- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Cerebral cavernous (or capillary-venous) malformations (CCM) have a prevalence of about 0.1–0.5% in the general population. Genes mutated in CCM encode proteins that modulate
junction formation between vascular endothelial cells. Mutations lead to the development of abnormal vascular structures. In this article, we review the clinical features, molecular and
genetic basis of the disease, and management. SIMILAR CONTENT BEING VIEWED BY OTHERS DEVELOPMENTAL VENOUS ANOMALIES ARE A GENETIC PRIMER FOR CEREBRAL CAVERNOUS MALFORMATIONS Article 14 March
2022 _PIK3CA_ AND CCM MUTATIONS FUEL CAVERNOMAS THROUGH A CANCER-LIKE MECHANISM Article 28 April 2021 GENETICS OF BRAIN ARTERIOVENOUS MALFORMATIONS AND CEREBRAL CAVERNOUS MALFORMATIONS
Article 13 July 2022 INTRODUCTION Cerebral cavernous (or capillary-venous) malformations (CCM; OMIM no. 116 860) are vascular malformations with a prevalence of 0.1–0.5% in the general
population, with a familial incidence close to 20%.1, 2, 3 CCM may occur sporadically, but most of the time it has an autosomal dominant inheritance pattern with variable expression and
incomplete penetrance.2, 3, 4, 5, 6 At least three genes have been associated with CCM: k-rev interaction trapped protein 1 (_KRIT1_) (_CCM1_; OMIM no. 604214), _MGC4607_ (_CCM2_; OMIM no.
603284) and programmed cell death 10 (_PDCD10_) (_CCM3_; OMIM no. 603285). These genes encode proteins that are involved in junction formation between vascular endothelial cells. Mutations
in the _CCM_ genes, which are in general loss-of-function mutations, lead to the development of abnormal vascular structures characterized by thin-walled, dilated blood vessels with gaps
between the endothelial cells.1, 7 The underlying genetic mechanism in CCM is partially understood. Second-site genetic mutations have been proposed as one of the possible molecular
mechanisms.1, 8 A total of 9% of individuals were symptomatic before age 10 years, 62–72% between 10 and 40 years, and 19% after age 40 years.9, 10 Up to 25% of individuals with CCM remain
symptom free throughout their lives.11 This percentage may be an underestimate because many asymptomatic persons go unrecognized. Otten _et al_12 reported an absence of symptoms in 90% of
individuals with CCM ascertained in autopsy. Approximately 50–75% of persons with CCM become symptomatic. Affected individuals most often present with seizures (40–70%), focal neurologic
deficits (35–50%), non-specific headaches (10–30%) and cerebral haemorrhage (41%).9, 11, 13, 14 In the most recent study, Denier _et al_15 found seizures in 55%, focal neurological deficits
in 9%, non-specific headaches in 4% and cerebral haemorrhage in 32%. In most cases, cavernous malformations (or cavernomas) are located within the brain, but in a small proportion of
patients with familial CCM, cavernomas may also be observed in the spinal cord, retina, skin or liver.2, 3, 16 Retinal cavernomas occur in about 5% of patients with familial CCM. They are
unilateral, generally stable and asymptomatic, and can be diagnosed by routine fundoscopy.2, 17 Cutaneous vascular malformations are seen in 9% of familial CCM patients. Three distinct major
phenotypes were identified: hyperkeratotic cutaneous capillary-venous malformations (39%), strongly associated with a _KRIT1_ mutation. Second, capillary malformations (34%) and finally,
venous malformations (21%) mostly seen in patients with a _PDCD10_ mutation. Patients with a _Malcavernin_ mutation are possibly less prone for cutaneous vascular malformations.2, 16, 18
MOLECULAR AND GENETIC BASIS OF CCMS MUTATED GENES AND NEW LOCI To date, three genes have been associated with the pathogenesis of CCM, including _KRIT1_ (also known as _CCM1_) located on
chromosome 7q11.2–21,19, 20 Malcavernin, murine OSM-osmosensing scaffold for MEKK3 (_MGC4607_, also known as _CCM2_) on chromosome 7p1319, 21 and _PDCD10_ (also known as _CCM3_), originally
identified as TF-1 cell apoptosis-related gene-15 (_TFAR15_) on chromosome 3q26.1 (Table 1).19, 22, 23 In addition, there is at least one further – as yet unspecified – gene that can cause
CCM, which has been mapped to chromosome 3q26.3–27.2. Gianfrancesco _et al_25 reported the zona pellucida-like domain containing 1 gene as possible candidate. This gene is also located on
the long arm of chromosome three centromeric of _PDCD10_.3, 24, 25, 26 DISTRIBUTION AND FREQUENCY OF GENE MUTATIONS Close to 100 mutations (88 germline mutations) have been identified in the
_KRIT1_ gene, representing about 40–53% of the CCM families. Mutations in the _MGC4607_ gene may account for 15–20% of familial CCM cases.3, 7, 26, 27, 28 The only missense mutation in the
_MGC4607_ gene reported so far, is a leucine to arginine substitution at amino acid 198 (L198R), located in the phosphotyrosine-binding domain (PTB) of Malcavernin.29 Approximately 10–40% of
CCM families have been linked to the _PDCD10_ gene.26, 28 With a single exception, mutations in the _PDCD10_ gene are either truncating or large genomic deletions of the entire gene. The
only known in-frame deletion of _PDCD10_ is located in exon 5, encompassing amino acids L33–K50, encoding the serine/threonine kinase binding and phosphorylation domain .22, 23, 30 In about
22% of CCM cases with multiple lesions no mutation is detected in the three _CCM_ genes.26 Although _de novo_ mutations have been reported for all three _CCM_ genes, they appear to be more
common in the _PDCD10_ gene.2, 31 The proportion of familial cases has been estimated approximately at 20% in the general population, and estimated to be as high as 50% in Hispanic-American
patients of Mexican descent. These families are all apparently related to the same founder mutation (Q455X) in the _KRIT1_ gene.2, 3, 5 GENOTYPE–PHENOTYPE RELATIONSHIP CCM is an autosomal
dominant disorder with a clinical penetrance of 88% in _CCM1_ families, 100% in _CCM2_ and 63% in _CCM3_ families.2, 32 Different explanations have been provided for the molecular
pathogenesis of lesion formation in CCM. First, a Knudsonian two-hit mechanism might be involved. According to this mechanism, CCM formation would require a complete loss of the two alleles
of a given _CCM_ gene within affected cells. Loss of one of the alleles (first hit) would be the result of a germline mutation, whereas loss of the second allele (or second hit) will occur
somatically. In this view, familial CCM exhibits an autosomal dominant mode of inheritance, but is likely recessive at the cellular level.2, 27, 33 On the basis of animal, as well as human
studies, evidence grows for the two-hit mechanism. For example, in Ccm heterozygous mice, homozygous knockout for _Msh2_, penetrance of CCM lesions has been increased. Even so, in surgically
resected mature lesions from CCM patients, mutations have been found in both alleles.34 Second, haploinsufficiency may also be an explanation in CCM pathophysiology. In this case, the
patient has only a single functional copy of one of the _CCM_ genes, due to mutational inactivation of the other. The single functional copy of the gene, however, does not result in
sufficient protein for, for example, an adequate functional junction formation between endothelial cells, which in turn leads to the development of abnormal vascular structures. Third,
paradominant inheritance might explain several CCM features. In paradominant inheritance, heterozygous individuals carrying a ‘paradominant’ mutation are phenotypically normal, but the trait
only becomes manifest when a somatic mutation occurs during embryogenesis, giving rise to loss of heterozygosity and formation of a mutant cell population that is homozygous for the
mutation. In addition, a second hit may be caused by environmental factors. The exposure of CCM mutated, presensitized microvascular regions to oxidative stress generated by endothelial
nitric oxide synthase uncoupling and reactive oxygen species formation could lead to perivascular astrocytosis.35, 36, 37 The localized nature and the number of lesions (usually a single one
in sporadic cases versus multiple lesions in familial cases), as well as the age of first presentation of the phenotype being earlier in familial cases and fits in this type of inheritance.
Finally, trans-heterozygosity, in which a patient has synergistic mutations in different genes of the CCM pathway (for example, a germline mutation in the _KRIT1_ gene with an additional
somatic mutation in the _MGC4607_ or _PDCD10_ gene), might also explain intrafamilial clinical variability. Indeed, it has been shown that a decrease in the _KRIT1_, _MGC4607_ or _PDCD10_
gene alone caused little or no effect independently, but when combined, resulted in very high incidence of intracranial haemorrhage.1, 3, 27, 38 BIOLOGY OF CCMS PROTEIN FUNCTION AND
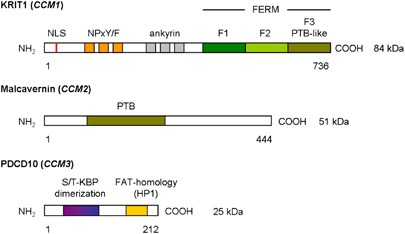
EXPRESSION PATTERN The _KRIT1_ gene contains 20 exons of which 16 encode a 736 amino acid protein containing three NPxY/F motifs and three ankyrin repeat domains at the N-terminus, and one
C-terminal band 4.1 ezrin radixin moesin domain (FERM) found in exons 14–20 (Figure 1).1, 2, 27, 39, 40, 41 The NPxY/F motifs may be involved in dimerization either intramolecular folding of
the KRIT1 protein, resulting in a closed and open conformation of KRIT1.41 After that, the first NPxY/F motif interacts with the α-isoform of the _β_1-integrin regulator integrin
cytoplasmic adaptor protein 1 (ICAP1_α_). ICAP1_α_ is a 200 amino acid protein containing a PTB domain and even as KRIT1 a nuclear localization signal motif in the N-terminus. There is
evidence that both KRIT1 and ICAP1_α_ can translocate into the nucleus, where they could cooperate in regulating gene expression. In particular, an open/closed conformation switch regulates
KRIT1 nucleocytoplasmic shuttling and molecular interactions.40, 41 The ankyrin repeats in KRIT1 are thought to be involved in protein–protein interaction and have been found in many
proteins. No partner interacting with KRIT1 ankyrin repeats has yet been found .27, 39, 41 The FERM domain in KRIT1 is composed of three subdomains, F1–F3, arranged in cloverleaf shape. The
F3 subdomain has a PTB-like domain, which recognizes the NPxY/F motif on the cytoplasmic tail of transmembrane receptors. Rap1a is also bind by the FERM domain, suggesting that KRIT1 may
function as a scaffold for transmembrane receptors and Rap1a.27, 29, 39, 41 The _MGC4607_ gene contains 10 exons encoding Malcavernin, a 444 amino acid protein containing a PTB domain
similar to that of ICAP1_α_. Malcavernin binds KRIT1 by the PTB domain and inhibits in this way nuclear translocation of the KRIT1–ICAP1_α_ complex.2, 7, 21, 41 The _PDCD10_ gene contains
seven exons encoding a 212 amino acid protein containing a dimerization domain at the N-terminus, and a C-terminal focal adhesion targeting-homology domain with a highly conserved HP1
surface.42 Previous studies suggested also the presence of an N-terminal serine/threonine kinase binding and phosphorylation domain, which binds proteins of the germinal centre kinase III
family (STK24, STK25, and Mst4).2, 30, 43, 44, 45, 46 The dimerization domain mediates dimerization of PDCD10. The Fat-homology domain is important for stabilization of the expressed PDCD10
protein and interacts with the PTB domain of Malcavernin and paxillin LD motifs.42 PDCD10 also binds Ptdlns(3,4,5)P3 and functions in this way in the (PI3k–)PIP3–PDPK1–Akt signalling
pathway.23, 47 He _et al_ suggested that the C-terminus of PDCD10 could be important in the stabilization of VEGFR2 signalling, which is crucial for vascular development.23, 48 Despite the
vascular nature of CCM, _in situ_ hybridization studies have shown _KRIT1_ mRNA and protein expression in astrocytes, neurons, and various epithelial cells. KRIT1 protein was also detected
in vascular endothelial cells during early angiogenesis, localized in the cell–cell junctions.3, 49 Guzeloglu-Kayisli _et al_50 demonstrate that KRIT1 is also present in endothelial cells
and cells involved in the formation of the blood-brain barrier, which implicates an important role for KRIT1 in intercellular communication and adherence. _MGC4607_ mRNA expression has been
detected in neurons and astrocytes, as well as in cerebral vessels. _PDCD10_ mRNA is expressed in neuronal cells at adult stages, but also during embryogenesis.3, 51 Additional, neural
expression of _KRIT1_, _MGC4607_ and _PDCD10_ imply that vascular malformations in CCM could also result from a defect in signalling between endothelial and neural cells, but it is still
unclear whether the primary defect is of vascular or neuronal origin.3 In spite of this, most research has been focused on endothelial cells. HISTOLOGY The vessel wall in CCM is
characterized by less and abnormal junction formation between endothelial cells. After that, the expression of intercellular junction proteins is increased to compensate for the loose of
cell contacts. Another characteristic of CCM is the lack of subendothelial support in the vessel wall of CCM made visible by decline in the presence of perivascular supporting cells
(pericytes) and deposition of a basal lamina with disorganized collagen bundles. In addition, the formation of microgaps at the interendothelial junction sites was observed using scanning
electron microscopy.52, 53 Zhao _et al_54 suggested that CCM may develop as a result of irregular organization of endothelial cells, as a consequence of an increased proliferation and
migration potential of these cells. In line with this hypothesis, increased migratory and proliferatory endothelial cell function would indeed require reduced cell–cell contact and reduced
presence of pericytes. MOLECULAR PATHOGENESIS OF CCMS At the molecular level CCM proteins regulate cell–cell adhesion (Figure 2a), cell polarity and most likely cell adhesion to the
extracellular matrix (Figure 2b).1, 46, 55 CELL–CELL ADHESION Initiation and maintenance of cell–cell adhesion require the assembly of adherence junctions. The formation of these adherence
junctions is stimulated by Rap1a, which forms a complex with KRIT1. Rap1a recruits KRIT1 to the plasma membrane, where it binds to the heart of glass 1 (HEG1) receptor to form a ternary
complex of HEG1, KRIT1 and Malcavernin.19, 41, 56 HEG1 is a transmembrane protein, expressed specifically in the endothelium and endocardium. No binding ligand for HEG1 is currently known,
although it has been suggested in previous studies that HEG1 may be involved in the Wnt/_β_-catenin signalling, possibly by binding KRIT1.41, 57, 58, 59 KRIT1 binds _β_-catenin and stimulate
the association of _β_-catenin with vascular endothelial-cadherin, required for adherence junction formation.60 _KRIT1_ may also function as a tumour suppressor gene; KRIT1-_β_-catenin
binding prevents _β_-catenin translocation to the nucleus where displacement of the transcriptional repressor Groucho from T-cell factor proteins by _β_-catenin would activate _Wnt_ target
gene expression.55, 61, 62 However, _β_-catenin activity in the nucleus is also vital for the blood-brain barrier, as many regulatory proteins involved in its development are under
Wnt/_β_-catenin control.41, 62 CELL POLARITY Adherence junctions also promote tight junction assembly. This takes place by the formation of a ternary complex of KRIT1, AF6/afadin and
claudin-5.55, 63 Tight junctions may function as a physical barrier along the cell surface. As a consequence of asymmetrical distribution of proteins and lipids across this barrier, cell
polarization takes place.41, 64 Cell polarity is important in the process of lumen formation.41 Except tight junction formation, cell polarity is also established through a reshaping of the
intracellular cytoskeleton organization. This is regulated by ROCK, a RhoA effector. Crose _et al_65 showed that Malcavernin regulates RhoA protein level. Malcavernin binding of Smurf1
increases Smurf1-mediated degradation of RhoA. After that, Borikova _et al_66 also showed that KRIT1 and PDCD10 in addition to Malcavernin are required for the regulation of RhoA protein
levels. KRIT1 is a negative regulator of RhoA activity. The functional mechanism of KRIT1 is not yet totally known. In contrast, some aspects of PDCD10 inhibition of RhoA activation has been
elucidated, as PDCD10 acts by stabilization of germinal centre kinase III proteins and subsequent activation of moesin, a RhoA inhibitor.67, 68, 69 Loss of the CCM proteins results in an
increase of RhoA activity and changes in regulation of ROCK and the cytoskeleton rigidity. CELL ADHESION TO THE EXTRACELLULAR MATRIX _β_1-integrin, essential for the control of the
intracellular cytoskeleton organization, regulates endothelial cell adhesion to the extracellular matrix.39, 70 It is proposed that _β_1-integrin signal to CDC42 and Rac1.41, 71, 72 Both are
required for the induction of vacuole and lumen formation in vascular endothelial cells.39, 43, 73, 74 In addition, _β_1-integrin also promotes blood vessel maturation by stimulating the
adhesion of mural cells to endothelial cells.41 _β_1-integrin function is inhibited by binding of ICAP1_α_. KRIT1 competes with _β_1-integrin for binding to ICAP1_α_, suggesting that KRIT1
may regulate the ICAP1_α_ inhibitory effect on _β_1-integrin.7, 27, 28, 29, 39, 41 Cell adhesion to the extracellular matrix induces formation of focal adhesion sites in which plaque
proteins, such as vinculin and paxillin, provide a bridge between _β_-integrins and the actin cytoskeleton. Subsequent activation of signalling cascades, regulated by focal adhesion kinase,
promote actin cytoskeleton plasticity.75 Malcavernin has been shown to be capable to regulate actin cytoskeleton plasticity. In response to hyperosmotic shock, restoration of cell volume and
cell shape is regulated by the p38 MAPK signalling cascade, controlled by Malcavernin. Malcavernin acts as a scaffold protein for Rac1 and the upstream kinases MEKK3 and MKK3. The p38 MAPK
signalling pathway leads to the activation of heat shock protein 27, which in turn activates actin polymerization and stabilization.29, 41, 55, 73, 76 CLINICAL MANAGEMENT OF CCMS GENETIC
COUNSELLING AND MOLECULAR DIAGNOSIS To estimate the genetic risk of CCM, three key points are essential (Figure 3):3 * a detailed three-generation family tree with specific enquiry about
seizures, cerebral haemorrhages, focal neurological deficits and (recurrent) headaches. * MRI of the brain to differentiate between solitary or multiple CCM lesions. * age of onset. Genetic
testing for _KRIT1_, _MGC4607_ and _PDCD10_ can confirm the clinical diagnosis in patients, and enables predictive and prenatal testing. The yield of mutation screening in CCM depends on
family history. If only a single lesion can be detected, familial transmission is extremely rare. In contrast, sporadic cases with multiple cerebral lesions are most likely to have a genetic
cause and need to be considered as familial cases. In these cases, genetic screening of all three _CCM_ genes is indicated. The sensitivity of this screen is estimated to be 57%; therefore,
the patient should be aware that a negative test does not exclude a genetic cause.2, 3 The explanation for a negative test may be a somatic mosaicism of a _de novo_ mutation during
gestation, which is not always detectable in DNA extracted from peripheral mononuclear blood cells. Also additional mutations outside the CCM coding exons may account for altered
transcription of CCM associated proteins and fail to be detected by conventional gene mapping techniques.2 In familial cases, sensitivity of genetic screening of all three _CCM_ genes in a
CCM proband with an affected relative is 96%. Once the mutation has been identified in a proband, sensitivity of screening of the relatives of this particular patient is 100%.2 Genetic
counselling is important to help patients and relatives to come to an informed choice. When mutation screening is negative, predictive testing of relatives is not an option, which precludes
the need for a magnetic resonance imaging (MRI). When mutation screening is positive, an additional MRI would be recommended. Although the sensitivity of MRI is very high, MRI as an initial
screening test does not exclude a predisposition for CCM, as the disease may be in its latent phase, devoid of CNS lesions.2, 27 Predictive testing of minors should not be performed, given
the possible psychological and socio-economic consequences of genetic testing, late onset, and reduced penetrance.2 PRENATAL DIAGNOSIS AND PREGNANCY Prenatal diagnosis or pre-implantation
genetic diagnosis is technically feasible in known familial mutations. Decisions about termination of pregnancy in case of familial mutation detection in a foetus might be difficult, because
of reduced penetrance and late onset of symptoms. There is no contra-indication for pregnancy and normal delivery in patients with identified small lesions, without recent clinical signs of
haemorrhage. Large lesions or recent symptomatic haemorrhages are a relative contraindication for pregnancy. In case of pregnancy, caesarean section should then be considered.2, 27 CLINICAL
MANAGEMENT OF CCMS Clinical monitoring of CCM depends on the presence of clinical manifestations. In asymptomatic individuals with an increased risk of CCM, a MRI analysis every 1 or 2
years should be considered. In our hospital MRI will be performed in carriers or at-risk persons. Only if neurological problems arise or increase, MRI will be repeated. The indication for
surgery should be discussed individually with the patient in an experienced neurosurgical centre. Thereby, patients clinical course in combination with MRI characteristics of the CCM lesion,
such as localization, size or new haemorrhage, are important factors for the decision of surgery. In case of deep-seated or brainstem lesions, surgery is associated with a morbidity rate of
30–70% and a mortality rate of 2%. Stereotactic radiosurgery for these lesions remains controversial.77, 78, 79 Medical treatment consists of inhibition of RhoA by simvastatin, or its
effector protein ROCK by fasudil. Also cyclic adenosine monophosphate-elevating drugs should be considered. All of them stabilize CCM lesions by improving vascular integrity.66, 79, 80, 81,
82 Preventing progression of CCM lesions could be reached by sorafenib, an anti-angiogenic drug, targeting VEGF receptors and ERK signalling, which is enhanced in the endothelium of CCM
lesions.83, 84 Treatment with antiplatelet drugs should be avoided, whereas anticoagulation with coumadin derivatives is contra-indicated .2, 19, 53 PROGNOSIS OF CCMS The long-term prognosis
of familial CCM is not well known, but the available data suggest that it is quite favourable after (surgical) treatment. MRI identified new lesions appear at a rate of 0.2–0.4 lesions per
patient year. The new onset seizure rate is 2.4% per patient year and the haemorrhage rate is 3.1%.2, 3, 10, 27, 85 CONCLUSIONS The pathogenesis of CCM remains to date incompletely
clarified. One theory is a perturbed relationship between adhesion and migration of endothelial precursor cells during the formation of the primary vascular plexus. Initiation, guidance and
termination of migration are precisely regulated by interaction with the extracellular matrix and neighbouring cells. Adhesion and migration are linked by the CCM pathway proteins. CCM
complex components function as bridging molecules between junctional and cytoplasmic proteins. Loss-of-function of one of the CCM proteins leads to a decrease in adhesion. This theory is
mainly based on research performed in endothelial cells. Additional studies to the effect of interaction between neural and endothelial cells are necessary, as it remains unclear whether the
primary defect of CCM is of vascular or neuronal origin. REFERENCES * Gore AV, Lampugnani MG, Dye L _et al_: Combinatorial interaction between CCM pathway genes precipitates hemorrhagic
stroke. _Dis Model Mech_ 2008; 1: 275–281. Article CAS PubMed PubMed Central Google Scholar * Labauge P, Denier C, Bergametti F _et al_: Genetics of cavernous angiomas. _Lancet Neurol_
2007; 6: 237–244. Article CAS PubMed Google Scholar * Revencu N, Vikkula M : Cerebral cavernous malformation: new molecular and clinical insights. _J Med Genet_ 2006; 43: 716–721.
Article CAS PubMed PubMed Central Google Scholar * Dashti SR, Hoffer A, Hu YC _et al_: Molecular genetics of familial cerebral cavernous malformations. _Neurosurg Focus_ 2006; 21: e2.
Article PubMed Google Scholar * Rigamonti D, Hadley MN, Drayer BP _et al_: Cerebral cavernous malformations. Incidence and familial occurrence. _N Engl J Med_ 1988; 319: 343–347. Article
CAS PubMed Google Scholar * Felbor U, Sure U, Grimm T _et al_: Genetics of cerebral cavernous angioma. _Zentralbl Neurochir_ 2006; 67: 110–116. Article CAS PubMed Google Scholar *
Brouillard P, Vikkula M : Genetic causes of vascular malformations. _Hum Mol Genet_ 2007; 16 (Spec. no. 2): R140–R149. Article CAS PubMed Google Scholar * Gault J, Shenkar R, Recksiek P
_et al_: Biallelic somatic and germ line CCM1 truncating mutations in a cerebral cavernous malformation lesion. _Stroke_ 2005; 36: 872–874. Article PubMed Google Scholar * Brunereau L,
Labauge P, Tournier-Lasserve E _et al_: Familial form of intracranial cavernous angioma: MR imaging findings in 51 families. _Radiology_ 2000; 214: 209–216. Article CAS PubMed Google
Scholar * Labauge P, Brunereau L, Laberge S _et al_: Prospective follow-up of 33 asymptomatic patients with familial cerebral cavernous malformations. _Neurology_ 2001; 57: 1825–1828.
Article CAS PubMed Google Scholar * Siegel AM : Familial cavernous angioma: an unknown, known disease. _Acta Neurol Scand_ 1998; 98: 369–371. Article CAS PubMed Google Scholar *
Otten P, Pizzolato GP, Rilliet B _et al_: [131 cases of cavernous angioma (cavernomas) of the CNS, discovered by retrospective analysis of 24,535 autopsies]. _Neurochirurgie_ 1989; 35:
82–83, 128–131. CAS PubMed Google Scholar * Zabramski JM, Wascher TM, Spetzler RF _et al_: The natural history of familial cavernous malformations: results of an ongoing study. _J
Neurosurg_ 1994; 80: 422–432. Article CAS PubMed Google Scholar * Rigamonti D, Drayer BP, Johnson PC _et al_: The MRI appearance of cavernous malformations (angiomas). _J Neurosurg_
1987; 67: 518–524. Article CAS PubMed Google Scholar * Denier C, Labauge P, Brunereau L _et al_: Clinical features of cerebral cavernous malformations patients with KRIT1 mutations. _Ann
Neurol_ 2004; 55: 213–220. Article CAS PubMed Google Scholar * Eerola I, Plate KH, Spiegel R _et al_: KRIT1 is mutated in hyperkeratotic cutaneous capillary-venous malformation
associated with cerebral capillary malformation. _Hum Mol Genet_ 2000; 9: 1351–1355. Article CAS PubMed Google Scholar * Labauge P, Krivosic V, Denier C _et al_: Frequency of retinal
cavernomas in 60 patients with familial cerebral cavernomas: a clinical and genetic study. _Arch Ophthalmol_ 2006; 124: 885–886. Article PubMed Google Scholar * Sirvente J, Enjolras O,
Wassef M _et al_: Frequency and phenotypes of cutaneous malformations in a consecutive series of 417 patients with familial cerebral cavernous malformations. _J Eur Acad Dermatol Venereol_
2009; 23: 1066–1072. Article CAS PubMed Google Scholar * Kleaveland B, Zheng X, Liu JJ _et al_: Regulation of cardiovascular development and integrity by the heart of glass-cerebral
cavernous malformation protein pathway. _Nat Med_ 2009; 15: 169–176. Article CAS PubMed PubMed Central Google Scholar * Laberge-le Couteulx S, Jung HH, Labauge P _et al_: Truncating
mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. _Nat Genet_ 1999; 23: 189–193. Article CAS PubMed Google Scholar * Liquori CL, Berg MJ, Siegel AM _et al_:
Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. _Am J Hum Genet_ 2003; 73: 1459–1464. Article CAS
PubMed PubMed Central Google Scholar * Bergametti F, Denier C, Labauge P _et al_: Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. _Am J Hum
Genet_ 2005; 76: 42–51. Article CAS PubMed Google Scholar * Dibble CF, Horst JA, Malone MH _et al_: Defining the functional domain of programmed cell death 10 through its interactions
with phosphatidylinositol-3,4,5,-trisposphate. _PLoS One_ 2010; 5: e11740. Article PubMed PubMed Central CAS Google Scholar * Liquori CL, Berg MJ, Squitieri F _et al_: Low frequency of
PDCD10 mutations in a panel of CCM3 probands: potential for a fourth CCM locus. _Hum Mutat_ 2006; 27: 118. Article PubMed Google Scholar * Gianfrancesco F, Esposito T, Penco S _et al_:
ZPLD1 gene is disrupted in a patient with balanced translocation that exhibits cerebral cavernous malformations. _Neuroscience_ 2008; 155: 345–349. Article CAS PubMed Google Scholar *
Riant F, Bergametti F, Ayrignac X _et al_: Recent insights into cerebral cavernous malformations: the molecular genetics of CCM. _FEBS J_ 2010; 277: 1070–1075. Article CAS PubMed Google
Scholar * Gault J, Sarin H, Awadallah NA _et al_: Pathobiology of human cerebrovascular malformations: basic mechanisms and clinical relevance. _Neurosurgery_ 2004; 55: 1–16; discussion
16–17. PubMed Google Scholar * Wang QK : Update on the molecular genetics of vascular anomalies. _Lymphat Res Biol_ 2005; 3: 226–233. Article CAS PubMed Google Scholar * Plummer NW,
Zawistowski JS, Marchuk DA : Genetics of cerebral cavernous malformations. _Curr Neurol Neurosci Rep_ 2005; 5: 391–396. Article CAS PubMed Google Scholar * Voss K, Stahl S, Hogan BM _et
al_: Functional analyses of human and zebrafish 18-amino acid in-frame deletion pave the way for domain mapping of the cerebral cavernous malformation 3 protein. _Hum Mutat_ 2009; 30:
1003–1011. Article CAS PubMed Google Scholar * Denier C, Labauge P, Bergametti F _et al_: Genotype-phenotype correlations in cerebral cavernous malformations patients. _Ann Neurol_ 2006;
60: 550–556. Article PubMed Google Scholar * Craig HD, Gunel M, Cepeda O _et al_: Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous
malformation, at 7p15-13 and 3q25.2-27. _Hum Mol Genet_ 1998; 7: 1851–1858. Article CAS PubMed Google Scholar * Akers AL, Johnson E, Steinberg GK _et al_: Biallelic somatic and germline
mutations in cerebral cavernous malformations (CCMs): evidence for a two-hit mechanism of CCM pathogenesis. _Hum Mol Genet_ 2009; 18: 919–930. Article CAS PubMed Google Scholar *
McDonald DA, Shenkar R, Shi C _et al_: A novel mouse model of cerebral cavernous malformations based on the two-hit mutation hypothesis recapitulates the human disease. _Hum Mol Genet_ 2011;
20: 211–222. Article CAS PubMed Google Scholar * Goitre L, Balzac F, Degani S _et al_: KRIT1 regulates the homeostasis of intracellular reactive oxygen species. _PLoS One_ 2010; 5:
e11786. Article PubMed PubMed Central CAS Google Scholar * Belik J, Jerkic M, McIntyre BAS _et al_: Age-dependent endothelial nitric oxide synthase uncoupling in pulmonary arteries of
endoglin heterozygous mice. _Am J Physiol Lung Cell Mol Physiol_ 2009; 297: L1170–L1178. Article CAS PubMed Google Scholar * Louvi A, Chen L, Two AM _et al_: Loss of cerebral cavernous
malformation 3 (Ccm3) in neuroglia leads to CCM and vascular pathology. _PNAS_ 2011; 108: 3737–3742. Article CAS PubMed PubMed Central Google Scholar * Knudson AG : Two genetic hits
(more or less) to cancer. _Nat Rev Cancer_ 2001; 1: 157–162. Article CAS PubMed Google Scholar * Marchuk DA, Srinivasan S, Squire TL _et al_: Vascular morphogenesis: tales of two
syndromes. _Hum Mol Genet_ 2003; 12 (Spec. no. 1): R97–112. Article CAS PubMed Google Scholar * Francalanci F, Avolio M, De Luca E _et al_: Structural and functional differences between
KRIT1A and KRIT1B isoforms: a framework for understanding CCM pathogenesis. _Exp Cell Res_ 2009; 315: 285–303. Article CAS PubMed Google Scholar * Faurobert E, Albiges-Rizo C : Recent
insights into cerebral cavernous malformations: a complex jigsaw puzzle under construction. _FEBS J_ 2010; 277: 1084–1096. Article CAS PubMed PubMed Central Google Scholar * Li X, Zhang
R, Zhang H _et al_: Crystal structure of CCM3, a cerebral cavernous malformation protein critical for vascular integrity. _J Biol Chem_ 2010; 285: 24099–24107. Article CAS PubMed PubMed
Central Google Scholar * Huang CY, Wu YM, Hsu CY _et al_: Caspase activation of mammalian sterile 20-like kinase 3 (Mst3). Nuclear translocation and induction of apoptosis. _J Biol Chem_
2002; 277: 34367–34374. Article CAS PubMed Google Scholar * Lu TJ, Lai WY, Huang CY _et al_: Inhibition of cell migration by autophosphorylated mammalian sterile 20-like kinase 3 (MST3)
involves paxillin and protein-tyrosine phosphatase-PEST. _J Biol Chem_ 2006; 281: 38405–38417. Article CAS PubMed Google Scholar * Ma X, Zhao H, Shan J _et al_: PDCD10 interacts with
Ste20-related kinase MST4 to promote cell growth and transformation via modulation of the ERK pathway. _Mol Biol Cell_ 2007; 18: 1965–1978. Article CAS PubMed PubMed Central Google
Scholar * Voss K, Stahl S, Schleider E _et al_: CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. _Neurogenetics_ 2007; 8: 249–256. Article CAS
PubMed Google Scholar * Schleider E, Stahl S, Wüstehube J _et al_: Evidence for anti-angiogenic and pro-survival functions of the cerebral cavernous malformation protein 3.
_Neurogenetics_ 2011; 12: 83–86. Article CAS PubMed Google Scholar * He Y, Zhang H, Yu L _et al_: Stabilization of VEGFR2 signalling by cerebral cavernous malformation 3 is critical for
vascular development. _Sci Signal_ 2010; 3: ra26. PubMed PubMed Central Google Scholar * Denier C, Gasc JM, Chapon F _et al_: Krit1/cerebral cavernous malformation 1 mRNA is
preferentially expressed in neurons and epithelial cells in embryo and adult. _Mech Dev_ 2002; 117: 363–367. Article CAS PubMed Google Scholar * Guzeloglu-Kayisli O, Amankulor NM,
Voorhees J _et al_: Krit1/cerebral cavernous malformation 1 protein localizes to vascular endothelium, astrocytes, and pyramidal cells of the adult human cerebral cortex. _Neurosurgery_
2004; 54: 943–949. Article PubMed Google Scholar * Petit N, Blecon A, Denier C _et al_: Patterns of expression of the three cerebral cavernous malformation (CCM) genes during embryonic
and postnatal brain development. _Gene Expr Patterns_ 2006; 6: 495–503. Article CAS PubMed Google Scholar * Wong JH, Awad IA, Kim JH : Ultrastructural pathological features of
cerebrovascular malformations: a preliminary report. _Neurosurgery_ 2000; 46: 1454–1459. Article CAS PubMed Google Scholar * Burkhardt JK, Schmidt D, Schoenauer R _et al_: Upregulation
of transmembrane endothelial junction proteins in human cerebral cavernous malformations. _Neurosurg Focus_ 2010; 29: E3. Article PubMed Google Scholar * Zhao Y, Tan YZ, Zhou LF _et al_:
Morphological observation and _in vitro_ angiogenesis assay of endothelial cells isolated from human cerebral cavernous malformations. _Stroke_ 2007; 38: 1313–1319. Article PubMed Google
Scholar * Dejana E, Tournier-Lasserve E, Weinstein BM : The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. _Dev Cell_ 2009; 16:
209–221. Article CAS PubMed Google Scholar * Whitehead KJ, Plummer NW, Adams JA _et al_: Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous
malformations. _Development_ 2004; 131: 1437–1448. Article CAS PubMed Google Scholar * Huang L, Ren J, Chen D _et al_: MUC1 cytoplasmic domain coactivates Wnt target gene transcription
and confers transformation. _Cancer Biol Ther_ 2003; 2: 702–706. CAS PubMed Google Scholar * Lang T, Hansson GC, Samuelsson T : An inventory of mucin genes in the chicken genome shows
that the mucin domain of Muc13 is encoded by multiple exons and that ovomucin is part of a locus of related gel-forming mucins. _BMC Genomics_ 2006; 7: 197. Article PubMed PubMed Central
CAS Google Scholar * Gopal U, Venkatraman J, Niranjali D _et al_: Interaction of MUC1 with beta-catenin modulates the Wnt target Gene cyclinD1 in _H. pylori_-induced gastric cancer. _Mol
Carcinog_ 2007; 46: 807–817. Article CAS Google Scholar * Glading AJ, Ginsberg MH : Rap1 and its effector KRIT1/CCM1 regulate beta-catenin signalling. _Dis Model Mech_ 2010; 3: 73–83.
Article CAS PubMed Google Scholar * Clevers H : Wnt/beta-catenin signalling in development and disease. _Cell_ 2006; 127: 469–480. Article CAS PubMed Google Scholar * Liebner S,
Corada M, Bangsow T _et al_: Wnt/beta-catenin signalling controls development of the blood-brain barrier. _J Cell Biol_ 2008; 183: 409–417. Article CAS PubMed PubMed Central Google
Scholar * Gonzalez-Mariscal L, Tapia R, Chamorro D : Crosstalk of tight junction components with signalling pathways. _Biochim Biophys Acta_ 2008; 1778: 729–756. Article CAS PubMed
Google Scholar * Lampugnani MG, Orsenigo F, Rudini N _et al_: CCM1 regulates vascular-lumen organization by inducing endothelial polarity. _J Cell Sci_ 2010; 123: 1073–1080. Article CAS
PubMed Google Scholar * Crose LE, Hilder TL, Sciaky N _et al_: Cerebral cavernous malformation 2 protein promotes smad ubiquitin regulatory factor 1-mediated RhoA degradation in
endothelial cells. _J Biol Chem_ 2009; 284: 13301–13305. Article CAS PubMed PubMed Central Google Scholar * Borikova AL, Dibble CF, Sciaky N _et al_: Rho kinase inhibition rescues the
endothelial cell cerebral cavernous malformation phenotype. _J Biol Chem_ 2010; 285: 11760–11764. Article CAS PubMed PubMed Central Google Scholar * Zheng X, Xu C, Di Lorenzo A _et al_:
CCM3 signalling through sterile 20-like kinases plays an essential role during zebrafish cardiovascular development and cerebral cavernous malformations. _J Clin Invest_ 2010; 120:
2795–2804. Article CAS PubMed PubMed Central Google Scholar * Preisinger C, Short B, De Corte V _et al_: YSK1 is activated by the Golgi matrix protein GM130 and plays a role in cell
migration through its substrate 14-3-3 {zeta}. _J Cell Biol_ 2004; 164: 1009–1020. Article CAS PubMed PubMed Central Google Scholar * Fidalgo M, Fraile M, Pires A _et al_: CCM3/PDCD10
stabilizes GCKIII proteins to promote Golgi assembly and cell orientation. _J Cell Sc_ 2010; 123: 1274–1284. Article CAS Google Scholar * Rupp PA, Little CD : Integrins in vascular
development. _Circ Res_ 2001; 89: 566–572. Article CAS PubMed Google Scholar * Bayless KJ, Davis GE : The Cdc42 and Rac1 GTPases are required for capillary lumen formation in
three-dimensional extracellular matrices. _J Cell Sci_ 2002; 115: 1123–1136. Article CAS PubMed Google Scholar * Iruela-Arispe ML, Davis GE : Cellular and molecular mechanisms of
vascular lumen formation. _Developmental Cell_ 2009; 16: 222–231. Article CAS PubMed PubMed Central Google Scholar * Whitehead KJ, Chan AC, Navankasattusas S _et al_: The cerebral
cavernous malformation signalling pathway promotes vascular integrity via Rho GTPases. _Nat Med_ 2009; 15: 177–184. Article CAS PubMed PubMed Central Google Scholar * Liu H, Rigamonti
D, Badr A _et al_: Ccm1 regulates microvascular morphogenesis during angiogenesis. _J Vasc Res_ 2011; 48: 130–140. Article CAS PubMed Google Scholar * Harburger DS, Calderwood DA :
Integrin signalling at a glance. _J Cell Sci_ 2009; 122: 159–163. Article CAS PubMed Google Scholar * Mudgett JS, Ding J, Guh-Siesel L _et al_: Essential role for p38alpha
mitogen-activated protein kinase in placental angiogenesis. _PNAS_ 2000; 97: 10454–10459. Article CAS PubMed PubMed Central Google Scholar * Pham M, Gross BA, Bendok BR _et al_:
Radiosurgery for angiographically occult vascular malformations. _Neurosurg Focus_ 2009; 26: E16. Article PubMed Google Scholar * Gross BA, Batjer HH, Awad IA _et al_: Brainstem cavernous
malformations. _Neurosurgery_ 2009; 64: E805–E818. Article PubMed Google Scholar * Yadla S, Jabbour PM, Shenkar R _et al_: Cerebral cavernous malformations as a disease of vascular
permeability: from bench to bedside with caution. _Neurosurg Focus_ 2010; 29: E4. Article PubMed PubMed Central Google Scholar * Krisht KM, Whitehead KJ, Niazi T _et al_: The
pathogenetic features of cerebral cavernous malformations: a comprehensive review with therapeutic implications. _Neurosurg Focus_ 2010; 29: E2. Article PubMed Google Scholar * Stockton
RA, Shenkar R, Awad IA _et al_: Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. _J Exp Med_ 2010; 207: 881–896. Article CAS PubMed PubMed
Central Google Scholar * Fukuhara S, Sakurai A, Sano H _et al_: Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function
through an Epac-Rap1 signalling pathway. _Mol Cell Biol_ 2005; 25: 136–146. Article CAS PubMed PubMed Central Google Scholar * Wilhelm S, Carter C, Lynch M _et al_: Discovery and
development of sorafenib: A multikinase inhibitor for treating cancer. _Nat Rev Drug Discov_ 2006; 5: 835–844. Article CAS PubMed Google Scholar * Wüstehube J, Bartol A, Liebler SS _et
al_: Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signalling. _PNAS_ 2010; 107: 12640–12645. Article PubMed PubMed Central Google
Scholar * Moriarity JL, Wetzel M, Clatterbuck RE _et al_: The natural history of cavernous malformations: a prospective study of 68 patients. _Neurosurgery_ 1999; 44: 1166–1173. CAS
PubMed Google Scholar Download references AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Molecular Cardiology Laboratory, Erasmus Medical Center Rotterdam, Rotterdam, The Netherlands Remco
A Haasdijk, Caroline Cheng & Henricus J Duckers * Department of Clinical Genetics, Erasmus Medical Center Rotterdam, Rotterdam, The Netherlands Anneke J Maat-Kievit Authors * Remco A
Haasdijk View author publications You can also search for this author inPubMed Google Scholar * Caroline Cheng View author publications You can also search for this author inPubMed Google
Scholar * Anneke J Maat-Kievit View author publications You can also search for this author inPubMed Google Scholar * Henricus J Duckers View author publications You can also search for this
author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Henricus J Duckers. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no conflict of interest. RIGHTS AND
PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Haasdijk, R., Cheng, C., Maat-Kievit, A. _et al._ Cerebral cavernous malformations: from molecular pathogenesis to
genetic counselling and clinical management. _Eur J Hum Genet_ 20, 134–140 (2012). https://doi.org/10.1038/ejhg.2011.155 Download citation * Received: 04 January 2011 * Revised: 14 June 2011
* Accepted: 05 July 2011 * Published: 10 August 2011 * Issue Date: February 2012 * DOI: https://doi.org/10.1038/ejhg.2011.155 SHARE THIS ARTICLE Anyone you share the following link with
will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative KEYWORDS * CCM * molecular mechanism * genetic counselling


