- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Access through your institution Buy or subscribe The receptor tyrosine kinase (RTK) FLT3 (Fms-like tyrosine kinase-3) is essential for the proliferation, differentiation and survival of
hematopoietic cells.1, 2 Mutations in the gene for _FLT3_ have been described in acute myeloid leukemia (AML) as internal tandem duplications (ITD) localized in the juxtamembrane (JM) domain
and _FLT3_ tyrosine kinase domain (TKD) mutations amounting to 2–30% and 7–10% of patients, respectively. In patients with AML, _FLT3_-ITD confers an unfavorable prognosis.3 In AML,
recently discovered _FLT3_ mutations involve point mutations as well as short deletions in the JM domain, highlighting the importance for a deeper understanding and further studies of these
aberrations.4, 5 The FLT3 JM domain (Supplementary Figure 1a) is indeed the critical regulator region of receptor autoinhibition.6 Kiyoi _et al._7 had shown constitutive activation of the
FLT3 receptor by deletion of a stretch of amino acids in the JM domain, generally duplicated in FLT3-ITD. Transient transfection into HEK293T cells confirmed activation of associated
signaling proteins by several deletion mutants. Even deletion of just one amino acid led to the activation of downstream signaling (Supplementary Figures 1b and c). When retrovirally
expressed in the IL3-dependent pro-B cell line Ba/F3, the _FLT3_ Stop and delLeu (Supplementary Table 1, Id 3 and Id 2, respectively) mutants were not further studied in this work, as we
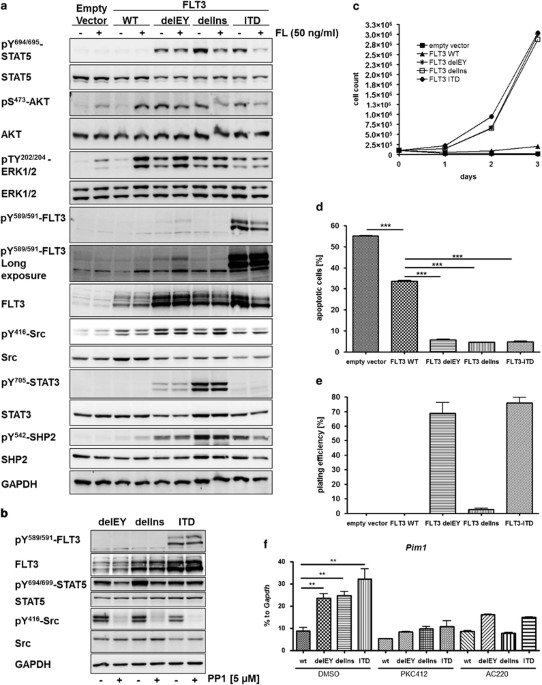
focused on activating mutations in the JM domain. The delEY and the delIns mutants led to constitutive activation of STAT3, ERK1/2, SFK (Src family kinases), SHP2 and AKT (Figure 1a).
