- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
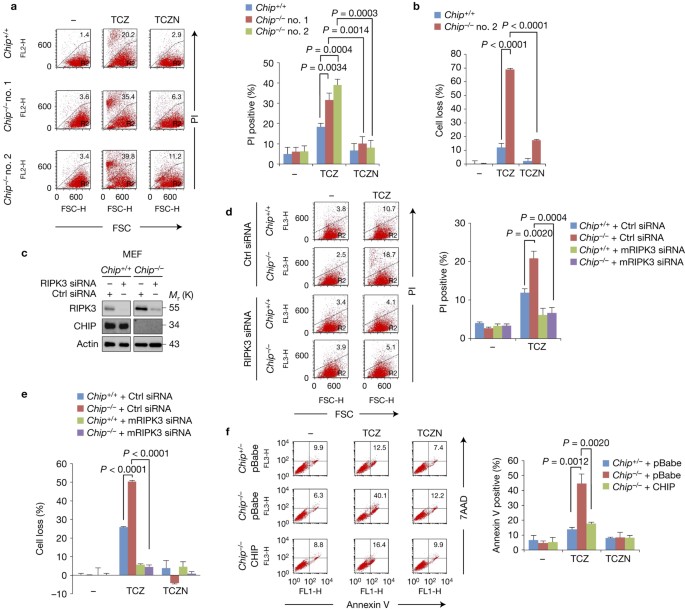
ABSTRACT Receptor-interacting protein kinase 3 (RIPK3) functions as a key regulator of necroptosis. Here, we report that the RIPK3 expression level is negatively regulated by CHIP (carboxyl
terminus of Hsp70-interacting protein; also known as STUB1) E3 ligase-mediated ubiquitylation. _Chip_−/− mouse embryonic fibroblasts and CHIP-depleted L929 and HT-29 cells exhibited higher
levels of RIPK3 expression, resulting in increased sensitivity to necroptosis induced by TNF (also known as TNFα). These phenomena are due to the CHIP-mediated ubiquitylation of RIPK3, which
leads to its lysosomal degradation. Interestingly, RIPK1 expression is also negatively regulated by CHIP-mediated ubiquitylation, validating the major role of CHIP in necrosome formation
and sensitivity to TNF-mediated necroptosis. _Chip_−/− mice (C57BL/6) exhibit inflammation in the thymus and massive cell death and disintegration in the small intestinal tract, and die
within a few weeks after birth. These phenotypes are rescued by crossing with _Ripk3_−/− mice. These results imply that CHIP is a bona fide negative regulator of the RIPK1–RIPK3 necrosome
formation leading to desensitization of TNF-mediated necroptosis. Access through your institution Buy or subscribe This is a preview of subscription content, access via your institution
ACCESS OPTIONS Access through your institution Subscribe to this journal Receive 12 print issues and online access $209.00 per year only $17.42 per issue Learn more Buy this article *
Purchase on SpringerLink * Instant access to full article PDF Buy now Prices may be subject to local taxes which are calculated during checkout ADDITIONAL ACCESS OPTIONS: * Log in * Learn
about institutional subscriptions * Read our FAQs * Contact customer support SIMILAR CONTENT BEING VIEWED BY OTHERS UBIQUITINATION OF RIPK1 REGULATES ITS ACTIVATION MEDIATED BY TNFR1 AND
TLRS SIGNALING IN DISTINCT MANNERS Article Open access 11 December 2020 RIPK1 DEPHOSPHORYLATION AND KINASE ACTIVATION BY PPP1R3G/PP1Γ PROMOTE APOPTOSIS AND NECROPTOSIS Article Open access 03
December 2021 RIPK3 SIGNALING AND ITS ROLE IN REGULATED CELL DEATH AND DISEASES Article Open access 29 April 2024 REFERENCES * Vanden Berghe, T., Linkermann, A., Jouan-Lanhouet, S.,
Walczak, H. & Vandenabeele, P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. _Nat. Rev. Mol. Cell Biol._ 15, 135–147 (2014). CAS PubMed Google Scholar
* Galluzzi, L. et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. _Cell Death Differ._ 22, 58–73 (2015). CAS PubMed Google Scholar * Mocarski, E.
