- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Up to 25% of colorectal cancer (CRC) may be caused by inherited genetic variants that have yet to be identified. Previous genome-wide linkage studies (GWLSs) have identified a new
loci postulated to contain novel CRC risk genes amongst affected families carrying no identifiable mutations in any of the known susceptibility genes for familial CRC syndromes. To undertake
a new GWLS, we recruited members from 54 non-syndromic families from Australia and Spain where at least two first-degree relatives were affected by CRC. We used single-nucleotide
polymorphism arrays to genotype 98 concordant affected relative pairs that were informative for linkage analyses. We tested for genome-wide significance (GWS) for linkage to CRC using a
quantile statistic method, and we found that GWS was achieved at the 5% level. Independently, using the PSEUDO gene-dropping algorithm, we also found that GWS for linkage to CRC was achieved
(_P_=0.02). Merlin non-parametric linkage analysis revealed significant linkage to CRC for chromosomal region 10p15.3–p15.1 and suggestive linkage to CRC for regions on 14q and 9q. The
10p15.3–p15.1 has not been reported to be linked to hereditary CRC in previous linkage studies, but this region does harbour the Kruppel-like factor 6 (_KLF6_) gene that is known to be
altered in common CRC. Further studies aimed at localising the responsible genes, and characterising their function will give insight into the factors responsible for susceptibility in such
families, and perhaps shed further light on the mechanisms of CRC development. SIMILAR CONTENT BEING VIEWED BY OTHERS FINE-MAPPING ANALYSIS INCLUDING OVER 254,000 EAST ASIAN AND EUROPEAN
DESCENDANTS IDENTIFIES 136 PUTATIVE COLORECTAL CANCER SUSCEPTIBILITY GENES Article Open access 26 April 2024 GENETIC RISK FACTORS FOR COLORECTAL CANCER IN MULTIETHNIC INDONESIANS Article
Open access 11 May 2021 EXOME SEQUENCING IDENTIFIES BREAST CANCER SUSCEPTIBILITY GENES AND DEFINES THE CONTRIBUTION OF CODING VARIANTS TO BREAST CANCER RISK Article Open access 17 August
2023 INTRODUCTION There is a good understanding of the molecular genetics underlying the major familial colorectal cancer (CRC) syndromes: Lynch syndrome or hereditary non-polyposis CRC
(HNPCC), familial adenomatous polyposis (FAP) and MYH-associated polyposis (MAP). However, these represent only ∼5% of CRC cases in the community.1, 2 Of the remainder, there is a clear
indication of inheritance suggesting that other predisposing genes are involved in up to 25% of all cases.3 The Amsterdam criteria4 are a set of diagnostic criteria used to help identify
families that are likely to have Lynch syndrome. Families meeting these criteria, but without evidence of mismatch repair (MMR) deficiency (eg, normal immunohistochemical staining for the
MMR genes and/or microsatellite stable cancers) and in the absence of any other identifiable mutations in any of the known familial CRC risk genes, have been termed familial CRC type X5 or
syndrome X6 families or more recently7 as hereditary non-syndromic CRC families. Various genome-wide linkage studies (GWLSs) have been undertaken to identify the underlying causative gene
variants in these and similar families. They have resulted in a number of different regions of linkage being reported,7, 8, 9, 10, 11, 12, 13, 14, 15 with regions on 3q7, 10, 12 and 9q8, 11,
15 being independently identified by different laboratories. It was the aim of our study to undertake a new genome-wide linkage study (GWLS) to investigate what region of the genome was
likely to contain genes conferring increased risk of CRC in a new set of families from Australia and Spain. MATERIALS AND METHODS ASCERTAINMENT OF FAMILIES AND EXCLUSION OF KNOWN SYNDROMES
We restricted our study to non-syndromic, high-risk CRC families, defined as those containing at least one affected person who has one or more first-degree affected relative(s) and where the
known causal mutations had been excluded. This study group was enriched for familial CRC type X families but also included other high-risk CRC families. Volunteers from 54 such families
were enrolled in our study by ascertaining the families with the following characteristics in order of priority: * Preference 1: Patients from families containing at least one affected
person who have had two or more first-degree affected relatives, and at least one affected person diagnosed before reaching 50 years of age (28 families). * Preference 2: Patients from
families containing at least one affected person who have had one or more first-degree affected relatives (26 families). Note that Preference 1 is identical to the Amsterdam I or familial
CRC type X criteria, except for the requirement of multiple generations in the latter. Affected status was defined as diagnosis with either colorectal carcinoma (CA) or advanced adenoma
(AA), where AA was defined as three or more synchronous or metachronous adenomas and/or adenoma(s) with villous morphology, and/or with severe dysplasia, and/or diameter ≥10 mm. Diagnoses
were confirmed by pathology reports. The study was reviewed and approved by the Human Research Ethics Committees of the three participating centres: Flinders Medical Centre, Adelaide (25
families), The Royal Melbourne Hospital, Melbourne (21 families), and Institut Català d’Oncologia, Barcelona (8 families), and informed consent was obtained from all participants. Families
with known colon cancer syndromes including HNPCC or Lynch syndrome, FAP, hereditary mixed polyposis, juvenile polyposis, Peutz–Jeghers, Cowden's syndrome or MYH-associated polyposis
were excluded by review of medical records. Persons for whom tumours were available underwent microsatellite instability (MSI) testing of BAT-25 and BAT-26, and/or immunohistochemistry (IHC)
of hMLHI, hMSH2, hMSH6 and hPMS2, and, if positive, their family was excluded from the study. In 48 families, one tumour was available and in four families two tumours were available. All
tested negative, and therefore these 52 families were included in the study. Two additional families satisfied the requirements for Preference 2, but did not have MSI or IHC data available.
These were also judged to be unlikely to be carrying any of the known mismatch repair gene mutations, as (1) no family member was clinically diagnosed with CRC earlier than 60 years, and (2)
identity-by-descent (IBD) sharing analysis, conducted on derived single-nucleotide polymorphism (SNP) data for each of the 2 Mb regions centred on _hMLHI_, _hMSH2_, _hMSH6_ and _hPMS2_,
indicated no inherited contribution. These two families were therefore included in the study to give a total of 54 families. DEMOGRAPHICS Summary statistics of families that met our
selection criteria were: * 27 familial CRC type X families; * 1 family meeting Preference 1, but failing to meet familial CRC type X requirements, as all affected individuals were in a
single generation; * 14 meeting Preference 1, except that all cases were diagnosed at an age ≥50 years; * 6 meeting Preference 2 and one reported family individual with an age of diagnosis
<50 years; * 6 meeting Preference 2 with minimum age of diagnosis ≥50 years. Genotypes of 132 individuals from these 54 families were used in the analyses. Of these, 98 were from
individuals with a diagnosis of CA with or without adenoma and 34 from individuals with AA but without CA. The number of available affected persons per family ranged from 2 to 11 and the
number of affected persons per family with DNA available ranged from 2 to 4. Of the 54 families, 16 were reported to contain affected persons in three or more generations, whereas 38 were
reported with affected family members in only one or two generations. As summarised in Table 1, the median age at diagnosis of CRC in the families was 59 years, significantly less than the
median ages of diagnosis of 70 years for men and 71 years for women observed in both the general Australian16 and Spanish populations17 (_χ_2=25.8, for females; _χ_2=21.4 for males; both on
1 d.f. and having _P_<10−6). The median age at diagnosis of colorectal adenomas was 56 years. FAMILIES USED IN OUR LINKAGE ANALYSES The Merlin non-parametric linkage (NPL) analyses used
complete pedigree information from all 54 families, as the _S_all statistic weights pedigrees containing more than two affected members. Concordant affected pairs used in the analyses are
shown in Table 1. GENOTYPING WITH THE 50K MAPPING ARRAY Blood samples obtained from consenting family members and DNAs were extracted and genotyped using the GeneChip Human Mapping 50K Array
Xba 240 assay (the 50K Array) (Affymetrix Inc., Santa Clara, CA, USA) as described in Supplementary data, to deliver non-redundant genotype data from 305 individuals. Annotation of SNPs was
conducted as described in Supplementary data. Also, as described in Supplementary data, pedigree checking was undertaken to account for the relatedness of the siblings and adjustments were
made to the data to take account of genotyping errors and linkage disequilibrium between SNPs. GENOME-WIDE SIGNIFICANCE Genome-wide significance (GWS) of the linkage with CRC was tested by
two independent methods: the quantile statistic (QS) described in Saunders _et al_18 and the gene-dropping algorithm in the PSEUDO package.19 The QS method is based on a summary statistic of
the set of likelihood ratio (LR) statistics computed for each SNP for sibling pairs.18 The 5% summary statistic was used here. Its GWS was determined from the results of Saunders _et al._18
IBD sharing was estimated for use in the QS method using an implementation in the _R_ package of the forward/backward algorithm20 with correction for 0.2% genotyping error rate (as found in
Saunders _et al_21). Merlin22 was used to generate NPL scores using the Kong and Cox23 linear model with the Whittemore and Halpern24 _S_all sharing statistic. Empirical GWS levels for the
NPL LOD scores were calculated using PSEUDO 0.3.5 (PSEUDO)19 generated from a pool of 100 random gene-dropping replicates. The mode of action of the gene is unknown so methods not heavily
reliant on a specific genetic model were used. As shown in Saunders _et al_,18 the alternative model used to calculate the LR statistic for the GWLS test does not have a large effect on the
power of the QS statistics. The calculations here used a dominant model with allele frequency 0.3 and penetrance 0.18 for carriers and 0.017 for non-carriers, which was found to perform well
across a range of alternative models.18 For locating the gene, a NPL approach was used, which is again insensitive to the exact inheritance model. EARLY ONSET FAMILIES The GWS of the
statistic was determined for data obtained from 42 families that contained suitable sibling pairs informative for linkage. It would be expected that an individual carrying an inherited
susceptibility allele would have an earlier age of onset than usual for the population. Therefore, additional analyses were carried out on three restricted data sets that included only
families reporting a case of CA or AA diagnosed either before age 55 years or 50 years or 45 years. However, each of these age restrictions reduced the number of individuals available for
analyses compared to the full data set and GWS was not achieved for any of these three additional data sets (data not shown). RESULTS ESTABLISHING GWS The GWS of linkage with CRC was
determined for data (Table 1) obtained from 54 families that contained suitable affected pairs, informative for linkage. Two independent methods were used: the QS,18 that is specific for
families containing sibling pairs and the gene-dropping algorithm in the PSEUDO package,19 that uses all relative pair data. For the subset of 42 families that contain concordant affected
sibling pairs, the 5% QS18 reached the 5% level of GWS. Supplementary Figure 1 shows the set of LR statistics for all SNPs used in the analysis. We also assessed GWS for subsets of earlier
onset families (see below). However, these groups proved to be too small to allow detection of linkage. For the full set of 54 families, GWS of linkage was also assessed by the Merlin
method22 of PSEUDO gene dropping and empirical _P_-values were computed. As summarised in Table 2, a genome-wide threshold of significance (_P_=0.02) was achieved for the highest
non-parametric linkage (NPL) score of 3.45 which occurred at cytoband 10p15.3–10p15.1. LOCATING CHROMOSOMAL REGIONS LINKED TO INCREASED CRC RISK For the genome-wide non-parametric analysis
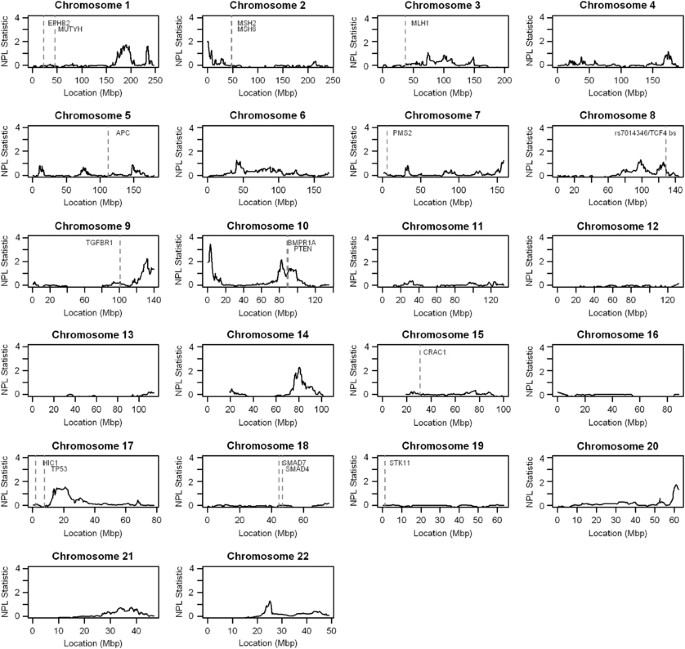
executed in the Merlin package, the results for all 54 families containing two or more genotyped affected members are shown in Figure 1. As summarised in Table 2, the strongest signal was
located on 10p15.3–p15.1 (maxNPL=3.45) for the full set of families. Although this did not quite satisfy a proposed generalised definition for significant linkage (maximal _S_all linear
model nonparametric LOD scores (maxNPL)>3.62),25 we found significant linkage using an empirical gene-dropping approach. Using a 1-LOD support interval approach, we estimated the size of
this region of linkage as 2 Mb and to be bounded by SNPs rs10489254 to rs10494827. Using the same approach, regions of suggestive linkage ((maxNPL)>2.2)25 were observed for a 6.5 Mb
region of chromosome 14q24.3–q31.1 (maxNPL=2.28) and for a 12 Mb region of 9q33.3–q34.3 (maxNPL=2.24). These three regions represent the most promising loci for further study. Regions of
weaker linkage were located across a 19 Mb region on 10q22.3–q24.1 (maxNPL=2.14), across 8 Mb on 2p25.3–p25.1 (maxNPL=2.04) and across 21 Mb on 1q25.2–q32.1 (maxNPL=1.71). SENSITIVITY
ANALYSIS To determine whether geographical differences in founder effect may be contributing to our analyses, we undertook a sensitivity analysis by separately analysing the samples from
Spain and Australia. It was found that the signals in the data from the two countries were consistent and jointly contributed to the overall significance of the linkage results (data not
shown). DISCUSSION In the present GWLS, using 54 Australian and Spanish non-syndromic CRC families, we identified promising regions for further study on chromosomes 1q, 2p, 9q, 10p, 10q and
14q. We also observe a minor linkage peak at 9q22 (Figure 1). The chromosomal region of linkage most strongly implicated to be harbouring a novel susceptibility gene for CRC was
10p15.3–p15.1. Although this region has not previously been implicated as a region of increased risk for hereditary CRC, an examination of current databases provides some evidence for such a
likelihood. This signal on 10p15 does lie in a region that has previously been associated26 with common or sporadic CRC. Interrogation of the RefSeq database27 revealed three protein coding
genes map to the 2 Mb region of 10p15.3–p15.1; _PFKP_, _PITRM1_ and Kruppel-like factor 6 (_KLF6_). Of these, _KLF6_ is a plausible candidate gene for increased risk of familial CRC. KLFs
are key transcription factors,28 and while there is debate26, 29 about the frequency of mutation activation in common CRC, there is good evidence that LOH of _KLF6_ is a feature of common
CRC.29 Interestingly, these inactivation events are rarely observed in HNPCC or FAP,26, 29 two hereditary syndromes that we excluded from our study. There is also biological plausibility for
the 14q and 9q regions to be harbouring new risk genes for hereditary CRC. In particular, it is interesting that the MMR gene, _MLH3_, is among the 84 protein coding genes that map to the
6.5 Mb region of 14q24.3–q31.1 (Supplementary Table 1) and is among the nine genes in this region that have been annotated in the Genecards database30 as being altered in CRC (Supplementary
Table 2). We also note that of the 152 protein coding genes that map to the 12 Mb region of 9q32–q34.13 (Supplementary Table 3), 22 have been annotated in the Genecards database,30 as being
altered in CRC (Supplementary Table 4) and that this includes three key enzymes in the prostaglandin biosynthesis pathway, _COX-1_, PTGES and _PTGES2_. Recently, increased CRC risk was
reported to be associated with a silent mutation in _COX-1_.31 Increased expression of _PTGES2_ and _PTGES_, that function downstream of _COX-2_, has also been correlated with prognosis in
CRC patients.32 It is likely that large scale DNA sequencing of these regions will be needed to determine the identity of the underlying causal variants for CRC in our non-syndromic CRC
families. Importantly, in this particular group of patients where we excluded families that fitted into the known syndromes, we found no linkage peaks near the genes known to cause familial
CRC syndromes including FAP, Lynch syndrome or HNPCC, hereditary mixed polyposis, juvenile polyposis, Peutz–Jeghers, Cowden's syndrome or MAP. Those genes and SNPs, either known to be
causative of established familial CRC syndromes or implicated in other GWLS or GWAS as being pre-disposing for CRC, are shown in Figure 1: _EPHB2_, _MUTYH_, _MSH2_, _MSH6_, _MLH1_, _APC_,
_PMS2_, rs7014346/TCF4-binding site, _TGFBR1_, _BMPR1A_, _PTEN_, _CRAC1_, _SMAD7_, _SMAD4_, _STK11_, _TP53_ and _HIC1_. Interestingly, none of these genes is located within the novel regions
of linkage described in this report, except for _BMPR1A_ and _PTEN_, which reside in the minor region of linkage on 10q22.3–q24.1. However, we also did not find any linkage to those regions
of hereditary but non-syndromic CRC that have been recently reported on 3q,7, 10, 12 7q14 or 9q22,8, 11, 15 and it is interesting to ask why. Similar to ours, these studies7, 8, 9, 10, 11,
12, 13, 14, 15 used similar sample size and also excluded families with mutations in known predisposition genes for CRC to maximise the likelihood of discovering novel CRC risk genes.
However, their experimental designs showed differences with each other and with ours. These included the degree to which families were stratified by clinical phenotypes,9 whether unaffected
relatives were included or excluded from the linkage analyses and whether subjects with AA but no adenocarcinoma were classified as affected.7, 8, 9, 10, 11, 12, 13, 14, 15 A feature of all
these studies, including ours, is that while promising candidate regions have been identified, there is limited concordance in the regions identified. Independent confirmation of common
regions between studies has only been achieved for regions on 3q7, 10, 12 and 9q22.8, 11, 15 Such observations may be explained in a number of ways. One developing view is that outside of
the mutations in the known syndromic genes, there are very few, if any, further high penetrance gene variants that predispose carriers to CRC. The bulk of familial CRC may result from the
co-inheritance of relatively common variants in multiple other genes. These may individually impart a small but finite risk of CRC but in combination are responsible for the observed
elevated risk in non-syndromic families. An alternative explanation is that, in addition to the well-characterised familial CRC genes, mutations in a number of different genes may still be
causative for familial CRC but that the frequency in the population of carriers of mutations in each of these genes is much lower. This could arise through the different target sizes of the
genes or the need to retain certain functions. Clustering of mutations in a single causative gene within any one study, leading to detection of significant linkage, may then arise randomly
through some hidden founder effect in a population group or through interaction of the causative mutation with other common genetic or environmental factors in that population group. A
recent paper33 identifying a causative gene for familial pancreatic cancer is illustrative. Originally identified through whole-genome exon sequencing of an individual with familial
pancreatic cancer, mutations in the same gene were subsequently identified as being causative in 3 of 90 families studied. If there are multiple different genes each contributing to a low
percentage of familial CRC cases, the current study's designs and sizes will continue to have difficulty consistently identifying equivalent genes or loci. We also considered whether
different founder mutations existed in the Spanish and Australian population and that by combining them, there had been a dilution of the linkage signal. In fact a sensitivity analysis
showed that the signals in the data from the two countries were consistent and jointly contributed to the overall significance of the results. Although this does not rule out different
founder effects contributing to the observed differences between studies, it does suggest that it is not likely to be major factor. This should, however, be further investigated by larger
inter-population comparisons. Combining data from across a number of similarly performed individual studies may improve the overall power particularly where accumulated numbers are
sufficient to stratify the data more rigorously. This could be based on any of a number of criteria but could include disease phenotype (either clinical or molecular), familial phenotype or
cancer genotype. To summarise, our data point to the likelihood that a mutation occurring in chromosomal region 10p15.3–p15.1 can cause an increased risk of CRC in these families. Next steps
include localising the responsible genes and characterising their function. This is likely to provide insights into the factors responsible for susceptibility in non-syndromic CRC families
and perhaps shed further light on the mechanisms of CRC development. REFERENCES * Johns LE, Houlston RS : A systematic review and meta-analysis of familial colorectal cancer risk. _Am J
Gastroenterol_ 2001; 96: 2992–3003. Article CAS Google Scholar * Lipton LR, Johnson V, Cummings C _et al_: Refining the Amsterdam Criteria and Bethesda Guidelines: testing algorithms for
the prediction of mismatch repair mutation status in the familial cancer clinic. _J Clin Oncol_ 2004; 22: 4934–4943. Article CAS Google Scholar * Terdiman JP, Conrad PG, Sleisenger MH :
Genetic testing in hereditary colorectal cancer: indications and procedures. _Am J Gastroenterol_ 1999; 94: 2344–2356. Article CAS Google Scholar * Vasen HF, Mecklin JP, Khan PM, Lynch HT
: The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). _Dis Colon Rectum_ 1991; 34: 424–425. Article CAS Google Scholar * Lindor NM, Rabe K,
Petersen GM _et al_: Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. _JAMA_ 2005; 293: 1979–1985. Article CAS
Google Scholar * Lipkin SM, Afrasiabi K : Familial colorectal cancer syndrome X. _Semin Oncol_ 2007; 34: 425–427. Article Google Scholar * Papaemmanuil E, Carvajal-Carmona L, Sellick GS
_et al_: Deciphering the genetics of hereditary non-syndromic colorectal cancer. _Eur J Hum Genet_ 2008; 16: 1477–1486. Article CAS Google Scholar * Wiesner GL, Daley D, Lewis S _et al_:
A subset of familial colorectal neoplasia kindreds linked to chromosome 9q22.2-31.2. _Proc Natl Acad Sci U S A_ 2003; 100: 12961–12965. Article CAS Google Scholar * Daley D, Lewis S,
Platzer P _et al_: Identification of susceptibility genes for cancer in a genome-wide scan: results from the colon neoplasia sibling study. _Am J Hum Genet_ 2008; 82: 723–736. Article CAS
Google Scholar * Kemp Z, Carvajal-Carmona L, Spain S _et al_: Evidence for a colorectal cancer susceptibility locus on chromosome 3q21-q24 from a high-density SNP genome-wide linkage scan.
_Hum Mol Genet_ 2006; 15: 2903–2910. Article CAS Google Scholar * Kemp ZE, Carvajal-Carmona LG, Barclay E _et al_: Evidence of linkage to chromosome 9q22.33 in colorectal cancer kindreds
from the United Kingdom. _Cancer Res_ 2006; 66: 5003–5006. Article CAS Google Scholar * Picelli S, Vandrovcova J, Jones S _et al_: Genome-wide linkage scan for colorectal cancer
susceptibility genes supports linkage to chromosome 3q. _BMC Cancer_ 2008; 8: 87. Article Google Scholar * Djureinovic T, Skoglund J, Vandrovcova J _et al_: A genome wide linkage analysis
in Swedish families with hereditary non-familial adenomatous polyposis/non-hereditary non-polyposis colorectal cancer. _Gut_ 2006; 55: 362–366. Article CAS Google Scholar * Neklason DW,
Kerber RA, Nilson DB _et al_: Common familial colorectal cancer linked to chromosome 7q31: a genome-wide analysis. _Cancer Res_ 2008; 68: 8993–8997. Article CAS Google Scholar *
Gray-McGuire C, Guda K, Adrianto I _et al_: Confirmation of linkage to and localization of familial colon cancer risk haplotype on chromosome 9q22. _Cancer Res_ 2010; 70: 5409–5418. Article
CAS Google Scholar * Weber MF, Banks E, Ward R, Sitas F : Population characteristics related to colorectal cancer testing in New South Wales, Australia: results from the 45 and Up Study
cohort. _J Med Screen_ 2008; 15: 137–142. Article Google Scholar * Ribes J, Navarro M, Cleries R _et al_: Colorectal cancer mortality in Spain: trends and projections for 1985–2019. _Eur J
Gastroenterol Hepatol_ 2009; 21: 92–100. Article Google Scholar * Saunders IW, Hannan GN, Brohede J _et al_: A range of simple summary genome-wide statistics for detecting genetic linkage
using high density marker data. _Genet Epidemiol_ 2007; 31: 565–576. Article Google Scholar * Wigginton JE, Abecasis GR : An evaluation of the replicate pool method: quick estimation of
genome-wide linkage peak p-values. _Genet Epidemiol_ 2006; 30: 320–332. Article Google Scholar * Baum L, Petrie T, Soules G, Weiss N : A maximization technique occuring in the statistical
analysis of probabilistic functions of Markov Chains. _Ann Math Statist_ 1970; 41: 164–171. Article Google Scholar * Saunders IW, Brohede J, Hannan GN : Estimating genotyping error rates
from Mendelian errors in SNP array genotypes and their impact on inference. _Genomics_ 2007; 90: 291–296. Article CAS Google Scholar * Abecasis GR, Cherny SS, Cookson WO, Cardon LR :
Merlin – rapid analysis of dense genetic maps using sparse gene flow trees. _Nat Genet_ 2002; 30: 97–101. Article CAS Google Scholar * Kong A, Cox NJ : Allele-sharing models: LOD scores
and accurate linkage tests. _Am J Hum Genet_ 1997; 61: 1179–1188. Article CAS Google Scholar * Whittemore AS, Halpern J : A class of tests for linkage using affected pedigree members.
_Biometrics_ 1994; 50: 118–127. Article CAS Google Scholar * Lander E, Kruglyak L : Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. _Nat
Genet_ 1995; 11: 241–247. Article CAS Google Scholar * Miyaki M, Yamaguchi T, Iijima T, Funata N, Mori T : Difference in the role of loss of heterozygosity at 10p15 (KLF6 locus) in
colorectal carcinogenesis between sporadic and familial adenomatous polyposis and hereditary nonpolyposis colorectal cancer patients. _Oncology_ 2006; 71: 131–135. Article CAS Google
Scholar * Pruitt KD, Tatusova T, Maglott DR : NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. _Nucleic Acids Res_ 2007;
35: D61–D65. Article CAS Google Scholar * Narla G, Heath KE, Reeves HL _et al_: KLF6, a candidate tumor suppressor gene mutated in prostate cancer. _Science_ 2001; 294: 2563–2566. Article
CAS Google Scholar * Reeves HL, Narla G, Ogunbiyi O _et al_: Kruppel-like factor 6 (KLF6) is a tumor-suppressor gene frequently inactivated in colorectal cancer. _Gastroenterology_ 2004;
126: 1090–1103. Article CAS Google Scholar * Safran M, Chalifa-Caspi V, Shmueli O _et al_: Human gene-centric databases at the Weizmann institute of science: GeneCards, UDB, CroW 21 and
HORDE. _Nucleic Acids Res_ 2003; 31: 142–146. Article CAS Google Scholar * Kury S, Buecher B, Robiou-du-Pont S _et al_: Low-penetrance alleles predisposing to sporadic colorectal cancers:
a French case-controlled genetic association study. _BMC Cancer_ 2008; 8: 326. Article Google Scholar * Seo T, Tatsuguchi A, Shinji S _et al_: Microsomal prostaglandin E synthase protein
levels correlate with prognosis in colorectal cancer patients. _Virchows Arch_ 2009; 454: 667–676. Article CAS Google Scholar * Jones S, Hruban RH, Kamiyama M _et al_: Exomic sequencing
identifies PALB2 as a pancreatic cancer susceptibility gene. _Science_ 2009; 324: 217. Article CAS Google Scholar * Kokko A, Laiho P, Lehtonen R _et al_: EPHB2 germline variants in
patients with colorectal cancer or hyperplastic polyposis. _BMC Cancer_ 2006; 6: 145. Article Google Scholar * Tenesa A, Campbell H, Barnetson R, Porteous M, Dunlop M, Farrington SM :
Association of MUTYH and colorectal cancer. _Br J Cancer_ 2006; 95: 239–242. Article CAS Google Scholar * Vasen HF, Hendriks Y, de Jong AE _et al_: Identification of HNPCC by molecular
analysis of colorectal and endometrial tumors. _Dis Markers_ 2004; 20: 207–213. Article CAS Google Scholar * Bodmer W : Familial adenomatous polyposis (FAP) and its gene, APC. _Cytogenet
Cell Genet_ 1999; 86: 99–104. Article CAS Google Scholar * Tenesa A, Farrington SM, Prendergast JG _et al_: Genome-wide association scan identifies a colorectal cancer susceptibility
locus on 11q23 and replicates risk loci at 8q24 and 18q21. _Nat Genet_ 2008; 40: 631–637. Article CAS Google Scholar * Tuupanen S, Turunen M, Lehtonen R _et al_: The common colorectal
cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. _Nat Genet_ 2009; 41: 885–890. Article CAS Google Scholar * Skoglund Lundin J,
Vandrovcova J, Song B _et al_: TGFBR1 variants TGFBR1(*)6A and Int7G24A are not associated with an increased familial colorectal cancer risk. _Br J Cancer_ 2009; 100: 1674–1679. Article CAS
Google Scholar * Daley D, Morgan W, Lewis S _et al_: Is TGFBR1*6A a susceptibility allele for nonsyndromic familial colorectal neoplasia? _Cancer Epidemiol Biomarkers Prev_ 2007; 16:
892–894. Article CAS Google Scholar * O’Riordan J, O’Donoghue D, Green A _et al_: Hereditary mixed polyposis syndrome due to a Bmpr1a mutation. _Colorectal Dis_ 2010; 12: 570–573. Article
Google Scholar * Nassif NT, Lobo GP, Wu X _et al_: PTEN mutations are common in sporadic microsatellite stable colorectal cancer. _Oncogene_ 2004; 23: 617–628. Article CAS Google
Scholar * Jaeger E, Webb E, Howarth K _et al_: Common genetic variants at the CRAC1 (HMPS) locus on chromosome 15q13.3 influence colorectal cancer risk. _Nat Genet_ 2008; 40: 26–28. Article
CAS Google Scholar * Gemignani F, Moreno V, Landi S _et al_: A TP53 polymorphism is associated with increased risk of colorectal cancer and with reduced levels of TP53 mRNA. _Oncogene_
2004; 23: 1954–1956. Article CAS Google Scholar * Russo A, Bazan V, Iacopetta B, Kerr D, Soussi T, Gebbia N : The TP53 colorectal cancer international collaborative study on the
prognostic and predictive significance of p53 mutation: influence of tumor site, type of mutation, and adjuvant treatment. _J Clin Oncol_ 2005; 23: 7518–7528. Article CAS Google Scholar *
Iacopetta B : TP53 mutation in colorectal cancer. _Hum Mutat_ 2003; 21: 271–276. Article CAS Google Scholar * Pittman AM, Naranjo S, Webb E _et al_: The colorectal cancer risk at 18q21
is caused by a novel variant altering SMAD7 expression. _Genome Res_ 2009; 19: 987–993. Article CAS Google Scholar * Royce SG, Alsop K, Haydon A _et al_: The role of SMAD4 in early onset
colorectal cancer. _Colorectal Dis_ 2010; 12: 213–219. Article CAS Google Scholar * Resta N, Simone C, Mareni C _et al_: STK11 mutations in Peutz-Jeghers syndrome and sporadic colon
cancer. _Cancer Res_ 1998; 58: 4799–4801. CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We thank the patients and their families for their participation in the study,
Bernadette Viney and Kerry Phillips for clinical research nurse support and Drs Konsta Duesing and Mike Buckley for critically reviewing the manuscript. AUTHOR INFORMATION Author notes * Ian
W Saunders & Jason Ross Present address: Current address: Karolinska Institutet, Department NVS, KI-Alzheimer Disease Research Center, Novum, Huddinge, Stockholm, Sweden., * Jesper
Brohede: These authors contributed equally to the data analysis. AUTHORS AND AFFILIATIONS * CSIRO Preventative Health Flagship, North Ryde, New South Wales, Australia Ian W Saunders, Jason
Ross, Jesper Brohede, Glenn Brown, Diana Brookes, Trevor Lockett, Peter L Molloy & Garry N Hannan * CSIRO Mathematics, Informatics and Statistics, Glen Osmond, South Australia, Australia
Ian W Saunders * CSIRO Food and Nutritional Sciences, North Ryde, New South Wales, Australia Jason Ross, Glenn Brown, Diana Brookes, Trevor Lockett, Peter L Molloy & Garry N Hannan *
Department of Medicine, University of Melbourne, and Colorectal Medicine and Genetics, The Royal Melbourne Hospital, Melbourne, Victoria, Australia Finlay Macrae * Flinders Centre for Cancer
Prevention and Control, Flinders University, Bedford Park, Adelaide, South Australia, Australia Graeme P Young * Hereditary Cancer Program, Institut Catala d'Oncologia-IDIBELL,
Barcelona, Spain Ignacio Blanco & Gabriel Capella * Department of Clinical Sciences, Faculty of Medicine, University of Barcelona, Barcelona, Spain Ignacio Blanco & Victor Moreno *
Cancer Prevention and Control Program, Institut Catala d'Oncologia-IDIBELL and CIBERESP, Barcelona, Spain Victor Moreno Authors * Ian W Saunders View author publications You can also
search for this author inPubMed Google Scholar * Jason Ross View author publications You can also search for this author inPubMed Google Scholar * Finlay Macrae View author publications You
can also search for this author inPubMed Google Scholar * Graeme P Young View author publications You can also search for this author inPubMed Google Scholar * Ignacio Blanco View author
publications You can also search for this author inPubMed Google Scholar * Jesper Brohede View author publications You can also search for this author inPubMed Google Scholar * Glenn Brown
View author publications You can also search for this author inPubMed Google Scholar * Diana Brookes View author publications You can also search for this author inPubMed Google Scholar *
Trevor Lockett View author publications You can also search for this author inPubMed Google Scholar * Peter L Molloy View author publications You can also search for this author inPubMed
Google Scholar * Victor Moreno View author publications You can also search for this author inPubMed Google Scholar * Gabriel Capella View author publications You can also search for this
author inPubMed Google Scholar * Garry N Hannan View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Garry N Hannan.
ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no conflict of interest. ADDITIONAL INFORMATION Supplementary Information accompanies the paper on European Journal of Human
Genetics website SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION (DOC 799 KB) RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Saunders, I., Ross, J.,
Macrae, F. _et al._ Evidence of linkage to chromosomes 10p15.3–p15.1, 14q24.3–q31.1 and 9q33.3–q34.3 in non-syndromic colorectal cancer families. _Eur J Hum Genet_ 20, 91–96 (2012).
https://doi.org/10.1038/ejhg.2011.149 Download citation * Received: 05 January 2011 * Revised: 03 June 2011 * Accepted: 24 June 2011 * Published: 10 August 2011 * Issue Date: January 2012 *
DOI: https://doi.org/10.1038/ejhg.2011.149 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * colorectal cancer * linkage * 10p * 14q * 9q