Similar to _FLT3_-ITD, they also activated STAT5. As seen in HEK293T cells, only FLT3-ITD induced upregulated phosphorylation of Y589/Y591. These tyrosines were previously thought to be
essential for STAT5 activation and transforming capacity of the RTK.9 Our data show that STAT5 was partly activated by SFKs in deletion mutant expressing cells (Figure 1b), whereas JAK2
inhibition had no effect on STAT5 phosphorylation (Supplementary Figure 2a). A similarity between FLT3 deletion mutants and the TKD mutant _FLT3_ D835Y (p.Asp835Tyr) is conceivable, as both
do not show an increase in Y589/Y591 phosphorylation.9 Nevertheless, the missing phosphorylation of Y589/591 was associated with low STAT5 activation in D835Y expressing cells but not for
_FLT3_ deletion mutants (Figure 1a). Immunoprecipitation experiments confirmed the phosphorylation of additional tyrosines independent from Y598/Y591 in the deletion mutants, as was already
shown for _FLT3_-ITD (Supplementary Figure 2b).9 FLT3 ligand stimulation of the WT receptor led to the phosphorylation of ERK1/2 and AKT but not of STAT5. Interestingly, STAT3 was not
activated by _FLT3_-ITD, suggesting that the deletion mutants and ITD induced different patterns of downstream signaling. These results were in line with those of Frohling _et al._10 of
STAT3 phosphorylation driven by FLT3 JM point mutations. We found that endoplasmic reticulum targeting or hypoglycosylation of FLT3 delEY mutant by brefeldin A and tunicamycin treatment,
respectively, clearly diminished STAT5 and STAT3 phosphorylation, whereas there was no change with FLT3-ITD (Supplementary Figure 2c), as published.11 The same was seen for the delIns mutant
only with tunicamycin treatment. All mutants exhibited decreased phosphorylation of AKT after brefeldin A and tunicamycin treatment, confirming receptor surface expression as a prerequisite
for PI3K/AKT pathway activation. This is a preview of subscription content, access via your institution ACCESS OPTIONS Access through your institution Subscribe to this journal Receive 12
print issues and online access $259.00 per year only $21.58 per issue Learn more Buy this article * Purchase on SpringerLink * Instant access to full article PDF Buy now Prices may be
subject to local taxes which are calculated during checkout ADDITIONAL ACCESS OPTIONS: * Log in * Learn about institutional subscriptions * Read our FAQs * Contact customer support
REFERENCES * McKenna HJ, Stocking KL, Miller RE, Brasel K, De Smedt T, Maraskovsky E _et al_. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells,
dendritic cells, and natural killer cells. _Blood_ 2000; 95: 3489–3497. CAS PubMed Google Scholar * Mackarehtschian K, Hardin JD, Moore KA, Boast S, Goff SP, Lemischka IR . Targeted
disruption of the flk2/flt3 gene leads to deficiencies in primitive hematopoietic progenitors. _Immunity_ 1995; 3: 147–161. Article CAS Google Scholar * Kayser S, Schlenk RF, Londono MC,
Breitenbuecher F, Wittke K, Du J _et al_. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome.
_Blood_ 2009; 114: 2386–2392. Article CAS Google Scholar * Reindl C, Bagrintseva K, Vempati S, Schnittger S, Ellwart JW, Wenig K _et al_. Point mutations in the juxtamembrane domain of
FLT3 define a new class of activating mutations in AML. _Blood_ 2006; 107: 3700–3707. Article CAS Google Scholar * Deeb KK, Smonskey MT, DeFedericis H, Deeb G, Sait SNJ, Wetzler M _et
al_. Deletion and deletion/insertion mutations in the juxtamembrane domain of the FLT3 gene in adult acute myeloid leukemia. _Leuk Res Rep_ 2014; 3: 86–89. PubMed PubMed Central Google
Scholar * Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F _et al_. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. _Mol Cell_ 2004; 13: 169–178. Article
CAS Google Scholar * Kiyoi H, Ohno R, Ueda R, Saito H, Naoe T . Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. _Oncogene_
2002; 21: 2555–2563. Article CAS Google Scholar * Li L, Bailey E, Greenblatt S, Huso D, Small D . Loss of the wild-type allele contributes to myeloid expansion and disease aggressiveness
in FLT3/ITD knockin mice. _Blood_ 2011; 118: 4935–4945. Article CAS Google Scholar * Rocnik JL, Okabe R, Yu J, Lee BH, Giese N, Schenkein DP _et al_. Roles of tyrosine 589 and 591 in
STAT5 activation and transformation mediated by FLT3-ITD. _Blood_ 2006; 108: 1339–1345. Article CAS Google Scholar * Fröhling S, Scholl C, Levine RL, Loriaux M, Boggon TJ, Bernard OA _et
al_. Identification of driver and passenger mutations of FLT3 by high-throughput DNA sequence analysis and functional assessment of candidate alleles. _Cancer Cell_ 2007; 12: 501–513.
Article Google Scholar * Choudhary C, Olsen JV, Brandts C, Cox J, Reddy PNG, Böhmer FD _et al_. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. _Mol Cell_
2009; 36: 326–339. Article CAS Google Scholar * Rahmani M, Aust MM, Attkisson E, Williams DC, Ferreira-Gonzalez A, Grant S . Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K
inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. _Cancer Res_ 2013; 73: 1340–1351. Article CAS Google Scholar * Armstrong SA,
Mabon ME, Silverman LB, Li A, Gribben JG, Fox EA . FLT3 mutations in childhood acute lymphoblastic leukemia. _Blood_ 2004; 103: 3544–3546. Article CAS Google Scholar * Chang P, Kang M,
Xiao A, Chang J, Feusner J, Buffler P _et al_. FLT3 mutation incidence and timing of origin in a population case series of pediatric leukemia. _BMC Cancer_ 2010; 10: 513. Article Google
Scholar * Kim TW, Lee H, Kang Y, Choe MS, Ryu M, Chang HM _et al_. Prognostic significance of c-kit mutation in localized gastrointestinal stromal tumors. _Clin Cancer Res_ 2004; 10:
3076–3081. Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank Kristina Feldberg for expert technical assistance. We acknowledge Frank D Böhmer for providing the
pcDNA3-FLT3 (WT)-HA vector and M-D Hagel for initial cloning of vectors. This work was in part supported by the Novartis Foundation. AUTHOR CONTRIBUTION NC, RCP, GR, JR, PC, MS, TH, THB, SS
and SK designed research. NC, GR, RCP and JR performed research. NC, GR, RCP, PC, TH, SS and SK analyzed data. NC, RCP, GR, SS and SK wrote the paper. AUTHOR INFORMATION AUTHORS AND
AFFILIATIONS * Department of Hematology, Oncology, Hemostaseology and Stem Cell Transplantation, Faculty of Medicine, RWTH Aachen University, Aachen, Germany N Chatain, J Rossa, M
Schemionek, T H Brümmendorf & S Koschmieder * Computational Biophysics, German Research School for Simulation Sciences and Computational Biomedicine, Institute for Advanced Simulation
IAS-5 and Institute of Neuroscience and Medicine INM-9, Forschungszentrum Jülich, Jülich, Germany R C Perera, G Rossetti & P Carloni * Jülich Supercomputing Centre, Forschungszentrum
Jülich, Jülich, Germany G Rossetti * MLL Munich Leukemia Laboratory, Munich, Germany T Haferlach & S Schnittger Authors * N Chatain View author publications You can also search for this
author inPubMed Google Scholar * R C Perera View author publications You can also search for this author inPubMed Google Scholar * G Rossetti View author publications You can also search for
this author inPubMed Google Scholar * J Rossa View author publications You can also search for this author inPubMed Google Scholar * P Carloni View author publications You can also search
for this author inPubMed Google Scholar * M Schemionek View author publications You can also search for this author inPubMed Google Scholar * T Haferlach View author publications You can
also search for this author inPubMed Google Scholar * T H Brümmendorf View author publications You can also search for this author inPubMed Google Scholar * S Schnittger View author
publications You can also search for this author inPubMed Google Scholar * S Koschmieder View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING
AUTHOR Correspondence to S Koschmieder. ETHICS DECLARATIONS COMPETING INTERESTS SK has received honoraria for consultancy from Novartis, Pfizer, Brystol-Meyers Squibb, and Ariad. TH and SS
have equity ownership of MLL. THB has received honoraria for consultancy from Novartis, Pfizer, Brystol-Meyers Squibb. The remaining authors declare no conflict of interest. ADDITIONAL
INFORMATION Supplementary Information accompanies this paper on the Leukemia website SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION 1 (DOC 5166 KB) SUPPLEMENTARY INFORMATION 2 (DOC 146
KB) RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Chatain, N., Perera, R., Rossetti, G. _et al._ Rare FLT3 deletion mutants may provide additional
treatment options to patients with AML: an approach to individualized medicine. _Leukemia_ 29, 2434–2438 (2015). https://doi.org/10.1038/leu.2015.131 Download citation * Published: 27 May
2015 * Issue Date: December 2015 * DOI: https://doi.org/10.1038/leu.2015.131 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link
Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative