriuretic peptide (_BNP_), atrial natriuretic peptide (_ANP_) and β-myosin heavy chain (_β_-_MHC_)13,14. Consistent
with the notion that Klotho deficiency accelerates ISO-induced pathological cardiac remodelling, the expression of cardiac fetal genes was increased by ISO in WT mice, and such increase in
gene expression was augmented in Klotho-deficient mice (Fig.1d–f). Increased expression of these cardiac fetal genes is mediated by activation of the calcineurin-NFAT pathway14. The _Trpc6_
gene contains NFAT-responsive elements in the promoter and its expression is upregulated in several human and rodent models of heart failure16,17,20. We therefore measured the expression of
_Trpc6_ in ISO-treated WT and Klotho-deficient hearts. _Trpc6_ mRNA levels were increased in WT hearts after ISO treatment (Supplementary Fig. S3a). For comparison, ISO treatment did not
alter the expression of _Trpc6_ in other tissues including the blood vessels, lung, kidney and liver. As was observed for cardiac fetal genes, ISO-induced increases in _Trpc6_ mRNA were
enhanced in Klotho-deficient relative to WT mice (Supplementary Fig. S3b). Interstitial fibrosis is another consequence of pathological cardiac hypertrophy and remodelling16. Trichrome
staining of heart sections revealed fibrosis in WT hearts after ISO treatment, and Klotho deficiency worsened ISO-induced cardiac fibrosis (Fig. 1g). In support of these results from
morphometric and gene expression studies, functional analysis of hearts using MRI showed that ISO treatment decreased the ejection fraction of WT hearts, and Klotho-deficiency markedly
aggravated the ISO-induced decline in the ejection fraction (Fig. 1h). Left ventricular end-systolic and end-diastolic volumes were markedly increased, and stroke volumes were decreased in
Klotho-deficient mice after ISO treatment (Supplementary Fig. S4a,b), indicating chamber dilatation as well as impaired contractility of the left ventricle (LV). Severe heart failure with
lung oedema developed in some Klotho-deficient mice after ISO treatment (Supplementary Fig. S4c,d). Thus, Klotho deficiency does not cause baseline cardiac abnormalities but renders the
heart more susceptible to stress-induced pathological cardiac remodelling. KLOTHO ATTENUATES STRESS-INDUCED CARDIAC HYPERTROPHY To further corroborate the above experimental data indicating
that Klotho protects the heart against stress-induced cardiac remodelling, we examined ISO-induced cardiac changes in transgenic mice that overexpress Klotho (KL-Tg). These mice live ~20–30%
longer than WT littermates, and the circulating level of soluble Klotho in transgenic mice is ~100% higher than WT (~200 pM in transgenic mice versus ~100 pM in WT mice)27. Klotho
overexpression in mice did not cause detectable changes in heart mass index and the heart size at baseline (Fig. 2a), and nor did it alter the systemic blood pressure (systolic BP: 103±7 mm
Hg and 103±4 mm Hg, WT versus KL-Tg, _n_=4 each). Klotho overexpression yet blunted the ISO-induced cardiac hypertrophic responses (Fig. 2a). Consistent with the notion that Klotho protects
against stress-induced cardiac remodelling, Klotho overexpression did not alter _BNP_ and _Trpc6_ mRNA levels at baseline, but attenuated ISO-induced increases in _BNP_ and _Trpc6_ mRNA
expression (Fig. 2c). It has been reported that elevated serum FGF23 promotes cardiac hypertrophy28. Because Klotho and FGF23 work in the same pathway to regulate phosphate metabolism, we
measured serum phosphate and FGF23 levels. Klotho overexpression in mice did not alter serum phosphate or FGF23 levels (Fig. 2e), indicating that the cardioprotective effect of Klotho was
not mediated by serum FGF23. As Klotho overexpression mice and control WT littermates were fed normal phosphate diets, these studies also exclude the role of dietary phosphate restriction in
cardioprotection by Klotho. KLOTHO PROTECTS THE HEART BY DOWNREGULATION OF TRPC6 Next, we investigated the mechanism by which Klotho protects against stress-induced cardiac hypertrophy and
remodelling. We have observed that Klotho-deficiency aggravated ISO-induced increases in _Trpc6_ expression, and conversely Klotho overexpression attenuated the ISO-induced increases in
cardiac _Trpc6_ mRNA expression (Supplementary Fig. S3b and Fig. 2d). Inhibition of cardiac TRPC6 by gene silencing or by dominant-negative expression of mutant channels confers
cardioprotection17,20. We examined whether Klotho may protect the heart by inhibiting TRPC6 by crossing Klotho-deficient mice with global _Trpc6_-knockout mice29. Mice with global deletion
of _Trpc6_ grow normally and have no apparent defects in major organ systems at baseline30. Consistently, we found that the baseline heart mass index was not different between mice with
global deletion of _Trpc6_ and control WT littermates (Fig. 3a). Deletion of _Trpc6_ partially protected against ISO-induced cardiac remodelling, but completely prevented the exaggerated
ISO-induced cardiac hypertrophy in Klotho-deficient mice. Consistent with these results, deletion of _Trpc6_ attenuated ISO-induced increases in _ANP_ and _BNP_ mRNA, and abolished the
exaggerated ISO-induced increases in the mRNA in Klotho-deficient mice (Fig. 3b). We further investigated cardioprotection by Klotho using transgenic mice that overexpress TRPC6 in hearts.
Mice with cardiac-specific overexpression of TRPC6 develop spontaneous cardiac hypertrophy and also have heightened sensitivity to stress-induced hypertrophy16. We crossed mice with cardiac
TRPC6 overexpression with transgenic mice overexpressing Klotho to create double transgenic mice, and compared the survival rate, heart mass index and cardiac fetal gene expression of double
transgenic mice with those having cardiac overexpression of TRPC6 and with WT littermates. Compared with WT littermates at 24 months of age, cardiac TRPC6-overexpressing mice had decreased
survival and increased heart mass index and cardiac fetal gene expression without ISO treatment (Fig. 3d–f). Klotho overexpression tended to improve the survival of cardiac
TRPC6-overexpressing mice, prevented the increase in heart mass, and markedly diminished the increase in fetal gene expression induced by overexpression of TRPC6 in hearts. These studies
also support the notion that Klotho protects the heart by inhibiting cardiac TRPC6. SOLUBLE KLOTHO INHIBITS TRPC6 IN ISOLATED CARDIAC MYOCYTES Klotho is not expressed in the heart. We tested
the hypothesis that soluble Klotho present in the systemic circulation mediates the inhibition of cardiac TRPC6. TRPC6 is activated by DAG15. We examined TRPC6 channel activity in freshly
isolated ventricular myocytes by whole-cell patch-clamp recording using stimulation by endothelin-1 (ET1) to release DAG (Fig. 4a). Ventricular myocytes from WT mice without ISO treatment
showed baseline ET-1-activated TRPC-like currents presumably mediated by non-TRPC6 channels, as currents were not different between _Trpc6_-knockout mice (C6−/−) and WT littermates (Fig.
4b). ISO treatment increased TRPC6-mediated currents in WT hearts (Fig. 4a); the role of TRPC6 is supported by the facts that the increase was eliminated in _Trpc6_-knockout mice (Fig. 4b),
and identical currents were seen in TRPC6-overexpressing hearts (Fig. 4a and Supplementary Fig. S5). Cell capacitance (a measurement of surface area of cells) of ventricular myocytes was
increased in WT hearts after ISO and in TRPC6-overexpressing hearts (Fig. 4c), supporting the conclusion that myocyte hypertrophy occurred under these conditions. Klotho overexpression in
mice prevented the ISO-induced increases in currents (Fig. 4b), and acute addition of soluble Klotho to culture media decreased TRPC6-mediated currents in myocytes isolated from WT mice
after ISO treatment (Fig. 4d). Similarly, direct addition of soluble Klotho inhibited the currents in myocytes isolated from TRPC6-overexpressing mice (Fig. 4e). It is theoretically possible
that Klotho inhibits TRPC6 by decreasing the production of DAG. However, soluble Klotho decreased TRPC6 currents in cardiomyocytes directly activated by membrane-permeant DAG (Fig. 4f),
indicating that it inhibits cardiac TRPC6 channel function acting downstream of DAG. KLOTHO BLOCKS IGF AND PI3K-DEPENDENT EXOCYTOSIS OF TRPC6 Because isolated mouse cardiomyocytes cannot be
cultured continuously and the low abundance of endogenous TRPC6 in hearts, we further investigated the mechanism of regulation by soluble Klotho using HEK cells expressing recombinant TRPC6
as well as isolated cardiac myocytes. As in cardiomyocytes, soluble Klotho decreased DAG-activated TRPC6 channels in HEK cells (Fig. 5a). Soluble Klotho treatment decreased cell surface
abundance of TRPC6 measured by biotinylation assays (Fig. 5b). Soluble Klotho exhibits sialidase (Sial) activity and increases cell surface abundance of TRPV5 channels by cleaving sialic
acids in the N-glycans of channels11. The Sial activity of Klotho is not responsible for the regulation of TRPC6, as purified Sial had no effect on TRPC6 whereas it stimulated TRPV5 (Fig.
5c). The above results also indicate that soluble Klotho decreases cell surface expression of TRPC6 via a mechanism not restricted to cardiomyocytes. The decrease in cell-surface abundance
of TRPC6 by soluble Klotho may be caused by decreased exocytosis and/or increased endocytosis of the channel. Blocking exocytosis by v-SNARE inhibitor tetanus toxin31 decreased TRPC6
currents, and prevented further inhibition by soluble Klotho (Fig. 5e). Because tetanus toxin completely prevented the effect by Klotho, the major (if not the sole) action of Klotho on TRPC6
is by blocking exocytosis. Consistent with this notion, we found that blocking endocytosis using a dominant-negative dynamin did not affect the ability of Klotho to inhibit TRPC6:
Coexpression with dominant-negative dynamin increased the basal (that is, without KL) TRPC6 currents, indicating inhibition of endocytosis of channels, but soluble Klotho decreased TRPC6
currents similarly in cells expressing dominant-negative dynamin and in cell expressing the control WT dynamin (Fig. 5f). Phosphoinositide-3-kinase (PI3K)-activating growth factors increase
cell surface abundance of TRPC channels by stimulating exocytosis32. Soluble Klotho inhibits PI3K signalling by insulin and insulin-like growth factors (IGF), which contributes to the
antiaging and tumor suppression effects of Klotho27,33,34,35. We therefore tested the hypothesis that soluble Klotho inhibits TRPC6 by interfering with IGF1 activation of PI3K to promote
exocytosis of channels. To allow for studying the effect of IGF1, we first examined the effect of serum deprivation. Serum deprivation lowered TRPC6 cell surface abundance and activities,
and prevented the inhibition by soluble Klotho (Fig. 6a). The role of IGF1 is demonstrated by findings showing that physiological concentrations of IGF1 (10 nM) reproduced the effect of
serum to promote TRPC6 currents and that soluble Klotho inhibited IGF1-stimulated TRPC6 currents (Fig. 6c). Moreover, PI3K inhibitor wortmannin decreased TRPC6 currents stimulated by IGF1,
and prevented a further decrease of TRPC6 by soluble Klotho (Fig. 6d). These results support the notion that soluble Klotho inhibits IGF1 and PI3K-dependent exocytosis of TRPC6. Because
wortmannin abrogates its effect, soluble Klotho acts upstream of PI3K. Finally, we examined whether soluble Klotho regulates TRPC6 in hearts via the same mechanism. Wortmannin decreased
TRPC6 currents and prevented the inhibition by soluble Klotho in isolated cardiomyocytes (Fig. 6e). Furthermore, the effect of wortmannin to decrease cardiac TRPC6 currents and to prevent
further inhibition by soluble Klotho was reproduced by tetanus toxin, and the effects of wortmannin and tetanus toxin were not additive (Fig. 6f). Thus, tetanus toxin and wortmannin inhibit
cardiac TRPC6 via the same mechanism; that is, by blocking exocytosis of channels. Collectively, these data strongly support the hypothesis that soluble Klotho inhibits TRPC6 by blocking
PI3K-dependent exocytosis of channels (Fig. 7). DISCUSSION The data presented in this study provide compelling evidence indicating that soluble Klotho protects the heart against
stress-induced cardiac hypertrophy and remodelling. Klotho expression is decreased in aging23, thus decline in circulating soluble Klotho may contribute to age-related cardiomyopathy in
humans. One consequence of cardiac aging is increased sensitivity to stress-induced heart failure36 similar to the changes in Klotho-deficient mice we observed here. Many Klotho-mediated
aging phenotypes, such as vascular calcification, growth defects and premature death, are attributed to defects in the function of membrane Klotho as co-receptors for FGF23 and phosphate
retention3,4,5,6,7,8. Our results indicate that the cardioprotective effect of soluble Klotho is independent of FGF23 and phosphate metabolism. First, Klotho overexpression in mice confers
cardioprotection without altering serum phosphate and FGF23 levels. Second, dietary phosphate restriction normalizes serum phosphate levels of Klotho-deficient mice to the level of WT mice,
excluding hyperphosphatemia as the culprit of cardiac dysfunction in these mice. Moreover, deletion of _Trpc6_ completely prevents exaggerated stress-induced cardiac hypertrophy and
remodelling in Klotho-deficient mice, indicating that cardioprotection by Klotho is mediated by downregulation of TRPC6. TRPC6 is broadly expressed in tissues15,37. Although it is possible
that the effect of Klotho on TRPC6 in other tissues also contributes to cardioprotection, the following results indicate that Klotho inhibition of cardiac TRPC6 has a critical role in the
process. First, Klotho ameliorates cardiac hypertrophy and remodelling induced by heart-specific overexpression of TRPC6, and Klotho inhibits TRPC6 in isolated cardiomyocytes. Second,
Klotho-deficient mice have no cardiac dysfunction at baseline, but develop exaggerated cardiomyopathy in response to ISO treatment. ISO treatment causes upregulation of _Trpc6_ mRNA in the
heart, but not in other tissues that influence cardiac function, such as the blood vessels, lung, and kidney. The effect of _Trpc6_ deletion to prevent ISO-induced exaggerated cardiomyopathy
in Klotho-deficient mice is therefore most likely due to abolition of ISO-induced increases of cardiac TRPC6. Our findings also have important implications in chronic kidney disease (CKD),
a disease that affects ~10% of the general population38,39. The prevalence of cardiac hypertrophy in patients of advanced stages of CKD is estimated as high as 90%, and cardiac dysfunction
is the main cause of death for the patients39,40,41. Klotho is predominantly produced in the kidney, and circulating levels of soluble Klotho are reportedly decreased in CKD42,43. Our study
supports that decreased levels of soluble Klotho contribute to the pathogenesis of cardiac hypertrophy in CKD. Recently, Faul _et al._ reported that FGF23 stimulates cardiomyocyte growth and
increased serum FGF23 contributes to cardiac hypertrophy in CKD28. Interestingly, FGF23 appears to induce cardiac hypertrophy independently of stress factors, whereas Klotho deficiency
predisposes the heart to stress-induced pathological hypertrophy. Thus, increased FGF23 and Klotho deficiency may synergistically contribute to cardiac hypertrophy in CKD by participating at
different stages of pathogenesis. The physiological role of TRPC6 in hearts is elusive. Its function appears to be dispensable, as mice with deletion of _Trpc6_ have no apparent cardiac
dysfunction. Consistent with this observation, we found that TRPC6 channel activity is undetectable in hearts at baseline. Overstimulation by ISO leads to increase in _Trpc6_ mRNA levels and
functional TRPC6 currents in mouse hearts. Increased expression of cardiac TRPC6 has also been reported in mouse models of cardiac hypertrophy induced by calcineurin gene overexpression, by
overstimulation by neuroendocrine hormones including ET-1, phenylephrine and angiotensin II, by thoracic aortic banding pressure overload, and in human failing hearts16,17,20. Thus, soluble
Klotho protects the heart by acting on a molecule that is normally quiescent but activated during stresses. Mechanistically, we propose that IGFs such as IGF1 provide a tonic stimulation
for exocytosis of TRPC6 via PI3K, and soluble Klotho exerts a tonic inhibition to the system (model in Fig. 7). Cardiac stresses increase the intracellular Ca2+ concentration from multiple
mechanisms44. The abnormal intracellular calcium signalling in the heart activates calcineurin and NFAT to initiate fetal gene expression and pathological cardiac hypertrophy and
remodelling. TRPC6 contains NFAT-responsive elements in its promoter and is also upregulated by stress. The increased Ca2+ influx through TRPC6 causes a feed-forward cycle and further
amplifies and sustains the process. By placing a brake on the system, Klotho protects the heart. Conversely, Klotho deficiency accelerates stress-induced cardiac remodelling. Without stress
signals to upregulate TRPC6, neither Klotho deficiency nor overexpression in mice affects cardiac function at baseline. Multiple studies have reported that soluble Klotho inhibits
intracellular signalling by insulin and IGF127,33,34,35. Kurosu _et al._27 first reported that soluble Klotho inhibits insulin and IGF-mediated activation of PI3K pathway by inhibiting
activation of receptors and repressing activated receptors. This antiinsulin/IGF effect contributes to aging suppression by Klotho in mice. Wolf _et al._ further found that soluble Klotho
suppresses the growth of human breast and pancreatic cancer cells33,34. They also found that soluble Klotho coimmunoprecipitated with IGF1 receptors, and suggested that soluble Klotho
inhibits the intracellular signalling by IGF1 at least partly by direct interactions with receptors33. It was also reported that Klotho prolongs lifespan and stress resistance in _C.
elegans_ by blocking insulin and IGF-like signalling in worms35. Our results support these previous reports and extend the antiinsulin/IGF role of Klotho to cardioprotection.
Cardioprotection by Klotho may contribute to the antiaging effect of Klotho in mice. Activation of PI3K and downstream Akt signalling cascade in the heart is important for physiological
cardiac growth, but it can also lead to pathological cardiac hypertrophy13,45. Inhibition of TRPC6 by soluble Klotho may be a mechanism for preventing PI3K to cause pathological cardiac
hypertrophy in the normal heart. Interestingly, shedding of soluble Klotho from membranous Klotho is mediated by metalloproteinases ADAM 10 and 17, and insulin stimulates the shedding
through the PI3K pathway46. Whether the regulation of shedding of soluble Klotho by insulin/IGF1 has any roles in the control of cardiac functions awaits future investigation. Other TRPC
channels including TRPC1, 3, 4 and 5 are also present in the heart. Increased expression of these channels is also associated with cardiac hypertrophy induced by pathological stimuli and
downregulation confers the protection17,18,19,20. Cardiac TRPC channels are likely heteromultimers of different TRPC members15,22,23, which may partly explain why inhibition of different
TRPC channels can confer cardioprotection. The exact molecular composition and stoichiometry of TRPC channels that form heteromultimers with TRPC6 in the heart, however, remains unknown. In
this study, deletion of _Trpc6_ totally abolishes exaggerated ISO-induced cardiac hypertrophy and remodelling in Klotho-deficient mice, indicating that inhibition of TRPC6 alone is
sufficient for cardioprotection. Our study yet does not exclude the possibility that soluble Klotho also exerts inhibition on other TRPC channels that form multimers with TRPC6. Of note,
_Trpc6_ deletion only partially blunts ISO-induced hypertrophy, and other factors besides TRPC6 (such as other TRPC channels) are also involved in the hypertrophic response to ISO treatment.
Pharmacological TRPC antagonism is in development as a potential treatment of cardiac hypertrophy21,22,47. As an endogenous hormone that may extend human lifespan, soluble Klotho or its
analogues or activators48 may prove to be important therapeutic agents. The involvement of TRPC6 in multiple models of cardiac hypertrophy and heart failure16,17,20 and the ability of Klotho
to protect against cardiac hypertrophy induced by heart-specific overexpression of TRPC6 suggest that Klotho-based therapeutic strategies may be applicable to diverse cardiac diseases.
TRPC6 is also expressed in the kidney, and systemic and pulmonary vasculature, and increased TRPC6 function in these tissues leads to pathologies15,37,49,50. Klotho-based therapeutics may
also be valuable in treating TRPC6-related pathologies in other organs. One major unanswered question in the pathogenesis of cardiac hypertrophy and remodelling is how the heart
distinguishes between overwhelming intracellular calcium transients during each normal cardiac cycle and the abnormal calcium signalling induced by stress signals44. Mice with Klotho
deficiency or overexpression have no apparent cardiac abnormalities at baseline. The selective targeting to the stress-induced abnormal calcium signalling by Klotho may provide clues to
answer this question in the future. METHODS GENERAL EXPERIMENTAL PROCEDURES OF MICE _Klotho_-hypomorphic, KL-Tg, _Trpc6_-knockout, and TRPC6-Tg (line L16) mice have been described1,16,27,29.
Each mouse line was backcrossed to 129/SvJ mice for >6 generations to achieve congenic background. For dietary phosphate restriction, mice were fed a purified diet containing with 0.2%
(wt/wt) inorganic phosphate (TD-09073, Harlan Teklad, Madison, WI) from weaning at ~3 weeks of age. Normal phosphate diets contain 0.35% inorganic phosphate. All mice subjected to
experiments were males at ~3 months of age, unless otherwise specified. Blood pressure was measured in WT, _Klotho_-hypomorphic and KL-Tg mice using a tail-cuff sphygmomanometer as
previously described51. For induction of cardiac hypertrophy, ISO (2 mg kg−1 per day diluted in PBS) was injected subcutaneously to mice once per day for 10 consecutive days52. Control mice
received PBS injection. At day 11, mice were euthanized and hearts were isolated. After measurement of weight, a portion of the hearts was snap-frozen in liquid N2 and saved for RNA
isolation, and the remainder fixed and stored for histology. All animal protocols were approved by the University of Texas Southwestern Institutional Animal Care and Use Committee. REAL-TIME
QUANTITATIVE RT–PCR ANALYSIS OF MRNA RNA was extracted from heart samples with trizol (Invitrogen), reverse-transcribed into cDNA (Taqman reverse transcription reagents, Applied
Biosystems-Roche), and mRNA abundance was analysed by real-time PCR with SYBR-green (iTaq or iQ SYBR-green Supermix, BioRad). Primers: _GAPDH_, 5′-tgcaccaccaactgcttagc-3′,
5′-ggcatggactgtggtcatgag-3′; _ANP_, 5′-gccatattggagcaaatcct-3′, 5′-gcaggttcttgaaatccatca-3′; _BNP_, 5′-ccaaggcctcacaaaagaac-3′, 5′-agacccaggcagagtcagaa-3′; _β-MHC_,
5′-ttggatgagcgactcaaaaa-3′, 5′-gctccttgagcttcttctgc-3′; _Trpc6_, 5′-cgctgccaccgtatgg-3′, 5′-ccgccggtgagtcagt-3′. HISTOLOGICAL ANALYSIS Dissected hearts were rinsed in PBS and incubated in
Krebs–Henseleit solution lacking Ca2+ for 30 min, and were then fixed in 4% paraformaldehyde overnight at room temperature. Samples were dehydrated and stored in 50% ethanol, mounted in
paraffin and sectioned. Sections were then stained with haematoxylin and eosin or with Masson’s Trichrome stain. SERUM COLLECTION AND MEASUREMENT Blood was drawn from mice using
retro-orbital bleeding method. Samples were immediately centrifuged, and supernatant collected and stored. Serum phosphate and FGF23 levels were measured using a phosphate assay kit (Stanbio
labs, San Antonio, TX) and FGF23 ELISA kit (Kainos lab, Japan), respectively. CARDIAC MRI Cardiac MRI of mice was performed in the Mouse MRI Core Facility of UT Southwestern Medical Center
as previously described53. To determine the LV volume, multiple parallel slices of 1-mm thickness perpendicular to the long heart axis were imaged. The area of LV of each slice at both
end-diastolic and end-systolic phases was measured using ImageJ software. LV volume was calculated as the sum of area of all slices at either phase. Stroke volume is the difference between
end-diastolic and end-systolic volumes. Ejection fraction is the percentage of stroke volume over end-diastolic volume. ISOLATION OF CARDIAC VENTRICULAR MYOCYTE Isolation of mouse
ventricular myocytes was performed per established procedure54. Briefly, mice received heparin (100 U per mouse) and anaesthesia. Hearts were quickly removed and perfused retrograde via the
aorta with a solution containing (in mM) 113 NaCl, 4.7 KCl, 1.2 MgSO4, 0.6 KH2PO4, 0.6 Na2HPO4, 10 NaHCO3, 30 taurine, 5.5 glucose, 10 2,3-butanedione monoxime, 10 HEPES (at pH 7.4) and
followed by a solution containing in addition 1 mg ml−1 type 2 collagenase (Worthinton) and 0.1 mg ml−1 protease XIV (Sigma). The perfusion solution was maintained at 37 °C and equilibrated
with 100% O2. Thereafter, the ventricle was removed, chopped into small pieces, and further digested in the enzyme solution. After stopping enzyme digestion by adding 2.5% bovine serum
albumin and 0.1 mM CaCl2, the tissue-cell suspension was filtered through a sterilized-gauze sponge, centrifuged using a tabletop centrifuge at 50_g_ for 1 min. The resulting cell pellet was
resuspended in the stopping buffer and [Ca2+] titrated to 0.5 mM by addition of 100 mM CaCl2 stock solution in four steps over 20 min. Isolated myocytes were stored at room temperature
until use. WHOLE-CELL RECORDING For recording of isolated myocytes, cells were transferred into a perfusion chamber mounted on an inverted microscope and continually perfused at the rate of
1 ml min−1 with bath solution. Whole-cell currents were recorded under voltage-clamp using an Axopatch 200B patch-clamp amplifier (Axon instruments Inc., Foster City, CA, USA)55. Voltage
protocol consists of holding at −40 mV and repetitive descending ramp pulses from +120 to −120 mV for 500 ms applied every 10 sec. The pipette solution contained (in mM) 9.4 NaCl, 120 CsCl,
1 MgCl2, 3.5 CaCl2, 10 BAPTA, 10 HEPES, 0.2 NaGTP (pH 7.2) (calculated ionized [Ca2+]~80 nM) and the bath solution contained 140 NaCl, 5 CsCl, 1 MgCl2, 1.2 CaCl2, 10 glucose, 10 HEPES (pH
7.4). Bath solution also contained 1 μM of nifedipine and 3 mM of NiCl2 to block current flow through L-type Ca2+ channel and Na+/Ca2+ exchanger, respectively. The pipette resistance was
~2–3 MΩ when filled with the pipette solution. Whole-cell access resistance was <10 MΩ. ET-1 (20 nM) was administrated using focal application method. The distance between the tip of the
applicator and myocyte was <50 μm. Currents were low-pass filtered at 2 kHz and sampled every 0.1 ms. Data acquisition was performed using pClamp9.2 program (Axon Instrument, Inc.) and
analysis using Prism (V3.0) software (GraphPad Software, San Diego, CA, USA). For whole-cell recording of recombinant TRPC6 channels in HEK cells, the pipette and bath solution contained (in
mM) 120 Cs-aspartate (Cs-Asp), 10 CsCl, 1 MgCl2, 2 MgATP, 5 EGTA, 1.5 CaCl2 (free [Ca2+]=70 nM) and 10 CsHEPES (pH 7.2) and 140 NaCl, 5 KCl, 0.5 EGTA and 10 NaHEPES (pH 7.4), respectively.
SURFACE BIOTINYLATION ASSAY HEK cells expressing haemagglutinin (HA)-tagged TRPC6 (in 35-mm culture dish) were incubated with or without soluble Klotho, washed with 1 ml of ice-cold PBS
three times, and incubated with 1 ml of PBS containing 1.5 mg ml−1 EZ-link-NHS-SS-biotin (Thermo Scientific) for 2 h at 4 °C. After quenching with glycine-containing PBS for 20 min, cells
were lysed in a buffer (150 mm NaCl, 50 mm Tris–HCl, 5 mm EDTA, 1% Triton X-100, 0.5% deoxycholate and 0.1% SDS) containing protease inhibitor mixture for 30 min. For detection of
biotinylated proteins, lysates were precipitated by streptavidin-agarose beads (Thermo Scientific) for 2 h at 4 °C. Beads were subsequently washed three times with TBS containing 1% Triton
X-100. Biotin-labelled proteins were eluted in sample buffer, separated by SDS–PAGE, and transferred to nitrocellulose membranes for western blotting using mouse monoclonal anti-HA antibody
(Sigma-Aldrich; 1:250 dilution) or anti-α-tubulin antibody (Sigma-Aldrich; 1:500 dilution). STATISTICAL ANALYSIS Statistical comparison was made between control and experimental groups
conducted during the same time period. Each experiment was repeated at least once at separate times and with similar results. Data are presented as means±s.e.m. Statistical comparison
between two groups of data were made using two-tailed unpaired Student’s _t_-test. Multiple comparisons were determined using one-way analysis of variance followed by Tukey's multiple
comparison tests. Statistical comparison of Kaplan–Meier cumulative survival curves was made using ‘log-rank’ analysis ( http://bioinf.wehi.edu.au/software/russell/logrank/). ADDITIONAL
INFORMATION HOW TO CITE THIS ARTICLE: Xie J. _et al._ Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. _Nat. Commun._ 3:1238 doi: 10.1038/ncomms2240
(2012). REFERENCES * Kuro-o M. et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. _Nature_ 390, 45–51 (1997). Article CAS ADS Google Scholar * ADHR
Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. _Nat. Genet._ 26, 345–348 (2000). * Kurosu H. et al. Regulation of fibroblast growth factor-23
signaling by Klotho. _J. Biol. Chem._ 281, 6129–6123 (2006). Article Google Scholar * Urakawa I. et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. _Nature_
444, 770–774 (2006). Article CAS ADS Google Scholar * Razzaque M. S. The FGF23-Klotho axis: endocrine regulation of phosphate homeostasis. _Nat. Rev. Endocrinol._ 5, 611–619 (2009).
Article CAS Google Scholar * Morishita K. et al. The progression of aging in klotho mutant mice can be modified by dietary phosphorus and zinc. _J. Nutr._ 131, 3182–3188 (2001). Article
CAS Google Scholar * Yoshida T., Fujimori T. & Nabeshima Y. Mediation of unusually high concentrations of 1,25-dihydroxyvitamin D in homozygous klotho mutant mice by increased
expression of renal 1α-hydroxylase gene. _Endocrinology_ 143, 683–689 (2002). Article CAS Google Scholar * Nakatani T. et al. _In vivo_ genetic evidence for klotho-dependent, fibroblast
growth factor 23 (Fgf23) -mediated regulation of systemic phosphate homeostasis. _FASEB J_ 23, 433–441 (2009). Article CAS Google Scholar * Imura A. et al. Secreted Klotho protein in sera
and CSF: implication for post-translational cleavage in release of Klotho protein from cell membrane. _FEBS Lett._ 565, 143–147 (2004). Article CAS Google Scholar * Chang Q., Hoefs S.,
van der Kemp A. W., Topala C. N., Bindels R. J. & Hoenderop J. G. The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. _Science_ 310, 490–493 (2005). Article CAS
ADS Google Scholar * Cha S. K., Ortega B., Kurosu H., Rosenblatt K. P., Kuro-o M. & Huang C.-L. Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel
via binding to galectin-1. _Proc. Natl Acad. Sci. USA_ 105, 9805–9810 (2008). Article CAS ADS Google Scholar * Hu M. C. et al. Klotho: a novel phosphaturic substance acting as an
autocrine enzyme in the renal proximal tubule. _FASEB J._ 24, 3438–3450 (2010). Article CAS Google Scholar * Frey N., Katus H. A., Olson E. N. & Hill J. A. Hypertrophy of the heart: a
new therapeutic target? _Circulation_ 109, 1580–1589 (2004). Article Google Scholar * Vega R. B., Bassel-Duby. R. & Olson E. N. Control of cardiac growth and function by calcineurin
signaling. _J. Biol. Chem._ 278, 36981–36984 (2003). Article CAS Google Scholar * Nilius B., Owsianik. G., Voets. T. & Peters J. A. Transient receptor potential cation channels in
disease. _Physiol. Rev._ 87, 165–217 (2007). Article CAS Google Scholar * Kuwahara K. et al. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. _J. Clin.
Invest._ 116, 3114–3126 (2006). Article CAS Google Scholar * Onohara N. et al. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. _EMBO J._ 25, 5305–5316
(2006). Article CAS Google Scholar * Bush E. W. et al. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. _J.
Biol. Chem._ 281, 33487–33496 (2006). Article CAS Google Scholar * Ohba T. et al. Upregulation of TRPC1 in the development of cardiac hypertrophy. _J. Mol. Cell Cardiol._ 42, 498–507
(2007). Article CAS Google Scholar * Wu. X., Eder P., Chang B. & Molkentin J. D. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. _Proc. Natl Acad. Sci. USA_
107, 7000–7005 (2010). Article CAS ADS Google Scholar * Rowell J., Koitabashi N. & Kass D. A. TRP-ing up heart and vessels: canonical transient receptor potential channels and
cardiovascular disease. _J. Cardiovasc. Transl. Res._ 3, 516–524 (2010). Article Google Scholar * Eder P. & Molkentin J. D. TRPC6 channels as effectors of cardiac hypertrophy.
_Circulation_ 108, 265–272 (2011). Article CAS Google Scholar * Yamazaki Y. et al. Establishment of sandwich ELISA for soluble alpha-Klotho measurement: Age-dependent change of soluble
alpha-Klotho levels in healthy subjects. _Biochem. Biophys. Res. Commun._ 398, 513–518 (2010). Article CAS Google Scholar * Lakatta E. G. & Levy D. Arterial and cardiac aging: major
shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. _Circulation_ 107, 346–354 (2003). Article Google Scholar * Boluyt M. O. et
al. Isoproterenol infusion induces alterations in expression of hypertrophy-associated genes in rat heart. _Am. J. Physiol._ 269, H638–H647 (1995). CAS PubMed Google Scholar * Takaki M.
Cardiac mechanoenergetics for understanding isoproterenol-induced rat heart failure. _Pathophysiology_ 19, 163–170 (2012). Article CAS Google Scholar * Kurosu H. et al. Suppression of
aging in mice by the hormone Klotho. _Science_ 309, 1829–1833 (2005). Article CAS ADS Google Scholar * Faul C. et al. FGF23 induces left ventricular hypertrophy. _J. Clin. Invest._ 121,
4393–4408 (2011). Article CAS Google Scholar * Dietrich A. et al. Increased vascular smooth muscle contractility in TRPC6−/− mice. _Mol. Cell Biol._ 25, 6980–6989 (2005). Article CAS
Google Scholar * Eckel J. et al. TRPC6 enhances angiotensin II-induced albuminuria. _J. Am. Soc. Nephrol._ 22, 526–535 (2011). Article CAS Google Scholar * Mendez M., Gross K. W., Glenn
S. T., Garvin J. L. & Carretero O. A. Vesicle-associated membrane protein-2 (VAMP2) mediates cAMP-stimulated renin release in mouse juxtaglomerular cells. _J. Biol. Chem._ 286,
28608–28618 (2011). Article CAS Google Scholar * Bezzerides V. J., Ramsey I. S., Kotecha S., Greka A. & Clapham D. E. Rapid vesicular translocation and insertion of TRP channels.
_Nat. Cell Biol._ 6, 709–720 (2004). Article CAS Google Scholar * Wolf I. et al. Klotho: a tumor suppressor and a modulator of the IGF-1 and FGF pathways in human breast cancer.
_Oncogene_ 27, 7094–7105 (2008). Article CAS Google Scholar * Abramovitz L. et al. KL1 internal repeat mediates klotho tumor suppressor activities and inhibits bFGF and IGF-I signaling in
pancreatic cancer. _Clin. Cancer Res._ 17, 4254–4266 (2011). Article CAS Google Scholar * Château M. T., Araiz C., Descamps S. & Galas S. Klotho interferes with a novel
FGF-signalling pathway and insulin/Igf-like signalling to improve longevity and stress resistance in _Caenorhabditis elegans_. _Aging_ 2, 567–581 (2010). Article Google Scholar * Liles J.
T., Ida K. K., Joly K. M., Chapo J. & Plato C. F. Age exacerbates chronic catecholamine-induced impairments in contractile reserve in the rat. _Am. J. Physiol._ 301, R491–R499 (2011).
CAS Google Scholar * Dietrich A. & Gudermann T. TRP channels in the cardiopulmonary vasculature. _Adv. Exp. Med. Biol._ 704, 781–810 (2011). Article CAS Google Scholar * Coresh J.
et al. Prevalence of chronic kidney disease in the United States. _JAMA_ 298, 2038–2047 (2007). Article CAS Google Scholar * Go A. S., Chertow G. M., Fan D., McCulloch C. E. & Hsu C.
Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. _N. Engl J. Med._ 351, 1296–1305 (2004). Article CAS Google Scholar * Taddei S., Nami R.,
Bruno. R. M., Quatrini. I. & Nuti R. Hypertension, left ventricular hypertrophy and chronic kidney disease. _Heart Fail. Rev._ 16, 615–620 (2010). Article Google Scholar * Glassock R.
J., Pecoits-Filho R. & Barberato S. H. Left ventricular mass in chronic kidney disease and ESRD. _Clin. J. Am. Soc. Nephrol._ 4(Suppl 1): S79–S91 (2009). Article Google Scholar * Hu M.
C. et al. Klotho deficiency causes vascular calcification in chronic kidney disease. _J. Am. Soc. Nephrol._ 22, 124–136 (2011). Article CAS Google Scholar * Shimamura Y. et al. Serum
levels of soluble secreted α-Klotho are decreased in the early stages of chronic kidney disease, making it a probable novel biomarker for early diagnosis. _Clin. Exp. Nephrol_ 16, 722–729
(2012). Article CAS Google Scholar * Heineke J. & Ritter O. Cardiomyocyte calcineurin signaling in subcellular domains: from the sarcolemma to the nucleus and beyond. _J. Mol. Cell
Cardiol._ 52, 62–73 (2012). Article CAS Google Scholar * Heineke J. & Molkentin J. D. Regulation of cardiac hypertrophy by intracellular signalling pathways. _Nat. Rev. Mol. Cell.
Biol._ 7, 589–600 (2006). Article CAS Google Scholar * Chen C. D., Podvin S., Gillespie E. & Leeman S. E., Abraham C. R. Insulin stimulates the cleavage and release of the
extracellular domain of Klotho by ADAM10 and ADAM17. _Proc. Natl Acad. Sci. USA_ 104, 19796–19801 (2007). Article CAS ADS Google Scholar * Koitabashi N. et al. Cyclic GMP/PKG-dependent
inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation Novel mechanism of cardiac stress modulation by PDE5 inhibition. _J. Mol. Cell
Cardiol._ 48, 713–724 (2010). Article CAS Google Scholar * King G. D. et al. Identification of novel small molecules that elevate Klotho expression. _Biochem. J._ 441, 453–461 (2012).
Article CAS Google Scholar * Winn M. P. et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. _Science_ 308, 1801–1804 (2005). Article CAS
ADS Google Scholar * Reiser J. et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. _Nat. Genet._ 37, 739–744 (2005). Article CAS Google
Scholar * Liu Z., Xie J., Wu T., Truong T., Auchus R. J. & Huang C. L. Downregulation of NCC and NKCC2 cotransporters by kidney-specific WNK1 revealed by gene disruption and transgenic
mouse models. _Hum. Mol. Genet._ 20, 855–866 (2011). Article CAS Google Scholar * Zou Y. et al. Isoproterenol activates extracellular signal–regulated protein kinases in cardiomyocytes
through calcineurin. _Circulation_ 104, 102–108 (2001). Article CAS Google Scholar * Aoyagi T. et al. Cardiac mTOR protects the heart against ischemia-reperfusion injury. _Am. J. Physiol.
Heart Circ. Physiol._ 303, H75–H85 (2012). Article CAS Google Scholar * O’Connell T. D., Ni Y. G., Lin K. M., Han H. & Yan Z. _Isolation and Culture of Adult Mouse Cardiac Myocytes
for Signaling_ Online at www.signaling-gateway.org/reports/v1/CM0005/CM0005.htm (2003). * An S. W., Cha S. K., Yoon J., Chang S., Ross E. M. & Huang C. L. WNK1 promotes PIP2 synthesis to
coordinate growth factor and GPCR-Gq signaling. _Curr. Biol._ 21, 1979–1987 (2011). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank Eric Olson for TRPC6-Tg mice,
Jyothsna Gattineni for assistance with measurements of serum FGF23 and phosphate, Masaya Takahashi and Kim Kangasniemi for cardiac MRI, and Peter Igarashi, Orson Moe and Aylin Rodan for
discussions and comments. This work was supported by NIH (DK59530, DK85726, DK79328, DK91392), the Intramural Research Program of the NIH (ZO1-ES-101684) and by a GRIP grant from Genzyme,
Inc. C.-L.H. holds the Jacob Lemann Professorship in Calcium Transport of University of Texas Southwestern Medical Center. AUTHOR INFORMATION Author notes * Seung-Kuy Cha Present address:
Present address: Department of Physiology and Institute of Life Style Medicine, Yonsei University Wonju College of Medicine, Ilsan-Dong 162, Wonju, Kangwondo 220-701, Republic of Korea, *
Seung-Kuy Cha and Sung-Wan An: These authors contributed equally to this work AUTHORS AND AFFILIATIONS * Department of Medicine, UT Southwestern Medical Center, Dallas, 75390, Texas, USA
Jian Xie, Seung-Kuy Cha, Sung-Wan An & Chou-Long Huang * Department of Pathology, UT Southwestern Medical Center, Dallas, 75390, Texas, USA Makoto Kuro-o * Laboratory of Neurobiology,
National Institute of Environmental Health Sciences, Research Triangle Park, 27709, North Carolina, USA Lutz Birnbaumer Authors * Jian Xie View author publications You can also search for
this author inPubMed Google Scholar * Seung-Kuy Cha View author publications You can also search for this author inPubMed Google Scholar * Sung-Wan An View author publications You can also
search for this author inPubMed Google Scholar * Makoto Kuro-o View author publications You can also search for this author inPubMed Google Scholar * Lutz Birnbaumer View author publications
You can also search for this author inPubMed Google Scholar * Chou-Long Huang View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS J.X.,
S.-K.C. and S.-W.A. designed the study, conducted the experiments, analysed the data, and participated in writing the paper; M.K. contributed Klotho-deficient and overexpression mice, and
analysis of Klotho expression in Klotho-deficient mice; L.B. contributed Trpc6-deleted mice. C.-L.H. supervised the entire project and wrote the final paper. All authors read, commented and
approved the paper. CORRESPONDING AUTHOR Correspondence to Chou-Long Huang. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing financial interests. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY INFORMATION Supplementary Figures S1-S5 and Supplementary Table S1 (PDF 795 kb) RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE
Xie, J., Cha, SK., An, SW. _et al._ Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. _Nat Commun_ 3, 1238 (2012). https://doi.org/10.1038/ncomms2240
Download citation * Received: 23 May 2012 * Accepted: 31 October 2012 * Published: 04 December 2012 * DOI: https://doi.org/10.1038/ncomms2240 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative







