- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Lateral gene transfer by phages has contributed significantly to the genetic diversity of bacteria. To accurately determine the frequency and range of phage-mediated gene transfer,
it is important to understand the movement of DNA among microbes. Using an _in situ_ DNA amplification technique (cycling primed _in situ_ amplification-fluorescent _in situ_ hybridization;
CPRINS-FISH), we examined the propensity for phage-mediated gene transfer in freshwater environments at the single-cell level. Phage P1, T4 and isolated _Escherichia coli_ phage EC10 were
used as vectors. All _E. coli_ phages mediated gene transfer from _E. coli_ to both plaque-forming and non-plaque-forming _Enterobacteriaceae_ strains at frequencies of 0.3–8 × 10−3 per
plaque-forming unit (PFU), whereas culture methods using selective agar media could not detect transductants in non-plaque-forming strains. The DNA transfer frequencies through phage EC10
ranged from undetectable to 9 × 10−2 per PFU (undetectable to 2 × 10−3 per total direct count) when natural bacterial communities were recipients. Direct viable counting combined with
CPRINS-FISH revealed that more than 20% of the cells carrying the transferred gene retained their viability in most cases. These results indicate that the exchange of DNA sequences among
bacteria occurs frequently and in a wide range of bacteria, and may promote rapid evolution of the prokaryotic genome in freshwater environments. SIMILAR CONTENT BEING VIEWED BY OTHERS
DIVERSE AND ABUNDANT PHAGES EXPLOIT CONJUGATIVE PLASMIDS Article Open access 12 April 2024 INTERACTIONS BETWEEN BACTERIAL AND PHAGE COMMUNITIES IN NATURAL ENVIRONMENTS Article 09 August 2021
MUTATION-INDUCED INFECTIONS OF PHAGE-PLASMIDS Article Open access 12 April 2023 INTRODUCTION Bacteriophages are abundant and ubiquitous in the natural environment. They induce bacterial
mortality, contribute to the carbon cycle and also affect host diversity (Fuhrman, 1999). Moreover, they mediate gene transfer between prokaryotes. Recent whole-genome analyses suggest that
lateral gene transfer by phages has contributed significantly to the acquisition of new genetic traits, the ability of bacteria to exploit new environments and the genetic diversity of many
bacteria (Ochman et al., 2000; Brüssow et al., 2004; Bordenstein and Reznikoff, 2005; Pallen and Wren, 2007). In gene transfer by phages, phage particles accidentally incorporate a piece of
the bacterial DNA into a phage head in place of phage DNA during the propagation. As phage capsids prevent nuclease digestion, phages may serve as reservoirs for foreign genes. Potential
gene transfer via phages has been documented in soil, freshwater and marine water environments (Saye et al., 1987; Zeph et al., 1988; Jiang and Paul, 1998). In these studies, researchers
inoculated well-known or isolated phages packed with indicator genes into environmental samples and performed transduction assays using natural bacterial communities as recipients. They have
suggested that transduction could be an important mechanism for lateral gene transfer in natural environments. Culture methods using selective agar media have a leading role in the study of
transduction (Zinder and Lederberg, 1952; Ogunseitan, 2008). However, many environmental bacteria are resistant to culture on conventional media (Amann et al., 1995). In addition, the
genetic characteristics used as an indicator for transduction are found in indigenous bacteria. For instance, there are antibiotic-resistant bacteria in the natural environment; their
occurrence makes it difficult to distinguish indigenous antibiotic-resistant bacteria from transductants (Teuber, 2001). Transfer of foreign DNA molecules (DNA entry) into a recipient
bacterium is an important first step in genetic diversification through lateral gene transfer, but the expression level of the transferred gene and cell growth on media may differ for each
recipient cell. Culture methods have a limited ability to quantify the genetic material introduced into individual cells at the DNA level. Thus, our current knowledge of gene transfer via
phages in the environment is rather limited because of such methodological constraints. Detection of single-copy genes in individual cells is necessary to determine the frequency and range
of DNA transfer in environmental bacteria, and to understand gene flow among microorganisms. _In situ_ DNA amplification methods allow the visualization of specific DNA sequences inside
bacterial cells (Kenzaka et al., 2005; Maruyama et al., 2005), clarifying the movement of a specific gene among _Escherichia coli_ cells at the single-cell level (Kenzaka et al., 2007). The
purpose of this study was to investigate the propensity for DNA transfer via phages in natural freshwater habitats at the single-cell level. We used an _in situ_ DNA amplification technique
(cycling primed _in situ_ amplification-fluorescent _in situ_ hybridization; CPRINS-FISH; Kenzaka et al., 2005), and both frequency and possible range of DNA transfer via three _E. coli_
phages (P1, T4 and isolated phage) were first examined at the single-cell level using both plaque-forming and non-plaque-forming _Enterobacteriaceae_ strains as recipients. To explore the
viability of cells that acquired the gene from phage, direct viable counting (DVC) was carried out after DNA was transferred by phages (Kogure et al., 1979). The propensity of DNA transfer
obtained by this method was compared with values determined by conventional methods. Green fluorescent protein gene (_gfp_) was used as an indicator of gene transfer because the freshwater
samples used in this study did not contain the _gfp_ gene sequence. MATERIALS AND METHODS BACTERIAL STRAINS Bacterial strains used in this study are described as follows: _Citrobacter
freundii_ IFO 12681, _Enterobacter aerogenes_ BM 2688, _E. coli_ C600 RK2, _E. coli_ NBRC 12713 KEN1, which carries transposon _Tn1_ (4951 bp) including an ampicillin resistance gene (beta
lactamase gene; _bla_) on the chromosome, _E. coli_ NBRC 12713 with or without RK2∷_gfp_, _E. coli_ W3110 with or without plasmid RK2∷_gfp_, _Pseudomonas putida_ ATCC 12633, _Proteus
mirabilis_ clinical isolate, _Salmonella enteritidis_ IID 640, _Serratia marcescens_ clinical isolate, _Yersinia enterocolitica_ IID 981. The _gfp_-tagged broad host-range plasmid RK2
(RK2∷_gfp_) was constructed using RK2 and pGFPuv (Clontech Laboratories, Palo Alto, CA, USA) as described by Jorquera et al. (2006). _E. coli_ strains were grown in Luria–Bertain (LB) medium
(1% tryptone, 0.5% yeast extract, 0.5% NaCl) at 37 °C. Strains with the _bla_ gene were cultured in LB broth containing 50 μg ml−1 ampicillin. Other strains were grown in LB medium at 30
°C. The absence of _bla_ gene and _gfp_ gene in the genomic DNA of all recipient strains was confirmed by PCR targeting the genes before transduction and DNA transfer experiments were
performed. BACTERIOPHAGE Phage P1_kc_ NBRC 20008, a derivative of phage P1 was obtained from National Institute of Technology and Evaluation, Japan (Enomoto and Stocker, 1974). Phage T4GT7,
a derivative of phage T4 was obtained from National Institute of Genetics, Japan (Wilson et al., 1979). Transducing _E. coli_ phage EC10 was isolated from eutrophic river (Kenzaka et al.,
2007). Phages were propagated with appropriate donor _E. coli_ strains (NBRC 12713 KEN1 for P1_kc_ and T4GT7, _E. coli_ W3110 RK2∷_gfp_ or _E. coli_ NBRC 12713 RK2∷_gfp_ for EC10) in 1 l LB
broth containing 0.2% MgSO4 and 10 mM CaCl2 overnight at 37 °C. Purification of phages by ultracentrifugation was performed as described by Kenzaka et al. (2007). DNase treatment was
performed before phages were purified by ultracentrifugation to prevent transformation. Before transduction and DNA transfer experiments, DNA was extracted from each phage (P1, T4 and EC10)
infecting _E. coli_ strains lacking the _bla_ and _gfp_ genes using the Wizard Lambda Preps DNA Purification System (Promega, Madison, WI, USA), and the absence of the _bla_ and _gfp_ genes
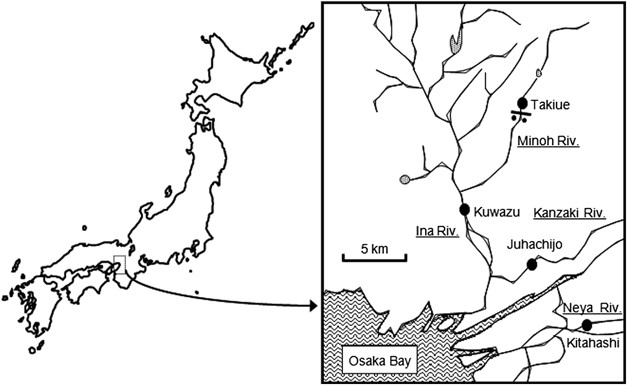
was confirmed by PCR. ENVIRONMENTAL SAMPLES Surface river water samples were taken from Kitahashi in the Neyagawa River, Juhachijo in the Kanzakigawa River, Kuwazu in the Inagawa River and
Takiue in the Minohgawa River, in the northern part of Osaka, Japan (Figure 1). At the sites, water samples were collected twice in early spring of 2005 (February–March) and three times in
autumn of 2009 (October–November). Kitahashi is located in a commercial area, Osaka Business Park. Kuwazu is located in an industrial area. These sites were considered to be polluted by
organic carbon (Tani et al., 1996; Yamaguchi and Nasu, 1997; Kenzaka et al., 2001). Takiue is surrounded by forest and is an oligotrophic site. At the site, the river is narrow and shallow,
and the water is not exposed to domestic or industrial effluents (Tani et al., 1996; Kenzaka et al., 1998, 2001). The water samples were collected in sterile 500-ml glass bottles and carried
to the laboratory on ice. The absence of _gfp_ gene in environmental samples was confirmed by PCR before DNA transfer experiments were performed. PLAQUE ASSAYS A volume of 100 μl of
stationary-phase cultures of bacterial strains was incubated with 10 μl of diluted phage (10−1–10−12) in 90 μl of SM buffer (50 mmol l−1 Tris–HCl (pH7.5), 100 mmol l−1 NaCl, 8 mmol l−1 MgSO4
and 0.01% gelatin) at 30 °C for 10 min. After incubation, samples were poured with LB soft agar (0.8% agar) on LB plate and incubated at 30 °C overnight to determine the plaque formation.
TRANSDUCTION ASSAYS For cultured recipients, 200 ml of stationary-phase cultures were collected by centrifugation at 8000 _g_ for 10 min at 4 °C and suspended in 10 ml of LB broth. A volume
of 3 ml of the suspended cultures was incubated with phages in 3 ml of SM buffer at 37 °C for 10 min at multiplicities of infection ranging from 0.2 to 2. Each control contained an equal
volume of the recipient cell culture and SM buffer. After a 10-min adsorption period at 37 °C for _E. coli_ and at 30 °C for other strains, cells were placed onto selective LB plates
containing 50 μg ml−1 ampicillin and incubated at 30 °C for 2 days. The transducing phage lysate (containing no recipient) and recipient (containing no transducing phage) were also plated
onto selective plates as a control. The frequencies of transduction were represented as the number of colonies on the selective LB plates per initial number of colonies on the non-selective
LB plates (colony-forming unit; CFU) of recipient or per initial plaque-forming unit (PFU) of phage. Results presented are averages of three transduction experiments. DNA TRANSFER
EXPERIMENTS A volume of 200 μl of stationary-phase culture was incubated with 200 μl of SM buffer containing each phage at 37 °C for 10 min at multiplicities of infection ranging from 0.2 to
2. The concentration of bacterial cells was adjusted to approximately 1 × 109 cells per ml. For other strains used as recipients, cultures were incubated with phages at 30 °C for 10 min.
For gene transfer assays with recipients at low concentration, concentration of _E. coli_ was adjusted to approximately 1 × 105, 106, 107 and 108 cells per ml, and 1% of _E. coli_ was added
to _P. putida_ at the concentration ranging from 4 × 105 to 107 cells per ml. The diluted cultures were incubated with phage EC10 at 25 °C for 10 min at the multiplicity of infection 2. For
gene transfer experiments in which indigenous river bacterial communities were used as recipients, 10–100-ml portions of river water samples were incubated with phage EC10 at 25 °C for 20
min at mixing ratio ranging from 2 to 200. The mixing ratio was calculated on the basis of the number of bacteria stained by SYBR Gold-labeled phage EC10 in river water samples as described
below. After the mixtures of recipients and phage were incubated under the conditions, as mentioned above, samples were fixed with 4% paraformaldehyde in phosphate-buffered saline (0.13 mol
l−1 NaCl, 7 mmol l−1 Na2HPO4 and 3 mmol l−1 NaH2PO4 (pH 7.2)) at 4 °C for 16 h. After fixation, 1 μl–30-ml portions were filtered through gelatin (0.1% gelatin, 0.01% CrK(SO4)2)-coated
polycarbonate white filters (0.2 μm pore size, 25 mm diameter, ADVANTEC, Tokyo, Japan) and rinsed twice with filtered deionized water. Then samples were stored at −20 °C. VIABILITY OF
RECIPIENT CELLS DETERMINED BY DVC To explore the viability of the recipient cells carrying the transferred gene, DVC was carried out. After recipient cell culture was mixed with each phage
under the conditions, as mentioned above, 20 μl of the mixture was added to 180 μl of LB broth containing an antibiotic cocktail (final concentration: 20 μg ml−1 nalidixic acid, 10 μg ml−1
piromidic acid, 10 μg ml−1 pipemidic acid, 10 μg ml−1 cephaloxin and 0.1 μg ml−1 ciprofloxacin; Joux and Lebaron, 1997) and incubated at 37 °C for _E. coli_ or 30 °C for other bacteria for 3
h. For river water samples, after the samples were mixed with the phage EC10 under conditions mentioned above, antibiotic cocktail was added to the mixture at the above final
concentrations, and incubated at 25 °C for 18 h (30 h for sample from oligotrophic site, Takiue). After incubation for DVC, samples were fixed with 4% paraformaldehyde in phosphate-buffered
saline at 4 °C for 16 h. After fixation, samples were filtered and were stored at −20 °C as described above. OLIGONUCLEOTIDES AND POLYNUCLEOTIDES Oligonucleotide primers and polynucleotide
probes for CPRINS-FISH used in this study were described as follows. AmpR840r primer and three probes for the _bla_ gene were reported in the study by Kenzaka et al. (2005). The nucleotide
sequences of _gfp_ gene were obtained from GenBank (Release 138.0), and gfp717r primer (5′-TTATTTGTAGAGCTCATCCA-3′) and probes (5′-GATGGTGATGTTAATGGGCACAAATTTTCTGTCAGTGGAGAGGGTGAAGG-3′;
5′-GACTTTTTCAAGAGTGCCATGCCCGAAGGTTATGTACAGGAACGC-3′; 5′-GATGACGGGAACTACAAGACGCGTGCTGAAGTCAAGTTTGAAGG-3′; 5′-ATTGGCGATGGCCCTGTCCTTTTACCAGACAACCATTACCTGTCCA-3′;
5′-CGAAAGATCCCAACGAAAAGCGTGACCACATGGTCCTTCTTGAG-3′) for the _gfp_ gene were designed in this study. The specificities of the primer and probe sequences were verified against National Center
for Biotechnology Information nucleotide databases using the Basic Local Alignment Search Tool program (Altschul et al., 1997). All primers and polynucleotide probes were purchased from
Texas Genomics (Tokyo, Japan), and probes were labeled with Alexa Fluor 546 at guanine using ULYSIS Alexa Fluor 546 Nucleic Acid Labeling Kit (Invitrogen, Tokyo, Japan) and the
manufacturer’s recommended procedures. CPRINS-FISH To detect cells carrying the _bla_ or _gfp_ gene transferred by phage, CPRINS-FISH was performed. Permeabilization for CPRINS-FISH was
carried out as described in the study by Kenzaka et al. (2005). Filters with bacterial cells were coated in gelatin to avoid cell loss during extensive cell wall permeabilization. After
lysozyme treatment, each filter was cut into 16 sections and subjected to CPRINS-FISH. A one-sixteenth section of the filter was transferred to a microtube (volume: 0.2 ml) and immersed in
100 μl of the CPRINS buffer (Kenzaka et al., 2007). Cycling primed _in situ_ amplification cycles consisted of a hot start at 95 °C for 9 min, denaturation at 94 °C for 1 min, annealing at
62 °C for 30 s and 72 °C for 1.5-min extension for AmpR840r primer or annealing at 58 °C for 30 s and 72 °C for 2-min extension for gfp717r primer. Amplification was repeated for 30 cycles
with a thermal cycler (PTC-200, Bio-Rad Laboratories, Hercules, CA, USA). After amplification, filters were rinsed with 0.1% Nonidet P40 and sterile deionized water, dehydrated in 99%
ethanol and vacuum dried. Hybridization, washing and 4′,6-diamidino-2-phenylindole (DAPI) staining were performed as described in the study by Kenzaka et al. (2007). To exclude the
possibility of nonspecific probe binding to cell structures other than target DNA in the target cells, (i) FISH using laboratory strains and environmental samples without amplification of
target DNA, (ii) CPRINS-FISH targeting the _bla_ or _gfp_ gene using _E. coli_ strains that did not carry the genes, and (iii) CPRINS-FISH targeting chloramphenicol acetyltransferase gene
using laboratory strains that did not carry the chloramphenicol acetyltransferase gene (Kenzaka et al., 2005), were performed. EPIFLUORESCENCE MICROSCOPY Filters were observed under an
epifluorescence microscope (E-400; Nikon, Tokyo, Japan) with the Nikon filter sets UV-2A (EX300-350, DM400 and BA420) for DAPI, B-2A (EX450/490, DM505 and BA520) for SYBR Gold and HQ-CY3
(G535/50, FT565 and BP610/75) for Alexa Fluor 546, respectively. Images were acquired by a cooled charge-coupled device camera (Cool Snap; Roper Photometrics, Tuscon, AZ, USA) and stored as
digital files. Exposure time was 0.1 s for DAPI, 0.5 s for SYBR Gold and 1 s for Alexa Fluor 546, respectively. A total of 3000–30 000 DAPI-stained objects were counted per sample.
Frequencies of gene transfer determined by CPRINS-FISH were represented as the number of CPRINS-FISH-positive cells per initial total direct counts (TDCs) of recipient or per initial PFU of
phage. Frequencies were determined in triplicate for each sample. Differences between means of frequencies were tested by the Student's _t_-test with Microsoft Excel XP software
(Microsoft, Redmond, WA, USA). FLUORESCENT BACTERIOPHAGE ASSAYS Fluorescently stained bacteriophages were produced by using a modification of procedure described by Maniatis et al. (1982).
The phage, EC10, was propagated with _E. coli_ C600 RK2 in 250 ml LB broth containing 0.2% MgSO4 overnight at 37 °C. The cell culture was centrifuged at 8000 _g_, 10 min, 4 °C, and the
supernatant was withdrawn. After nuclease treatment with DNase and RNase at 37 °C for 1 h, 0.2 μl ml−1 SYBR Gold (10 000 × , Invitrogen) was added and stained at 37 °C for 1 h. The stained
sample was subjected to polyethylene glycol precipitation. Stained phage particles were further purified by ultracentrifugation. A volume of 10 μl of both log-phase and stationary-phase
cultures were incubated with 10 μl of the purified SYBR Gold-labeled phage at 37 °C for 20 min. For estimating the bacterial population to which phage EC10 could inject their DNA in river
water samples, 1 ml of water samples were mixed with 20 μl of the SYBR Gold-labeled phage at 30 °C for 20 min. After incubation, samples were fixed in 4% paraformaldehyde in
phosphate-buffered saline at room temperature (ca. 20 °C) for 10 min. After fixation, samples were stained with 1 μg ml−1 DAPI for 5 min, and filtered through a polycarbonate filter (pore
size: 0.2 μm; ADVANTEC). Filters were mounted in immersion oil for observation by epifluorescence microscopy. RESULTS GENE TRANSFER VIA PHAGE P1 AND T4 Phage P1 and T4GT7 are commonly used,
generalized transducing phages for _E. coli_. They have the ability to mediate gene transfer from _E. coli_ to not only _E. coli_ but to also some plaque-forming _Enterobacteriaceae_ strains
(Goldberg et al., 1974; Tetart et al., 1996). The frequency and possible range of transfer of the _bla_ gene on the NBRC 12713 KEN1 chromosome of _E. coli_ via the two phages was first
examined by both a culture-based method using selective medium containing ampicillin and a culture-independent method with CPRINS-FISH targeting the _bla_ gene. Plaque-forming strains and
non-forming _Enterobacteriaceae_ strains were used as recipients. Transduction was observed in _E. aerogenes_ and _E. coli_, which were plaque-forming strains, and the transduction
frequencies for the _bla_ gene on selective medium were 0.6–2 × 10−6 transductants per PFU for P1_kc_ (Table 1), and undetectable to 2 × 10−8 transductants per PFU for T4GT7 (Table 2) at a
multiplicity of infection 1. Although non-plaque-forming strains did not have the _bla_ gene, they could grow on selective medium containing ampicillin perhaps due to overproduction of
multidrug-resistant efflux pump when they were plated at high density (ca. >107 CFUs per ml). Thus it was hard to determine the transduction frequency in the non-plaque-forming strains by
conventional methods. The frequencies of DNA transfer determined by CPRINS-FISH were 3–8 × 10−3 per PFU for P1_kc_ (Table 1) and 0.3–2 × 10−3 per PFU for T4GT7 (Table 2) in plaque-forming
strains. The CPRINS-FISH analysis revealed that both P1_kc_ and T4GT7 transferred the _bla_ gene to non-plaque-forming strains. Phage P1_kc_ transferred the _bla_ gene to _C. freundii_ and
_S. enteritidis_ at similar frequencies (0.8–4 × 10−3 per PFU), and phage T4GT7 transferred the _bla_ gene to _C. freundii_, _P. mirabilis_, _S. enteritidis_ and _Y. enterocolitica_ at
similar frequencies (0.5–2 × 10−3 per PFU). Viable bacteria carrying transferred genes may be infected by other phages and the transferred gene could be further transferred to other
bacteria. They may also maintain the transferred gene. Thus, DVC and CPRINS-FISH were combined to estimate viable cells carrying the transferred _bla_ gene. Representative photographs of
viable _C. freundii_ cells to which the _bla_ gene was transferred by phage P1_kc_ are shown in Figures 2a and b. Direct viable counting combined with CPRINS-FISH visualized the target
viable recipients as elongated cells that showed bright Alexa Fluor 546 fluorescence under green excitation (Figure 2b). More than 30% of the remaining cells carrying the _bla_ gene became
elongated and/or fattened, that is, possessed protein synthesis activity. The transfer frequencies determined by DVC combined with CPRINS-FISH were at least three (P1_kc_) or five (T4GT7)
orders of magnitude higher than for colony-forming bacteria on selective medium in plaque-forming strains. Our results show that DNA transfer mediated by phage P1_kc_ and T4GT7 happened in
more divergent strains than those estimated by conventional methods. GENE TRANSFER VIA ISOLATED PHAGE EC10 To examine the potential for gene transfer by phages in a freshwater environment,
we prepared isolated transducing phage EC10 packed with the _gfp_ gene, and DNA transfer experiments were performed using the _gfp_ gene as an indicator of gene transfer. Phage EC10 that was
propagated with _E. coli_ NBRC 12713 RK2∷_gfp_ was mixed with _Enterobacteriaceae_ strains. Transfer of the _bla_ and _gfp_ gene on the plasmid RK2∷_gfp_ via phage EC10 was examined by both
selective agar plating and CPRINS-FISH targeting the _bla_ and _gfp_ genes (Table 3). Frequencies on selective media containing ampicillin were undetectable in this study using _E. coli_
NBRC 12713 RK2∷_gfp_ as a donor, although phage EC10 has the ability to transfer _E. coli_ genes to both plasmids and chromosomes (Kenzaka et al., 2007). Frequencies of DNA transfer
determined by CPRINS-FISH targeting the _bla_ gene were 7–10 × 10−3 per PFU. Frequencies of DNA transfer of the _gfp_ gene were slightly lower than those of the _bla_ gene (_P_<0.05).
Most gene-positive cells determined by CPRINS-FISH retained protein synthesis activity. Next, the possible range of transfer of the _gfp_ gene via EC10 was investigated using other
_Enterobacteriaceae_ strains as recipients (Table 4). Frequencies of DNA transfer determined by CPRINS-FISH targeting the _gfp_ gene in the strains ranged from 3 to 4 × 10−3 per PFU. Viable
cells carrying the transferred _gfp_ gene determined by DVC combined with CPRINS-FISH constituted more than 30% of the _gfp_ gene-positive cells determined by CPRINS-FISH. These frequencies
were similar to plaque-forming _E. coli_ (Table 3). The results demonstrated that isolated phage EC10 also has the ability to transfer the gene from _E. coli_ to other _Enterobacteriaceae_
strains, and conventional methods underestimate the range of phage-mediated gene transfer. FREQUENCY OF DNA TRANSFER AT LOW BACTERIAL CONCENTRATIONS In natural aquatic environments,
bacterial concentration is significantly lower than in laboratory conditions. Thus, DNA-transfer frequencies at low bacterial concentration were examined by CPRINS-FISH (Table 5). Although
it was expected that phage absorption by recipients was reduced by the low bacterial concentration, DNA-transfer frequencies were similar at a concentration of 105–108 _E. coli_ cells per
ml. Usually bacterial communities in aquatic environments consist of a small number of hosts and a large number of non-hosts. As phage EC10 did not transfer the _gfp_ gene into _P. putida_,
_E. coli_ was added to _P. putida_ and DNA transfer experiments were performed to examine the effect of a small population size on DNA-transfer frequency. The CPRINS-FISH analysis on a
polycarbonate filter made it possible to concentrate target cells through filtration, and has advantages in estimating the frequency of gene transfer in samples with low bacterial
concentrations, such as natural aquatic environments. GENE TRANSFER WITH INDIGENOUS BACTERIAL COMMUNITIES AS RECIPIENTS Potential gene transfer by phages in freshwater environments was
examined with bacterial communities from oligotrophic and eutrophic rivers as recipients. Four samples were collected from different rivers including the Kanzakigawa River from which phage
EC10 was isolated. Plasmid RK2∷_gfp_ encodes ampicillin, kanamycin and tetracycline resistance genes (Thomas, 1981), but a significant part of indigenous bacteria from these sites (10−5–10−4
per TDC) were resistant to these antibiotics. The presence of antibiotic-resistant bacteria in the river samples made it difficult to detect transductants by culture-dependent methods using
selective medium. Both _E. coli_ W3110 RK2∷_gfp_ and NBRC 12713 RK2∷_gfp_ strains were used as donors. There was no difference in the DNA-transfer frequencies between these donor strains
when _E. coli_ NBRC 12713 was used as a recipient (data not shown). To assess the possible range of DNA injection by phage EC10, fluorescent bacteriophage assay was carried out first (Hennes
et al., 1995; Noble and Fuhrman, 2000). Although plaque formation was observed only in _E. coli_, phage DNA was injected by phage EC10 into _C. freundii_, _E. aerogenes_, _P. mirabilis_,
_S. enteritidis_ and _S. marcescens_. Fluorescent bacteriophage assay showed that phage EC10 could infect indigenous bacterial cells in river water samples (Figures 2c and d). Only cells
stained by fluorescently labeled-phage EC10 emitted the green fluorescence of SYBR Gold under blue excitation (Figure 2d). The frequency of cells stained by SYBR gold-labeled phage EC10
constituted 0.4–2 × 10−2 per TDC in the river water samples (Table 6). The abundance of _E. coli_ in these rivers determined by FISH with a ribosomal RNA-targeted probe for _E. coli_
(Kenzaka et al., 2001) was about 10−4 of TDC, which was lower than the numbers determined by fluorescent bacteriophage assay. Thus, phage EC10 was thought to inject DNA into non-_E. coli_
cells in the samples. The ‘mixing ratio’ was calculated on the basis of the number of cells stained by SYBR gold-labeled phage EC10 in the river water samples. Then the frequencies of the
_gfp_ gene transfers to indigenous bacterial communities were investigated by DVC combined with CPRINS-FISH (Table 6). The DVC-positive cells in river water samples accounted for 15–30% of
the total bacteria at Juhachijo, 9–34% at Kitahashi, 8–28% at Kuwazu and 17–24% at Takiue through the sampling periods. Independent of the mixing ratio (2, 20 and 200), the DNA-transfer
frequencies per TDC determined by CPRINS-FISH or DVC combined with CPRINS-FISH at Juhachijou were similar. Thus, the indigenous bacterial populations to which phage EC10 could inject the
_gfp_ gene in river water samples were confined to limited cells. Gene transfer by phage EC10 was also observed in other river water samples from oligotrophic (Takiue) and eutrophic
(Juhachijo, Kitahashi and Kuwazu) sites (Table 6). Indigenous bacteria from these four locations did not show positive signals with CPRINS-FISH before transducing phages were added, but they
were CPRINS-FISH positive after gene transfer experiments were performed with phage EC10. The frequencies of _gfp_ gene transfer in these bacterial communities ranged from undetectable to 2
× 10−3 cells per TDC, which correspond to undetectable to 9 × 10−2 cells per PFU. Viable cells carrying the transferred _gfp_ gene were detected by DVC combined with CPRINS-FISH (Figures 2e
and f), and they constituted more than 20% of the _gfp_ gene-positive cells in most cases. The frequencies of _gfp_ gene-positive viable cells ranged from undetectable to 9 × 10−2 cells per
PFU (undetectable to 1 × 10−3 cells per TDC). Although it was hard to detect transductants using the culture-dependent method, DVC combined with CPRINS-FISH clarified the DNA-transfer
frequencies through the _E. coli_ phage in the river water samples at the single-cell level. The frequencies of _gfp_ gene-positive viable cells per PFU at each site were compared between
early spring and autumn, and among three sampling dates in autumn. Overall, the obtained frequencies at each site were not significantly different among sampling dates (_P_>0.05) except
those at Juhachijo between 7 March 2005 and 18 October 2009 (_P_<0.05). DISCUSSION This study attempted to accurately determine the frequency and possible range of DNA transfer mediated
by _E. coli_ phages using a gene-targeting approach. Although advances in genomics and nucleotide sequence analysis have highlighted the significance of phages in lateral gene transfer as
possible contributors to bacterial evolution (Bordenstein and Reznikoff, 2005), transduction frequencies in laboratory experiments using selective media in most studies were generally
reported to be low and the range of phage-mediated gene transfer was considered narrow, although broad host-range phages were occasionally found in aquatic environments (Beumer and Robinson,
2005). To understand gene flow among bacteria, sensitive and reliable methods of detecting transferred DNA are necessary, which should have the following properties: (i) recognition of
transferred gene in individual cells without the requirement of cultivation or gene expression, (ii) effective concentration of target bacteria that acquire the indicator gene, for example,
by filtration, and (iii) identical conditions that can be applied to diverse bacteria because the gene is expected to be transferred to bacteria physiology of which is unclear. In this
study, we used a gene-targeting approach based on _in situ_ DNA amplification, which enabled us to determine the frequency and possible range of gene transfer at the single-cell level.
Previous studies show transduction frequencies determined by culture-dependent methods using selective agar media varied over orders of magnitude from 10−11 to 10−5 per PFU in freshwater and
marine systems (Weinbauer and Rassoulzadegan, 2004). In contrast, DNA-transfer frequencies determined by DVC combined with CPRINS-FISH for _gfp_ gene were up to 3 × 10−3 per PFU in
_Enterobacteriaceae_ strains, and up to 9 × 10−2 per PFU in natural bacterial communities; a few orders of magnitude greater than those previously measured throughout the studied periods.
Conventional methods confirmed that the range of gene transfer by phage P1_kc_, T4GT7 and EC10 was limited to plaque-forming strains (_E. coli_ and _E. aerogenes_), but CPRINS-FISH revealed
that the three _E. coli_ phages could mediate transfer of the gene from _E. coli_ to non-plaque-forming _Enterobacteriaceae_ strains. Specifically, some bacteria, which were not thought to
be a ‘host’ or ‘recipient’ when conventional methods were used, were able to receive the bacterial gene via the _E. coli_ phages. The phage life cycle consists of adsorption into a cell,
injection of nucleic acid, commandeering of host machinery, production of phage proteins and nucleic acid, assembly and release by either lysis or extrusion (Wommack and Colwell, 2000). For
visible plaques, it is necessary to complete these processes and disperse effectively within soft agar. Certain phages in natural environments do not form plaques on any known host because
they have the mechanism to suppress host lysis (lysogeny), have a mutation in the genes responsible for lysis or lack genetic material themselves (defective phage; Garro and Marmur, 1970).
The presence of such phages masks actual host range. The gene-targeting approach described here makes it possible to detect target DNA sequences in bacterial cells without cultivation or
gene expression, and is less likely to be biased by physiological or genetic (that is, marker expression) limitations. Consequently, our results suggest that DNA exchange among bacteria via
phage may occur in a more divergent range of bacteria than previously thought using conventional methods. The entry of DNA into a recipient bacterium is an important first step in genetic
diversification through lateral gene transfer. On DNA entry into a cell, binding between the phage receptor on the cell surface and the tail fiber on the phage is necessary. Our results show
that three _E. coli_ phages (P1_kc_, T4GT7 and EC10) attached on more divergent strains than those estimated by conventional methods, and transferred the _E. coli_ gene to the strains
(Table 1, 2, and 4). These results indicate that the binding between the phage receptor and the tail fiber might be more plastic than previously thought. In addition, the structure of the
phage receptor on the cell surface and the tail fiber on the phage could be changed by mutation (Yu and Mizushima, 1982; Tetart et al., 1996). This would allow the movement of DNA molecules
among more divergent genera mediated by phages more frequently. The fate of the transferred gene in recipient cells is of interest for genomic diversity studies. During the incubation for
DVC, a proportion of transferred genes inside recipient cells may be destroyed, but DVC combined with CPRINS-FISH showed that a significant proportion of the transferred gene remained in
viable recipient cells. Potential future study should examine the frequency and mechanism of the maintenance of transferred genes and aim to elucidate the mechanism of maintenance based on
DNA sequence. Previous studies have shown that a high degree of genomic diversity may occur among closely related genomes (Ohnishi et al., 2002). Genomes of _Vibrio splendidus_ in natural
coastal areas showed extensive allelic diversity, and the group consists of at least 1000 distinct genotypes in 1 ml of sea water (Thompson et al., 2005). Such variation might arise because
of DNA exchange among microorganisms at the single-cell level. Several prokaryotic genomes contain large fractions of foreign genes, for example, more than 15% of the genes of _E. coli_ have
been acquired by lateral gene transfer (Lawrence and Ochman, 1998). The events of gene transfer have significantly driven diversification in the bacterial genome. This study offers a way to
address the movement of a specific gene among bacterial cells _in situ_, regardless of culturability or gene expression. Our results suggest that DNA exchange among bacteria via phages in
natural aquatic environments may occur in a more divergent range of bacteria and more frequently than thought previously using conventional methods. ‘Lateral gene transfer’, in the general
meaning, consists of several steps: entry of the foreign gene into viable recipient cells, gene replication, maintenance during cell growth, gene expression and so forth. Reporter gene
technology, such as green fluorescent protein, allows the estimation of bacterial cells in which the reporter gene is expressed (Dahlberg et al., 1998; Hendrickx et al., 2003), although
potential limitations and advantages have been discussed (Sorensen et al., 2005; Maruyama et al., 2006). Our approach has a great potential to provide more quantitative information on gene
transfer steps in the natural environment. It will be important in the future to clarify whether such extensive gene transfer events via phages observed at the single-cell level are a
general feature of interactions between natural bacteria and phages using other phage-host systems. REFERENCES * Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W _et al_.
(1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. _Nucleic Acids Res_ 25: 3389–3402. Article CAS PubMed PubMed Central Google Scholar * Amann RI,
Ludwig W, Schleifer KH . (1995). Phylogenetic identification and _in situ_ detection of individual microbial cells without cultivation. _Microbiol Rev_ 59: 143–169. CAS PubMed PubMed
Central Google Scholar * Beumer A, Robinson JB . (2005). A broad-host-range, generalized transducing phage (SN-T) acquires 16S rRNA genes from different genera of bacteria. _Appl Environ
Microbiol_ 71: 8301–8304. Article CAS PubMed PubMed Central Google Scholar * Bordenstein SR, Reznikoff WS . (2005). Mobile DNA in obligate intracellular bacteria. _Nature Rev Microbiol_
3: 688–699. Article CAS Google Scholar * Brüssow H, Canchaya C, Hardt WD . (2004). Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion.
_Microbiol Mol Biol Rev_ 68: 560–602. Article PubMed PubMed Central Google Scholar * Dahlberg C, Bergstrom M, Hermansson M . (1998). _In situ_ detection of high levels of horizontal
plasmid transfer in marine bacterial communities. _Appl Environ Microbiol_ 64: 2670–2675. CAS PubMed PubMed Central Google Scholar * Enomoto M, Stocker BA . (1974). Transduction by phage
P1_kc_ in _Salmonella typhimurium_. _Virology_ 60: 503–514. Article CAS PubMed Google Scholar * Fuhrman JA . (1999). Marine viruses and their biogeochemical and ecological effects.
_Nature_ 399: 541–548. Article CAS PubMed Google Scholar * Garro AJ, Marmur J . (1970). Defective bacteriophages. _J Cell Physiol_ 76: 253–263. Article CAS PubMed Google Scholar *
Goldberg RB, Bender RA, Streicher SL . (1974). Direct selection for P1-sensitive mutants of enteric bacteria. _J Bacteriol_ 118: 810–814. CAS PubMed PubMed Central Google Scholar *
Hendrickx L, Hausner M, Wuertz S . (2003). Natural genetic transformation in monoculture _Acinetobacter_ sp. strain BD413 biofilms. _Appl Environ Microbiol_ 69: 1721–1727. Article CAS
PubMed PubMed Central Google Scholar * Hennes KP, Suttle CA, Chan AM . (1995). Fluorescently labeled virus probes show that natural virus populations can control the structure of marine
microbial communities. _Appl Environ Microbiol_ 61: 3623–3627. CAS PubMed PubMed Central Google Scholar * Jiang SC, Paul JH . (1998). Gene transfer by transduction in the marine
environment. _Appl Environ Microbiol_ 64: 2780–2787. CAS PubMed PubMed Central Google Scholar * Jorquera M, Yamaguchi N, Tani K, Nasu M . (2006). A combination of direct viable counting,
fluorescence _in situ_ hybridization, and green fluorescent protein gene expression for estimating plasmid transfer at the single cell level. _Microbes Environ_ 21: 101–111. Article Google
Scholar * Joux F, Lebaron P . (1997). Ecological implications of an improved direct viable count method for aquatic bacteria. _Appl Environ Microbiol_ 63: 3643–3647. CAS PubMed PubMed
Central Google Scholar * Kenzaka T, Tamaki S, Yamaguchi N, Tani K, Nasu M . (2005). Recognition of individual genes in diverse microorganisms by cycling primed _in situ_ amplification.
_Appl Environ Microbiol_ 71: 7236–7244. Article CAS PubMed PubMed Central Google Scholar * Kenzaka T, Tani K, Sakotani A, Yamaguchi N, Nasu M . (2007). High frequency phage-mediated
gene transfer among _Escherichia coli_ determined at the single cell level. _Appl Environ Microbiol_ 73: 3291–3299. Article CAS PubMed PubMed Central Google Scholar * Kenzaka T,
Yamaguchi N, Prapagdee B, Mikami E, Nasu M . (2001). Bacterial community composition and activity in urban rivers in Thailand and Malaysia. _J Health Sci_ 47: 353–361. Article CAS Google
Scholar * Kenzaka T, Yamaguchi N, Tani K, Nasu M . (1998). rRNA-targeted fluorescent _in situ_ hybridization analysis of bacterial community structure in river water. _Microbiology_ 144:
2085–2093. Article CAS PubMed Google Scholar * Kogure K, Simidu U, Taga N . (1979). A tentative direct microscopic method for counting living marine bacteria. _Can J Microbiol_ 25:
415–420. Article CAS PubMed Google Scholar * Lawrence JG, Ochman H . (1998). Molecular archaeology of the _Escherichia coli_ genome. _Proc Natl Acad Sci USA_ 95: 9413–9417. Article CAS
PubMed PubMed Central Google Scholar * Maniatis T, Fritsch EF, Sambrook J . (1982). _Molecular Cloning_. A laboratory manual. Isolation of bacteriophage Lambda and plasmid DNA. pp
75–85. * Maruyama F, Kenzaka T, Yamaguchi N, Tani K, Nasu M . (2005). Visualization and enumeration of bacteria carrying a specific gene sequence by _in situ_ rolling circle amplification.
_Appl Environ Microbiol_ 71: 7933–7940. Article CAS PubMed PubMed Central Google Scholar * Maruyama F, Tani K, Kenzaka T, Yamaguchi N, Nasu M . (2006). Quantitative determination of
free-DNA uptake in river bacteria at the single-cell level by _in situ_ rolling-circle amplification. _Appl Environ Microbiol_ 72: 6248–6256. Article CAS PubMed PubMed Central Google
Scholar * Noble RT, Fuhrman JA . (2000). Rapid virus production and removal as measured with fluorescently labeled viruses as tracers. _Appl Environ Microbiol_ 66: 3790–3797. Article CAS
PubMed PubMed Central Google Scholar * Ochman H, Lawrence JG, Groisman EA . (2000). Lateral gene transfer and the nature of bacterial innovation. _Nature_ 405: 299–304. Article CAS
PubMed Google Scholar * Ogunseitan OA . (2008). Genetic transduction in freshwater ecosystems. _Freshwater Biol_ 53: 1228–1239. Article Google Scholar * Ohnishi M, Terajima J, Kurokawa
K, Nakayama K, Murata T, Tamura K _et al_. (2002). Genomic diversity of enterohemorrhagic _Escherichia coli_ O157 revealed by whole genome PCR scanning. _Proc Natl Acad Sci USA_ 99:
17043–17048. Article CAS PubMed PubMed Central Google Scholar * Pallen MJ, Wren BW . (2007). Bacterial pathogenomics. _Nature_ 449: 835–842. Article CAS PubMed Google Scholar * Saye
DJ, Ogunseitan O, Sayler GS, Miller RV . (1987). Potential for transduction of plasmids in a natural freshwater environment: effect of plasmid donor concentration and a natural microbial
community on transduction in _Pseudomonas aeruginosa_. _Appl Environ Microbiol_ 53: 987–995. CAS PubMed PubMed Central Google Scholar * Sorensen SJ, Bailey M, Hansen LH, Kroer N, Wuertz
S . (2005). Studying plasmid horizontal transfer _in situ_: a critical review. _Nature Rev Microbiol_ 3: 700–710. Article CAS Google Scholar * Tani K, Chen JM, Yamaguchi N, Nasu M .
(1996). Estimation of bacterial volume and biomass by scanning electron microscopic image analysis. _Microbes Environ_ 11: 11–17. Article Google Scholar * Tetart F, Repoila F, Monod C,
Krisch HM . (1996). Bacteriophage T4 host range is expanded by duplications of a small domain of the tail fiber adhesion. _J Mol Biol_ 258: 726–731. Article CAS PubMed Google Scholar *
Teuber M . (2001). Veterinary use and antibiotic resistance. _Curr Opin Microbiol_ 4: 493–499. Article CAS PubMed Google Scholar * Thomas CM . (1981). Molecular genetics of broad host
range plasmid RK2. _Plasmid_ 5: 10–19. Article CAS PubMed Google Scholar * Thompson JR, Pacocha S, Pharino C, Klepac-Ceraj V, Hunt DE, Benoit J _et al_. (2005). Genotypic diversity
within a natural coastal bacterioplankton population. _Science_ 307: 1311–1313. Article CAS PubMed Google Scholar * Weinbauer MG, Rassoulzadegan F . (2004). Are viruses driving microbial
diversification and diversity? _Environ Microbiol_ 6: 1–11. Article PubMed Google Scholar * Wilson GG, Young KY, Edlin GJ, Konigsberg W . (1979). High-frequency generalised transduction
by bacteriophage T4. _Nature_ 280: 80–82. Article CAS PubMed Google Scholar * Wommack KE, Colwell RR . (2000). Virioplankton: viruses in aquatic ecosystems. _Microbiol Mol Biol Rev_ 64:
69–114. Article CAS PubMed PubMed Central Google Scholar * Yamaguchi N, Nasu M . (1997). Flow cytometric analysis of bacterial respiratory enzymatic activity in the natural aquatic
environment. _J Appl Microbiol_ 83: 43–52. Article CAS Google Scholar * Yu F, Mizushima S . (1982). Roles of lipopolysaccharide and outer membrane protein OmpC of _Escherichia coli_ K-12
in the receptor function for bacteriophage T4. _J Bacteriol_ 151: 718–722. CAS PubMed PubMed Central Google Scholar * Zeph LR, Onaga MA, Stotzky G . (1988). Transduction of _Escherichia
coli_ by bacteriophage P1 in soil. _Appl Environ Microbiol_ 54: 1731–1737. CAS PubMed PubMed Central Google Scholar * Zinder ND, Lederberg J . (1952). Genetic exchange in _Salmonella_.
_J Bacteriol_ 64: 679–699. CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This study was supported by the JSPS Grant-in-Aid for Young Scientists (B)
(18780055). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Environmental Science and Microbiology, Faculty of Pharmacy, Osaka Ohtani University, Nishikiori-kita, Tondabayashi, Japan Takehiko
Kenzaka & Katsuji Tani * Environmental Science and Microbiology, Graduate School of Pharmaceutical Sciences, Osaka University, Yamada–oka, Suita, Japan Takehiko Kenzaka, Katsuji Tani
& Masao Nasu Authors * Takehiko Kenzaka View author publications You can also search for this author inPubMed Google Scholar * Katsuji Tani View author publications You can also search
for this author inPubMed Google Scholar * Masao Nasu View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Katsuji Tani.
RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Kenzaka, T., Tani, K. & Nasu, M. High-frequency phage-mediated gene transfer in freshwater
environments determined at single-cell level. _ISME J_ 4, 648–659 (2010). https://doi.org/10.1038/ismej.2009.145 Download citation * Received: 28 July 2009 * Revised: 19 November 2009 *
Accepted: 22 November 2009 * Published: 21 January 2010 * Issue Date: May 2010 * DOI: https://doi.org/10.1038/ismej.2009.145 SHARE THIS ARTICLE Anyone you share the following link with will
be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative KEYWORDS * bacteriophage * freshwater * lateral gene transfer * transduction