S., Kaiser, W. J., Livingston-Rosanoff, D., Upton, J. W. & Daley-Bauer, L. P. True grit: programmed necrosis in antiviral host defense, inflammation, and immunogenicity. _J. Immunol._
192, 2019–2026 (2014). CAS PubMed Google Scholar * Linkermann, A. & Green, D. R. Necroptosis. _N. Engl. J. Med._ 370, 455–465 (2014). CAS PubMed PubMed Central Google Scholar *
Murphy, J. M. & Silke, J. Ars Moriendi; the art of dying well—new insights into the molecular pathways of necroptotic cell death. _EMBO Rep._ 15, 155–164 (2014). CAS PubMed PubMed
Central Google Scholar * Cho, Y. S. et al. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. _Cell_ 137, 1112–1123
(2009). CAS PubMed PubMed Central Google Scholar * He, S. et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. _Cell_ 137, 1100–1111 (2009). CAS
PubMed Google Scholar * Zhang, D. W. et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. _Science_ 325, 332–336 (2009). CAS PubMed
Google Scholar * Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. _Cell_ 148, 213–227 (2012). CAS PubMed Google Scholar *
Murphy, J. M. et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. _Immunity_ 39, 443–453 (2013). CAS PubMed Google Scholar * Wu, J. et al. Mlkl knockout
mice demonstrate the indispensable role of Mlkl in necroptosis. _Cell Res._ 23, 994–1006 (2013). CAS PubMed PubMed Central Google Scholar * Remijsen, Q. et al. Depletion of RIPK3 or MLKL
blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. _Cell Death Dis._ 5, e1004 (2014). CAS PubMed PubMed Central Google Scholar * Li, J. et
al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. _Cell_ 150, 339–350 (2012). CAS PubMed PubMed Central Google Scholar *
Dondelinger, Y. et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. _Cell Rep._ 7, 971–981 (2014). CAS PubMed Google Scholar * Wang, H. et al.
Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. _Mol. Cell_ 54, 133–146 (2014). CAS PubMed Google Scholar * Cai, Z. et al.
Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. _Nat. Cell Biol._ 16, 55–65 (2014). CAS PubMed Google Scholar * Chen, X. et al.
Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. _Cell Res._ 24, 105–121 (2014). CAS PubMed Google Scholar * Wu, X. N. et al.
Distinct roles of RIP1–RIP3 hetero- and RIP3–RIP3 homo-interaction in mediating necroptosis. _Cell Death Differ._ 21, 1709–1720 (2014). CAS PubMed PubMed Central Google Scholar * Cook,
W. D. et al. RIPK1- and RIPK3-induced cell death mode is determined by target availability. _Cell Death Differ._ 21, 1600–1612 (2014). CAS PubMed PubMed Central Google Scholar * Chen, W.
et al. Ppm1b negatively regulates necroptosis through dephosphorylating Rip3. _Nat. Cell Biol._ 17, 434–444 (2015). CAS PubMed PubMed Central Google Scholar * Ahmed, S. F. et al. The
chaperone-assisted E3 ligase C terminus of Hsc70-interacting protein (CHIP) targets PTEN for proteasomal degradation. _J. Biol. Chem._ 287, 15996–16006 (2012). CAS PubMed PubMed Central
Google Scholar * Esser, C., Scheffner, M. & Hohfeld, J. The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. _J. Biol. Chem._ 280,
27443–27448 (2005). CAS PubMed Google Scholar * Ko, H. R. et al. P42 Ebp1 regulates the proteasomal degradation of the p85 regulatory subunit of PI3K by recruiting a chaperone-E3 ligase
complex HSP70/CHIP. _Cell Death Dis._ 5, e1131 (2014). CAS PubMed PubMed Central Google Scholar * Lee, E. W., Seo, J., Jeong, M., Lee, S. & Song, J. The roles of FADD in extrinsic
apoptosis and necroptosis. _BMB Rep._ 45, 496–508 (2012). CAS PubMed Google Scholar * Vanden Berghe, T., Kalai, M., van Loo, G., Declercq, W. & Vandenabeele, P. Disruption of HSP90
function reverts tumor necrosis factor-induced necrosis to apoptosis. _J. Biol. Chem._ 278, 5622–5629 (2003). CAS PubMed Google Scholar * Park, S. Y., Shim, J. H. & Cho, Y. S.
Distinctive roles of receptor-interacting protein kinases 1 and 3 in caspase-independent cell death of L929. _Cell Biochem. Funct._ 32, 62–69 (2014). CAS PubMed Google Scholar *
Petrucelli, L. et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. _Hum. Mol. Genet._ 13, 703–714 (2004). CAS PubMed Google Scholar * Mollah, S. et al.
Targeted mass spectrometric strategy for global mapping of ubiquitination on proteins. _Rapid Commun. Mass Spectrom._ 21, 3357–3364 (2007). CAS PubMed Google Scholar * Kim, W. et al.
Systematic and quantitative assessment of the ubiquitin-modified proteome. _Mol. Cell_ 44, 325–340 (2011). CAS PubMed PubMed Central Google Scholar * Sahara, N. et al. _In vivo_ evidence
of CHIP up-regulation attenuating tau aggregation. _J. Neurochem._ 94, 1254–1263 (2005). CAS PubMed Google Scholar * Gunther, C. et al. Caspase-8 regulates TNF-α-induced epithelial
necroptosis and terminal ileitis. _Nature_ 477, 335–339 (2011). PubMed PubMed Central Google Scholar * Kaiser, W. J. et al. RIP3 mediates the embryonic lethality of caspase-8-deficient
mice. _Nature_ 471, 368–372 (2011). CAS PubMed PubMed Central Google Scholar * Oberst, A. et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis.
_Nature_ 471, 363–367 (2011). CAS PubMed PubMed Central Google Scholar * Welz, P. S. et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation.
_Nature_ 477, 330–334 (2011). CAS PubMed Google Scholar * Zhang, H. et al. Functional complementation between FADD and RIP1 in embryos and lymphocytes. _Nature_ 471, 373–376 (2011). CAS
PubMed PubMed Central Google Scholar * Dillon, C. P. et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. _Cell_ 157, 1189–1202 (2014). CAS PubMed PubMed
Central Google Scholar * Newton, K. et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. _Science_ 343, 1357–1360 (2014). CAS PubMed Google
Scholar * Rickard, J. A. et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. _Cell_ 157, 1175–1188 (2014). CAS PubMed Google Scholar * Dannappel,
M. et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. _Nature_ 513, 90–94 (2014). CAS PubMed PubMed Central Google Scholar * Takahashi, N. et al.
RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. _Nature_ 513, 95–99 (2014). CAS PubMed Google Scholar * Mandal, P. et al. RIP3 induces apoptosis
independent of pronecrotic kinase activity. _Mol. Cell_ 56, 481–495 (2014). CAS PubMed PubMed Central Google Scholar * Orozco, S. et al. RIPK1 both positively and negatively regulates
RIPK3 oligomerization and necroptosis. _Cell Death Differ._ 21, 1511–1521 (2014). CAS PubMed PubMed Central Google Scholar * Duprez, L. et al. RIP kinase-dependent necrosis drives lethal
systemic inflammatory response syndrome. _Immunity_ 35, 908–918 (2011). CAS PubMed Google Scholar * Onizawa, M. et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the
kinase RIPK3 and protects cells from necroptosis. _Nat. Immunol._ 16, 618–627 (2015). CAS PubMed PubMed Central Google Scholar * Petersen, S. L. et al. TRAF2 is a biologically important
necroptosis suppressor. _Cell Death Differ._ 22, 1846–1857 (2015). CAS PubMed PubMed Central Google Scholar * Kang, T. B., Yang, S. H., Toth, B., Kovalenko, A. & Wallach, D.
Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. _Immunity_ 38, 27–40 (2013). CAS PubMed Google Scholar * Lawlor, K. E. et al. RIPK3 promotes cell death and
NLRP3 inflammasome activation in the absence of MLKL. _Nat. Commun._ 6, 6282 (2015). CAS PubMed Google Scholar * Vince, J. E. et al. Inhibitor of apoptosis proteins limit RIP3
kinase-dependent interleukin-1 activation. _Immunity_ 36, 215–227 (2012). CAS PubMed Google Scholar * Moriwaki, K. et al. The Necroptosis Adaptor RIPK3 promotes injury-induced cytokine
expression and tissue repair. _Immunity_ 41, 567–578 (2014). CAS PubMed PubMed Central Google Scholar * Humphries, F., Yang, S., Wang, B. & Moynagh, P. N. RIP kinases: key decision
makers in cell death and innate immunity. _Cell Death Differ._ 22, 225–236 (2015). CAS PubMed Google Scholar * Silke, J., Rickard, J. A. & Gerlic, M. The diverse role of RIP kinases
in necroptosis and inflammation. _Nat. Immunol._ 16, 689–697 (2015). CAS PubMed Google Scholar * Min, J. N. et al. CHIP deficiency decreases longevity, with accelerated aging phenotypes
accompanied by altered protein quality control. _Mol. Cell. Biol._ 28, 4018–4025 (2008). CAS PubMed PubMed Central Google Scholar * Vanden Berghe, T. et al. Passenger mutations confound
interpretation of all genetically modified congenic mice. _Immunity_ 43, 200–209 (2015). CAS PubMed Google Scholar * Schisler, J. C. et al. CHIP protects against cardiac pressure overload
through regulation of AMPK. _J. Clin. Invest._ 123, 3588–3599 (2013). CAS PubMed PubMed Central Google Scholar * Lee, I. H. et al. Ahnak functions as a tumor suppressor via modulation
of TGF_β_/Smad signaling pathway. _Oncogene_ 33, 4675–4684 (2014). CAS PubMed PubMed Central Google Scholar * Oh, W. et al. PML-IV functions as a negative regulator of telomerase by
interacting with TERT. _J Cell Sci._ 122, 2613–2622 (2009). CAS PubMed Google Scholar * Lee, E. W. et al. USP11-dependent selective cIAP2 deubiquitylation and stabilization determine
sensitivity to Smac mimetics. _Cell Death Differ._ 22, 1463–1476 (2015). CAS PubMed PubMed Central Google Scholar * Lee, E. W. et al. Differential regulation of p53 and p21 by MKRN1 E3
ligase controls cell cycle arrest and apoptosis. _EMBO J._ 28, 2100–2113 (2009). CAS PubMed PubMed Central Google Scholar * Lee, E. W. et al. Ubiquitination and degradation of the FADD
adaptor protein regulate death receptor-mediated apoptosis and necroptosis. _Nat. Commun._ 3, 978 (2012). PubMed Google Scholar Download references ACKNOWLEDGEMENTS We thank S. Murata for
the _Chip_−/− mice and V. M. Dixit for the _Ripk3_−/− mice. We thank the Cancer Metabolism Interest Group (cMIG) led by NCC Korea for discussion and advice. This research was supported by a
grant from the National R&D Program for Cancer Control, Ministry of Health & Welfare, Republic of Korea (NCC-1420300) (to J.Song) and by a grant from the National Research Foundation
of Korea (NRF) funded by the Ministry of Education (2012R1A6A3A04040105) (to E.-W.L.) and by the Ministry of Science, ICT and Future Planning (NRF-2015R1A3A2066581) (to J.Song). Research in
the Vandenabeele group is supported by Flemish grants (Research Foundation Flanders: FWO G.0875.11, FWO G.0973.11, FWO G.0A45.12N, FWO G.0787.13N, FWO G0E04.16N), a Methusalem grant
(BOF16/MET_V/007), Ghent University grants (MRP, GROUP-ID consortium), a grant from the Foundation against Cancer (F94), and grants from VIB. Additionally, this research was partly supported
by the BK21 Plus project of the National Research Foundation of Korea Grant (to J.Seo, M.J., H.-K.L., D.S., J.-H.K. and S.Y.H.) and by the grants for KMPC (Korea Mouse Phenotype Center) (to
J.K.S.) from National Research Foundation of Korea (NRF), and C.L. acknowledges institutional support by KIST. AUTHOR INFORMATION Author notes * Jinho Seo and Eun-Woo Lee: These authors
contributed equally to this work. AUTHORS AND AFFILIATIONS * Department of Biochemistry, College of Life Science and Biotechnology, Yonsei University, Seoul 120-749, Korea Jinho Seo, Eun-Woo
Lee, Daehyeon Seong, Jihye Shin, Manhyung Jeong, Hae-Kyung Lee, Jung-Hoon Kim, Su Yeon Han & Jaewhan Song * Laboratory of Developmental Biology and Genetics, College of Veterinary
Medicine, BK21Plus Program for Creative Veterinary Science Research, Interdisciplinary Program for Bioinformatics, BIO-MAX institute, Korea Mouse Phenotyping Center, Seoul National
University, Seoul 151-742, South Korea Hyerim Sung & Je Kyung Seong * Department of Biomedical Molecular Biology, Ghent University, Technologiepark 927, 9052 Zwijnaarde-Ghent, Belgium
Yves Dondelinger & Peter Vandenabeele * Inflammation Research Center, VIB, Technologiepark 927, 9052 Zwijnaarde-Ghent, Belgium Yves Dondelinger & Peter Vandenabeele * BRI, Korea
Institute of Science and Technology, Seoul 136-791, Korea Jihye Shin & Cheolju Lee Authors * Jinho Seo View author publications You can also search for this author inPubMed Google
Scholar * Eun-Woo Lee View author publications You can also search for this author inPubMed Google Scholar * Hyerim Sung View author publications You can also search for this author inPubMed
Google Scholar * Daehyeon Seong View author publications You can also search for this author inPubMed Google Scholar * Yves Dondelinger View author publications You can also search for this
author inPubMed Google Scholar * Jihye Shin View author publications You can also search for this author inPubMed Google Scholar * Manhyung Jeong View author publications You can also
search for this author inPubMed Google Scholar * Hae-Kyung Lee View author publications You can also search for this author inPubMed Google Scholar * Jung-Hoon Kim View author publications
You can also search for this author inPubMed Google Scholar * Su Yeon Han View author publications You can also search for this author inPubMed Google Scholar * Cheolju Lee View author
publications You can also search for this author inPubMed Google Scholar * Je Kyung Seong View author publications You can also search for this author inPubMed Google Scholar * Peter
Vandenabeele View author publications You can also search for this author inPubMed Google Scholar * Jaewhan Song View author publications You can also search for this author inPubMed Google
Scholar CONTRIBUTIONS J.Seo, E.-W.L. and J.Song conceived and designed the study. J.Seo, E.-W.L., D.S. and M.J. performed most of the experiments. J.Shin and C.L. performed the mass
spectrometry analysis. J.Seo, H.S., D.S., H.-K.L., J.-H.K., S.Y.H. and J.K.S. performed the animal experiments and analysis. Y.D. and P.V. discussed the results, conceived some experiments,
and provided critical reagents and comments. J.Seo, E.-W.L. and J.Song wrote the manuscript. CORRESPONDING AUTHOR Correspondence to Jaewhan Song. ETHICS DECLARATIONS COMPETING INTERESTS The
authors declare no competing financial interests. INTEGRATED SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURE 1 ABLATION OF CHIP INCREASES RIPK3-MEDIATED NECROPTOSIS IN HUMAN HT-29 AND MOUSE
L929 CELLS. (A) HT-29 cells were transfected with the indicated siRNAs for 48 h and then treated with 30 ng/mL human TNF_α_ (T), 5 μg/mL cycloheximide (C), and 30 μM z-VAD-fmk (Z) in the
presence or absence of 30 μM Nec-1 (N) for 8 h. The cells were stained with Annexin V and 7-AAD, and then analysed by FACS. Representative plots of data are shown in left panel, and means ±
S.D. from _n_ = 3 independent experiments are presented in right panel. (B,C) HT-29 cells were transfected with siRNA against CHIP and/or RIPK3 and treated as in (A) for 10 h. Knockdown
efficiency was confirmed by WB (B) and necrotic cells were detected by FACS (C). Representative plots of data are shown in left panel, and means ± S.D. from _n_ = 3 independent experiments
are presented in right panel (C). (D) L929 cells were transfected with the indicated siRNAs for 48 h and then treated with 5 ng/mL TNF_α_ and 10 μM z-VAD-fmk in the presence or absence of
Nec-1 for 2.5 h. The cells were stained with Annexin V and analysed by FACS. Representative plots of data from 3 independent experiments are shown in left panel and summarized in right
panel. (E) L929 cells were transfected and treated as described in (D). Cell loss were determined using the Cell Titer Glo Luminescent Cell Viability Assay Kit. (F–H) L929 cells were
transfected with the indicated siRNAs and treated with TNF_α_ and z-VAD. The knockdown efficiencies of each siRNAs were shown by WB (F). Necrotic cells were determined by Annexin V staining
and analysed by FACS (G). Cell loss was also determined as described above (H). (I,J) L929 cells stably expressing retroviral human CHIP (human CHIP shares all amino acids except for one
amino acid with mouse CHIP) were transfected with the indicated siRNAs for 48 h. Cells were then treated with or without TNF_α_ and z-VAD for 2.5 h. Expression of CHIP was analysed by WB
(I). Cell death was determined by Annexin V staining (J). Data in (D,E,G,H,J) are means ± S, D.; _n_ = 3 independent experiments (J). _P_ values were determined by unpaired two-tailed
Student’s _t_-test. (*_P_ < 0.05, **_P_ < 0.01, ***_P_ < 0.001). Raw data from independent experiments are provided in Supplementary Table 1. SUPPLEMENTARY FIGURE 2 CHIP DOES NOT
AFFECT TNF-MEDIATED APOPTOSIS. (A) Primary _Chip_+/+ and _Chip_−/−MEFs were treated as indicated (T: mTNF_α_, 30 ng/mL; C: Cycloheximide, 2 μg/mL; Z: z-VAD-fmk, 30 μM; N: necrostatin-1, 30
μM) for 6 h or 12 h. The cells were stained with Annexin V and 7-AAD, and then analysed by FACS. Representative plots of data from _n_ = 3 independent experiments are shown in left panel,
and means ± S.D. are presented in right panel. (B) L929 cells were transfected with CHIP siRNA and treated as indicated for 2.5 h. Dead cells were determined by FACS analysis using Annexin V
and 7-AAD staining. Representative plots of data from _n_ = 3 independent experiments are shown in left panel, and means ± S.D. are presented in right panel. (C) HT-29 cells were
transfected with CHIP siRNA and treated as indicated (T: hTNF_α_, 30 ng/mL; C: Cycloheximide, 5 μg/mL; Z: z-VAD-fmk, 30 μM; N: necrostatin-1, 30 μM) for 10 h or 12 h. The cells were stained
with Annexin V and 7-AAD, and then analysed via FACS. Representative plots of data from _n_ = 3 independent experiments are shown in left panel, and means ± S.D. are presented in right
panel. _P_ values were determined by unpaired two-tailed Student’s _t_-test. (*_P_ < 0.05, **_P_ < 0.01, ***_P_ < 0.001). Raw data from independent experiments are provided in
Supplementary Table 1. SUPPLEMENTARY FIGURE 3 CHIP DEPLETION FACILITATES NECROSOME FORMATION BY STABILISING RIPK3 AND RIPK1 PROTEINS. (A,B) HT-29 and L929 cells transfected with the siRNA
against human CHIP or mouse CHIP for 48 h, respectively. HT-29 and L929 cells were then treated with hTNF, CHX, and zVAD (TCZ) or mTNF and CHX (TZ), respectively, as indicated. Cells were
lysed with lysis buffer and immunoprecipitated with anti-Caspase-8 (A) or FADD (B). Associated proteins were detected by WB using the indicated antibodies. (C–E) HT-29 and L929 cells were
transfected with control siRNA or siCHIP for 48 h. Expression of cell death related proteins were determined by WB in HT-29 and L929 cells with CHIP knockdown (C). Relative amounts of RIPK3
and RIPK1 were calculated and shown after normalising to Actin. mRNA levels were also measured by qRT-PCR (D,E). The data represent means ± S.D.; _n_ = 3 independent experiments; **_P_ <
0.01, ***_P_ < 0.001 (D, E). (F) The stability of RIPK1 and RIPK3 in CHIP-depleted L929 cells was measured by treating the cells with cycloheximide for the indicated period, followed by
WB analysis. Representative results of three independent experiments are shown. Relative amounts of RIPK3 and RIPK1 were calculated after normalising to Actin, and are shown in the graph.
Half-lives of RIPK3 and RIPK1 were also calculated and presented in the graph. SUPPLEMENTARY FIGURE 4 CHIP BINDS TO THE KINASE AND RHIM DOMAINS OF RIPK3 AND RIPK1, RESPECTIVELY. (A,B) 293FT
cells were transfected with plasmids expressing FLAG–CHIP and HA–RIPK3 or HA–RIPK1. The cell lysates were immunoprecipitated using an anti-FLAG or -HA antibodies, followed by WB using
anti-RIPK3, -RIPK1 and -FLAG antibodies. (C,D) L929 cells were immunoprecipitated using an anti-RIPK3 (C) or –RIPK1 (D) antibody. Interactions between endogenous CHIP and RIPK3 or RIPK1 were
then analysed by WB. (E,F) Bacterially produced GST and GST-CHIP were used for the _in vitro_ binding analysis. HA-RIPK3 or HA-RIPK1 was produced using the TNT T7 reticulocyte system. GST
or GST-CHIP was incubated with HA-RIPK3 or HA-RIPK1, and the sample was precipitated using Glutathione Sepharose beads. The interaction between CHIP and RIPKs was analysed by WB using
anti-RIPK3 and anti-RIPK1 antibodies. The asterisk indicates N-terminal cleaved form of RIPK3 because anti-HA antibody did not detect the lower band. Only full-length RIPK3 bound to
GST-CHIP, suggesting the N-terminal region of RIPK3 is required for the interaction with CHIP. (G,H) 293FT cells were transfected with the indicated plasmids. The cell lysates were
immunoprecipitated using an anti-Myc antibody, and the interaction between these two proteins was analysed by WB. (I,J) 293FT cells were transfected with a combination of plasmids expressing
of CHIP deficient mutants and RIPK3 or RIPK1. The cell lysates were immunoprecipitated using the indicated antibodies and then analysed via western blotting. (K,L) 293FT cells were
transfected with a combination of CHIP wild type and RIPK3 or RIPK1 domain expressing plasmids and then immunoprecipitated using the indicated antibodies. The interaction between the
peptides was analysed via western blotting using the indicated antibodies. (M) A schematic model of the interactions among RIPK1, RIPK3 and CHIP. SUPPLEMENTARY FIGURE 5 CHIP IS NOT INVOLVED
IN THE GELDANAMYCIN-MEDIATED DEGRADATION OF RIPK3 OR RIPK1 AND INDUCES THE DEGRADATION OF MUTANTS RIPK3 AND RIPK1 DEFECTIVE IN AGGRESOME FORMATION. (A) L929 cells were transfected with
siCtrl or siCHIP and then treated with 1 μM geldanamycin for 16 h. The proteins were detected by WB using the indicated antibodies. (B,C) H1299 cells were co-transfected with plasmids
expressing of FLAG-CHIP, HA-RIPK3 or HA-RIPK1 with GST, a transfection control, and then treated with geldanamycin for 12 h. The cells were analysed by WB. (D) H1299 cells were transfected
with GFP-RIPK3 or GFP-RIPK1 in the absence or presence of FLAG-CHIP or FLAG-CHIP H260Q mutant and then treated with E64d + Pepstatin A for 16 h. The lysosomes and nucleus were stained using
LysoTracker (red) and DAPI (blue). The cells were analysed via fluorescence microscopy. (E) H1299 cells were transfected with GFP-RIPK3 or GFP-RIPK1 in the absence or presence of WT or H260Q
mutant of FLAG-CHIP and then treated with E64d-Pepstatin A. The cells were stained with an anti-FLAG antibody (red). (F) H1299 cells were transfected with GFP-RIPK3 or GFP-RIPK1 in the
presence of FLAG-CHIP WT or mutant and then treated with E64d-Pepstatin A. Cells were stained with LysoTracker (red) and an anti-FLAG antibody (blue). (G) H1299 cells were transfected with
the indicated plasmids and then treated with E64d-Pepstatin A. Cells were stained with LysoTracker (red) and an anti-FLAG antibody (blue). (H) H1299 cells were transfected with HA-RIPK3
V460P in the absence and presence of FLAG-CHIP, followed by WB using anti-HA, -FLAG and-GST antibodies. (I) H1299 cells were transfected with HA-RIPK1 I539D in the absence and presence of
FLAG-CHIP, followed by WB using anti-HA, -FLAG and-GST antibodies. In D–G, a representative image is shown in the Fig. 5 and means from two independent experiments are presented in the
graph. In each independent experiment, _n_ = 100 cells were counted. All scale bars are 10 μm. SUPPLEMENTARY FIGURE 6 LYSINES OF 55 AND 363 OF RIPK3 ARE RESPONSIBLE TO CHIP DEPLETION-INDUCED
RIPK3 UP-REGULATION. (A) 293FT cells were transfected with plasmids expressing the HA-RIPK3 WT or 4KR, K55⋅363R, K89⋅501R mutants in the absence or presence of Myc-CHIP. The lysates were
immunoprecipitated using an anti-Myc antibody and then analysed via western blotting. (B) H1299 cells were transfected with HA-RIPK3 WT or K55⋅363R in the absence and presence of FLAG-CHIP,
and treated with CHX as indicated. The proteins were detected by WB using anti-HA, -FLAG, -Actin antibodies, quantified by Image J, and are shown in the graph. Half-life of RIPK3 are
presented in the graph. (C) H1299 cells were transfected with plasmids expressing GFP/RIPK3 K55⋅363R or K89⋅501R mutants in the absence or presence of FLAG-CHIP. Transfected cells were
treated with E64d-Pepstatin A and LysoTracker for lysosome staining. Stained cells were analysed using fluorescence microscopy. A representative image is shown in the upper panel and means
from two independent experiments are presented in the lower panel. In each independent experiment, _n_ = 100 cells were counted. (D) Degradation of WT and single point mutant RIPK3 by CHIP
was determined by WB in H1299 cells. (E,F) HeLa cells stably transfected with pBabe-RIPK3 WT, K55⋅363R and an empty vector were transfected with the indicated siRNAs for 48 h. Cells were
treated with 10 ng/mL hTNF_α_, 5 μg/mL cycloheximide, and 20 μM z-VAD-fmk (TCZ) for 4 h (E). Cell loss was determined by Cell Titer Glo Luminescent Cell Viability Assay Kit (E), and protein
levels were determined by western blot (F). Data are means ± S.D. _n_ = 3 independent experiments (*_P_ < 0.05, **_P_ < 0.01). Raw data from independent experiments are provided in
Supplementary Table 1. All scale bars are 10 μm. SUPPLEMENTARY FIGURE 7 THE K571⋅604⋅627R (3KR) RIPK1 ARE RESISTANT TO DEGRADATION AND LYSOSOMAL LOCALISATION BY CHIP. (A) H1299 cells were
transfected with HA-RIPK1 mutants with or without FLAG-CHIP as indicated. Degradation of each mutant by CHIP were determined by WB. (B) The amino acid sequence alignment of human and mouse
RIPK1. (C) 293FT cells were transfected with plasmids expressing the HA-RIPK1 WT or 3KR mutant in the absence or presence of Myc-CHIP. The lysates were immunoprecipitated using an anti-Myc
antibody and then analysed via western blotting. (D) 293FT cells transfected with the plasmid expression HA-RIPK1 (WT and 3KR) and His-Ub with or without FLAG-CHIP in the presence and
absence of E64d-Pepstatin A. A pull-down assay was performed using Ni2+-NTA beads. The level of RIPK1 ubiquitylation was determined by WB using an anti-RIPK1 antibody. (E) H1299 cells were
transfected with a combination of GFP/RIPK1 WT or 3KR and FLAG-CHIP and were treated with E64d/Pepstatin A and LysoTracker. The transfected cells were stained using an anti-FLAG antibody and
then analysed via fluorescence microscopy. A representative image is shown in the left panel and means from two independent experiments are presented in the right panel. In each independent
experiment, _n_ = 100 cells were counted. All scale bars are 10 μm. SUPPLEMENTARY FIGURE 8 THE ANALYSIS OF _CHIP_−/− AND CHIP−/− RIPK3−/− MICE IN SPLEEN, THYMUS AND SMALL INTESTINE. (A)
Genomic DNA was prepared via phenol-extraction. The genotypes were determined via PCR using specific primers for each gene. (B) The expected and observed frequencies and the ratio of each
genotype in the littermates from the crosses of _Chip_+/− x _Chip_+/− mice and _Chip_+/−_Ripk3_−/− x _Chip_+/−_Ripk3_−/− mice. (C) Representative spleen pictures and weight of WT (_n_ = 13;
male = 4, female = 9), _Chip_−/− (_n_ = 8; male = 3, female = 5) and DKO (_n_ = 9; male = 3, female = 6) at 4 weeks of age. Data are means ± S.D.; _n_ = number of mice analysed; *_P_ <
0.05. Raw data are presented in Supplementary Table 1. (D) Representative pictures of WT (_n_ = 3; female = 3) and DKO (_n_ = 3; female = 3) thymus and spleen at 16 weeks of age. n means
number of mice analysed. (E) Representative H&E-stained of sections of WT and DKO small intestine at 16 weeks of age (_n_ = 3 for each genotype). Scale bars, 100 μm. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY INFORMATION Supplementary Information (PDF 18363 kb) SUPPLEMENTARY TABLE 1 Supplementary Information (XLSX 45 kb) RIGHTS AND PERMISSIONS Reprints and permissions
ABOUT THIS ARTICLE CITE THIS ARTICLE Seo, J., Lee, EW., Sung, H. _et al._ CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. _Nat Cell Biol_ 18,
291–302 (2016). https://doi.org/10.1038/ncb3314 Download citation * Received: 18 September 2015 * Accepted: 18 January 2016 * Published: 22 February 2016 * Issue Date: March 2016 * DOI:
https://doi.org/10.1038/ncb3314 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently
available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative