- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The phagocytosis of apoptotic cells (efferocytosis) shifts macrophages to an anti-inflammatory state through a set of still poorly understood soluble and cell-bound signals.
Apoptosis is a common feature of inflamed tissues, and efferocytosis by tissue macrophages is thought to promote the resolution of inflammation. However, it is not clear how the exposure of
tissue macrophages to inflammatory cues (e.g., PAMPs, DAMPs) in the early stages of inflammation affects immune outcomes of macrophage-apoptotic cell interactions occurring at later stages
of inflammation. To address this, we used low-dose endotoxin conditioning (LEC, 1 ng/ml LPS 18 h) of mouse resident peritoneal macrophages (RPMФ) to model the effects of suboptimal (i.e.,
non-tolerizing), antecedent TLR activation on macrophage inflammatory responses to apoptotic cells. Compared with unconditioned macrophages (MФ), LEC-MФ showed a significant enhancement of
apoptotic cell-driven suppression of many inflammatory cytokines (e.g., TNF, MIP-1_β_, MCP-1). We then found that enzymatic depletion of adenosine or inhibition of the adenosine receptor A2a
on LEC-MФ abrogated apoptotic cell suppression of TNF, and this suppression was entirely dependent on the ecto-enzyme CD73 (AMP→adenosine) but not CD39 (ATP→AMP), both of which are highly
expressed on RPMФ. In addition to a requirement for CD73, we also show that _Adora2a_ levels in macrophages are a critical determinant of TNF suppression by apoptotic cells. LEC treatment of
RPMФ led to a ~3-fold increase in _Adora2a_ and a ~28-fold increase in adenosine sensitivity. Moreover, in RAW264.7 cells, ectopic expression of both A2a and CD73 was required for TNF
suppression by apoptotic cells. In mice, mild, TLR4-dependent inflammation in the lungs and peritoneum caused a rapid increase in macrophage _Adora2a_ and _Adora2b_ levels, and CD73 was
required to limit neutrophil influx in this peritonitis model. Thus immune signaling via the CD73–A2a axis in macrophages links early inflammatory events to subsequent immune responses to
apoptotic cells. SIMILAR CONTENT BEING VIEWED BY OTHERS LIGAND-DEPENDENT KINASE ACTIVITY OF MERTK DRIVES EFFEROCYTOSIS IN HUMAN IPSC-DERIVED MACROPHAGES Article Open access 25 May 2021
MACROPHAGES USE APOPTOTIC CELL-DERIVED METHIONINE AND DNMT3A DURING EFFEROCYTOSIS TO PROMOTE TISSUE RESOLUTION Article 31 March 2022 THE INFLAMMATORY RESPONSE INDUCED BY _PSEUDOMONAS
AERUGINOSA_ IN MACROPHAGES ENHANCES APOPTOTIC CELL REMOVAL Article Open access 27 January 2021 MAIN The phagocytic clearance of dead cells (efferocytosis) from inflamed tissues by resident
macrophages is important for the resolution of inflammation and the restoration of normal tissue function.1, 2, 3 Moreover, failure to promptly clear apoptotic cells can result in secondary
cellular necrosis and loss of membrane integrity that can provoke tissue inflammation and autoimmunity.4, 5, 6, 7, 8 Beyond the removal of cell corpses, efferocytosis also promotes
resolution by suppressing production of pro-inflammatory cytokines (e.g., TNF, IL-1_β_, IL-12) and stimulating anti-inflammatory cytokines and growth factors (e.g., IL-10, TGF_β_, VEGF) in
macrophages.9, 10, 11, 12 However, the molecular mechanisms that underlie the immunomodulatory effects of apoptotic cells on macrophages are incompletely understood. Apoptotic cells release
soluble factors that act as ‘find-me’ signals to stimulate recruitment of phagocytes, including phospholipids, chemokines, and nucleotides such as ATP and UTP.13 Recent reports have shown
that ATP and its catabolic derivatives ADP, AMP, and adenosine, contribute to the immunomodulatory effects of apoptotic cells on macrophages via activation of multiple G-protein coupled
adenosine receptors A2a, A2b and A3.14, 15 The breakdown of extracellular adenine nucleotides (ATP, ADP, AMP) to adenosine by hematopoietic cells is regulated by two key ecto-nucleotidases:
CD39 (ATP/ADP→AMP) and CD73 (AMP→adenosine).16, 17 In mice, CD39 is widely expressed on hematopoietic populations, whereas CD73 expression is restricted to specific immune cell populations,
including regulatory T cells, neutrophils and tissue-resident lung and peritoneal macrophages.18, 19, 20 A2a, A2b, and CD73 have key roles in preventing excessive inflammation and mortality
in response to inflammatory challenges such as influenza and sepsis in mice.21, 22 Although previous reports have shown that adenosine generated during efferocytosis regulates expression of
certain inflammatory factors in macrophages, including _Thbs1,_ CXCL1 (KC), and CXCL2 (MIP-2),14, 15 it is unclear what role the ecto-enzymes CD39 and CD73 have generating adenosine during
efferocytosis. Here we use a combination of _in vitro_ efferocytosis co-cultures and _in vivo_ analyses to show that CD73 has a vital role in generating adenosine during efferocytosis that
acts to mediate suppression of inflammatory responses by endotoxin-conditioned macrophages. RESULTS ENDOTOXIN CONDITIONING OF TISSUE MACROPHAGES ENHANCES THE ANTI-INFLAMMATORY EFFECTS OF
APOPTOTIC CELLS Much of what we know about the immunomodulatory effects of apoptotic cells on macrophages stems from studies using naive/resting macrophages. We reasoned that at the onset of
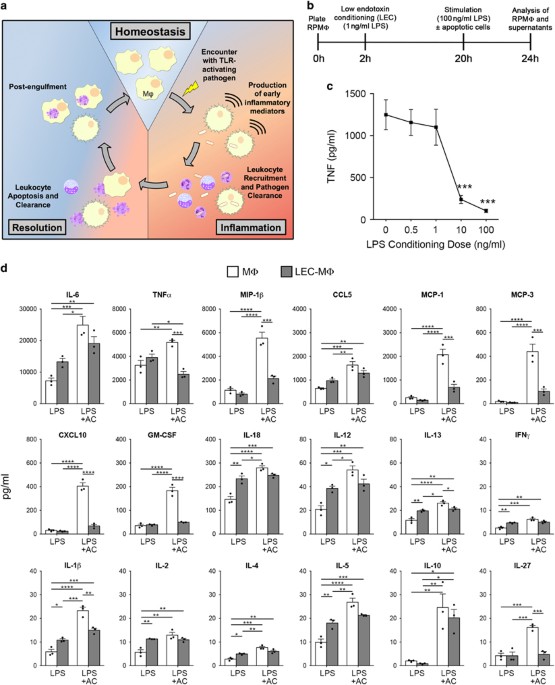
acute inflammation, tissue macrophages will be exposed to inflammatory cues such as TLR agonists before the accumulation of substantial numbers of apoptotic leukocytes (depicted in Figure
1a). To understand how early exposure to such inflammatory cues might affect subsequent responses of macrophages to apoptotic cells, we established an _in vitro_ model system wherein
macrophages harvested from the peritoneum of untreated mice were cultured in the presence or absence of a low dose of ‘ultrapure’ LPS (0.5–1 ng/ml) for 18 h and subsequently stimulated with
a high dose of LPS (100 ng/ml) in the presence or absence of apoptotic cells for 4–8 h and cytokines in the supernatants measured by ELISA and multiplex assays (Figure 1b). As shown in
Figure 1c, we chose 0.5–1 ng/ml LPS as our conditioning dose to avoid potential issues related to endotoxin tolerance.23 Indeed, of the 18 cytokines measured in these experiments we found
that low-dose endotoxin-conditioned macrophages (LEC-MФ) produced either similar or slightly elevated levels of these cytokines following high-dose LPS stimulation compared with
unconditioned macrophages (MФ) (Figure 1d, open _versus_ filled bars in ‘LPS’ condition). Thus, LEC-MФ showed no signs of endotoxin tolerance, enabling us to directly compare the effects of
apoptotic cells on cytokine production between normal and endotoxin-conditioned macrophages. We next compared the levels of 18 cytokines in the supernatants of LEC-MФ and MФ following
high-dose LPS stimulation in the absence or presence of apoptotic cells. The addition of apoptotic cells significantly increased MФ production of all 18 cytokines compared with LPS-only
treated MФ (Figure 1d, open bars). This was an unexpected finding based on previous reports showing apoptotic cells can suppress pro-inflammatory cytokines such as IL-6, TNF, MIP-1_β_, and
MCP-1 in other naive/resting macrophage populations.9, 10, 24 In contrast to MФ, the addition of apoptotic cells to LEC-MФ significantly affected levels of only 2 of 18 cytokines compared
with LPS-only treated LEC-MФ; TNF levels were inhibited and IL-10 levels increased by the addition of apoptotic cells to LEC-MФ (Figure 1d, filled bars). Also, compared with apoptotic cell
treatment of MФ, LEC-MФ treated with apoptotic cells showed significantly reduced levels of 8 pro-inflammatory cytokines, including TNF, MIP-1_β_, CXCL10, and IL-1_β_ (Figure 1d, open
_versus_ filled bars in ‘LPS+AC’ condition). These differences in cytokine production between MФ and LEC-MФ were not associated with changes in apoptotic cell phagocytosis, as the
efferocytic capacity of MФ and LEC-MФ was similar (Supplementary Figure 1a). Taken together, these results show that exposure of tissue-resident macrophages to a mild, suboptimal dose of
TLR4 agonist significantly enhances the anti-inflammatory effects of apoptotic cells on macrophages compared with naive/resting macrophages. SUPPRESSION OF MACROPHAGE INFLAMMATORY CYTOKINE
PRODUCTION BY APOPTOTIC CELLS AND ADENOSINE We and others have shown that apoptotic cells release ATP through pannexin-1 (PANX1) channels early in the apoptotic program.25, 26, 27 As ATP is
a major source of the potent immunosuppressive molecule adenosine,16, 28, 29 we compared the effects of exogenous adenosine (300 nM) and apoptotic cells on LEC-MФ cytokine production after
normalizing to cytokine levels of MФ treated in the same manner (Figure 2). We then calculated the similarity of LEC-MФ cytokine levels on treatment with either adenosine or apoptotic cells.
We found that of the 18 cytokines examined, only four (IL-10, MIP-1_β_, MCP-1, TNF) showed similarity scores of ⩽1.5 indicative of at least 50% similarity in responses generated by these
two treatments (Figure 2, right axis). Although previous reports have demonstrated a role for adenosine in modulating levels of multiple macrophage inflammatory factors, including _Nr4a1,
Thbs1,_ CXCL1, CXCL2, CXCL10, and IL-1,14,15 these studies did not report a role for apoptotic cell-derived adenosine in regulating TNF production during efferocytosis, despite strong
evidence that A2a and A2b activation in macrophages potently suppresses TNF production.30, 31, 32 Therefore, we sought to determine whether the adenosine signaling pathway was an important
mechanism of apoptotic cell suppression of TNF by LEC-MФ. ADENOSINE AND A2A MEDIATE SUPPRESSION OF LEC-MФ TNF PRODUCTION BY APOPTOTIC CELLS LEC-MФ from WT mice were stimulated with LPS (100
ng/ml) in the presence of increasing numbers of apoptotic cells or cell-free supernatants from equivalent numbers of apoptotic cells. Although both apoptotic cells and cell-free supernatants
suppressed TNF production by LEC-MФ in a dose-dependent manner, apoptotic supernatants were significantly more effective at inhibiting TNF (Figure 3a). By contrast, neither live cells nor
supernatants from live cells significantly affected TNF levels (Figure 3a). Nucleotide release by apoptotic lymphocytes is dependent on caspase-3/7 cleavage of hexameric PANX1 surface
channels.25, 26 Accordingly, apoptotic Jurkat cells stably transfected with a caspase-insensitive dominant-negative PANX1 mutant (PANX1DN) failed to release ATP (Supplementary Figure 1b) and
did not significantly suppress TNF production by LEC-MФ, despite similar rates of apoptosis as parental Jurkat cells (Figure 3b and Supplementary Figure 1b). To determine whether adenosine
in the apoptotic cell supernatants was responsible for inhibiting TNF production by LEC-MФ, we treated supernatants from apoptotic cells with adenosine deaminase (ADA) to deplete adenosine
before adding the supernatants to macrophages. The addition of active, but not denatured, ADA to the apoptotic supernatants, as well as the positive control 300 nM adenosine, abrogated the
suppressive effects of these treatments on TNF production by LEC-MФ (Figure 3c). Extracellular adenosine inhibits macrophage production of TNF by activating Gs-coupled A2a and A2b
receptors.30, 31, 32, 33 In our efferocytosis co-cultures, pretreatment of LEC-MФ with selective inhibitors of A2a abrogated the inhibitory effects of apoptotic cells and exogenous
adenosine, whereas A2b inhibition had a significant but modest effect on TNF suppression by apoptotic cells (Figure 3d). Altogether these data show that PANX1-dependent accumulation of
adenosine from apoptotic cells acts on A2a and, to a lesser extent, A2b receptors on LEC-MФ to modulate TNF expression by macrophages. ADENINE NUCLEOTIDES RELEASED BY APOPTOTIC CELLS ARE
CONVERTED TO ADENOSINE BY CELL-SURFACE CD73 Having found that adenosine derived from apoptotic cells suppresses LEC-MФ TNF production, we hypothesized that ATP released by apoptotic cells
was hydrolyzed to adenosine via the ecto-enzymes CD39 (ATP/ADP→AMP) and CD73 (AMP→adenosine) on macrophages. By flow cytometry, we found that both CD39 and CD73 are abundantly expressed on
the surface of the main population of homeostatic peritoneal macrophages in mice, CD11b+/F4/80hi ‘large’ peritoneal macrophages,34 as well as CD11Chi alveolar macrophages (Figure 4a).
Similar levels of CD39 and CD73 were seen on peritoneal cells obtained from three strains of mice: C57BL/6J, BALB/cJ and 129S1 (Supplementary Figure 2a). Mean fluorescence intensity (MFI)
analysis of anti-CD39 and anti-CD73 flow cytometry revealed that surface expression of these enzymes is ~10-fold higher on peritoneal macrophages than other peritoneal populations such as T
and B lymphocytes (Supplementary Figure 2b). Interestingly, BMDM and three other commonly used mouse macrophage cell lines (RAW264.7, J774A.1, BV-2) were all positive for CD39 but none
expressed CD73 (Figure 4b). Treatment of BMDM with various activation or polarization stimuli, including LEC conditioning, high-dose LPS stimulation, IL-4 or IFN_γ_, failed to induce surface
expression of CD73 on BMDM (Supplementary Figure 2c and data not shown). To determine if CD39 and CD73 are required for generating adenosine from apoptotic cell-derived ATP in these
efferocytosis cultures, we tested the effects CD39 and CD73 inhibitors on the ability of apoptotic cell supernatants to inhibit TNF production by WT LEC-MФ. We found that the CD73 inhibitor
AMPCP completely reversed the suppressive effects of apoptotic cell supernatants on LEC-MФ, whereas the CD39 inhibitor POM-1 had no effect on this suppression (Figure 4c). These results
suggest that extracellular AMP, rather than ATP or ADP, is the primary source of adenosine derived from apoptotic cells under these culture conditions. To confirm these findings, we carried
out similar experiments using LEC-MФ from WT, CD73−/− and CD39−/− mice. Although the loss of CD73 did not significantly affect TNF production following LPS-only treatment, CD73−/−
macrophages were insensitive to the inhibitory effects of apoptotic cell supernatants (Figures 4d and e). By contrast, suppression of TNF by apoptotic cell supernatants was similar between
WT and CD39−/− LEC-MФ (Figure 4f). These results show that apoptotic cell-mediated suppression of LEC-MФ TNF, under these conditions, is entirely dependent on CD73-mediated conversion of
AMP→adenosine. In support of this observation, we found that exogenous AMP, at doses ranging from 1 to 100 _μ_M, effectively suppressed LPS-stimulated, but not zymosan-induced, TNF
production by LEC-MФ (Supplementary Figure 3a). HPLC/MS analysis confirmed increased levels of ATP, ADP, and AMP in supernatants of apoptotic cells compared with live cells, with AMP (~600
nM) being two to three times more abundant than ATP or ADP (Figure 4g). Addition of exogenous ATP and AMP at levels comparable to those seen in apoptotic cell supernatants inhibited TNF
production by LEC-MФ ~50% as was seen with apoptotic cell supernatants (Supplementary Figure 3b). Thus, although Figures 4c and f show that CD39 on macrophages is dispensable for TNF
suppression in our macrophage cultures, it is possible that ATP and ADP released by apoptotic cells can contribute to the pool of available AMP through the actions of other apyrase-like
proteins associated with the macrophages or the culture medium in these experiments. CD73 ON MACROPHAGES OR APOPTOTIC CELLS CAN MEDIATE A2A-DEPENDENT TNF SUPPRESSION CD73 expression is
restricted to certain macrophage and myeloid cell populations in normal and inflamed tissues (Figures 4a and b; Supplementary Figures 4a and b; and refs 18, 19). Therefore, we asked if
extracellular adenosine generated by CD73+ cells could act in a paracrine manner to suppress TNF in CD73- macrophages. To test this, CD73−/− LEC-MФ were cultured with apoptotic Jurkat cells
stably transfected with human CD73 or CD39 (Figure 5a). Expression of CD73, but not CD39, on apoptotic cells inhibited TNF production by CD73-deficient LEC-MФ (Figure 5a). This _trans_
effect of CD73 was abrogated by inhibiting CD73 enzymatic activity with AMPCP (Figure 5a). Interestingly, ectopic expression of CD73 in RAW264.7 macrophages, which lack endogenous CD73
(Figure 4b), reduced TNF production only on co-expression of the high-affinity adenosine receptor A2a (Figure 5b). These results show that CD73 has a central role in converting extracellular
AMP from apoptotic cells into adenosine to stimulate A2a activation and TNF suppression during efferocytosis. LOW-DOSE ENDOTOXIN CONDITIONING ENHANCES THE ADENOSINE RESPONSIVENESS OF
MACROPHAGES We next sought to understand the molecular mechanisms underlying the effects of LEC conditioning on macrophage responses to apoptotic cell-derived adenosine. As shown in Figure
6a, LEC treatment enhanced the sensitivity of macrophages to exogenous adenosine by ~28-fold compared with unconditioned macrophages (IC50: MФ, 8.3 _μ_M and LEC-MФ, 0.3 _μ_M). Previous
studies have shown that TLR activation of macrophages can increase expression of the A2a and A2b-encoding genes _Adora2a_ and _Adora2b._31, 32, 35 We found that _Adora2a,_ but not _Adora2b,_
was significantly increased by the LEC regimen (Figure 6b), with _Adora2a_ levels peaking 2–4 h after LEC treatment and remaining elevated above MФ for 24 h (Figure 6c). Importantly,
LEC-induced upregulation of _Adora2a_ and adenosine responsiveness was similar in RPMФ from WT and CD73−/− mice (Supplementary Figures 4c and d). In addition, we found that LEC treatment did
not affect TNF suppression caused by forskolin, a direct activator of cAMP, or by adenosine or apoptotic cell supernatants in the presence of addition of two different ADA inhibitors,
erythro-9-(2-hydroxy-3-nonyl)adenine (EHNA) and pentostatin (Figures 6d and e; Supplementary Figure 4e). These results indicate that the increased sensitivity of LEC-MФ to adenosine was not
due to LEC-induced changes in cAMP signaling or differences in adenosine deaminase activity between MФ and LEC-MФ.36 Therefore, our findings show that increased expression of the
high-affinity adenosine receptor A2a, either ectopically (Figure 5b) or via LEC treatment (Figures 6a–c), is sufficient to sensitize macrophages to the sub-micromolar levels of adenosine
produced during efferocytosis (Figure 4g). We next asked whether a similar increase in _Adora2a_ levels occurs in tissue-resident macrophages in the lung and peritoneum during self-limiting
inflammation _in vivo_. Following i.p. injection of 1 × 106 heat-killed _E. coli_ particles, we found that _Adora2a_ levels were significantly increased in FACS-sorted F4/80+ tissue-resident
macrophages as early as 4 h and remained elevated above macrophages from untreated mice up to 72 h (Figure 6f). Interestingly, _Adora3_, which encodes the Gi/Gq-coupled A3 adenosine
receptor, was significantly increased over peritoneal macrophages from untreated mice at 48 h after stimulation (Figure 6f). Similarly, gene expression analysis of sorted alveolar
macrophages from mice following i.t. administration of LPS (20 _μ_g) also showed strikingly elevated levels of _Adora2a_ and _Adora2b_ in lung-resident macrophages (Figure 6g). These results
indicate that, like LEC-conditioned macrophages, elevated adenosine receptor levels are a common response by tissue-resident macrophages early after the onset of inflammation _in vivo_.
CD73 REGULATION OF MACROPHAGE RESPONSES TO APOPTOTIC CELL AND INFLAMMATION _IN VIVO_ Although mouse peritoneal and alveolar macrophages have previously been shown to express CD73,18, 19
presently very little is known about the functional significance of CD73 on these macrophage populations. To extend our investigation of CD73 beyond TNF regulation, we tested whether CD73
has a role in regulating other acute-phase, pro-inflammatory factors by macrophages during efferocytosis as described in Figure 1d. For this, we measured levels of MCP-1, MCP-3, MIP-1_β_ and
CXCL10 produced by WT and CD73−/− peritoneal macrophages following stimulation with LPS in the absence or presence of apoptotic cells. Compared with WT LEC-MФ, CD73−/− LEC-MФ produced
significantly higher levels of these four cytokines on stimulation with LPS+apoptotic cells (Figure 7a). Because efferocytosis can skew macrophages toward a pro-resolution or ‘M2-like’
phenotype,2 we compared the expression of several genes associated with either M1- or M2-like macrophage states in WT and CD73−/− LEC-MФ. As shown in Figure 7b, CD73-deficient macrophages
expressed higher levels of _Nos2_ and lower levels of _Tgfb_ in response to treatment with LPS+apoptotic cells compared with WT LEC-MФ. Although other genes implicated in M1/M2 polarization,
including _Nr4a1, Vegf_, and _Timp1_, did not reach statistical significance in these experiments (Figure 7b), the expression pattern of the five genes tested supports a potential role for
CD73 in efferocytosis-mediated programming of macrophage toward a pro-resolution phenotype. We then asked whether CD73 had a similar anti-inflammatory role _in vivo_ by comparing neutrophil
recruitment in WT and CD73−/− mice in the _E. coli_ peritonitis model described in Figure 6f. Compared with WT mice, the total number of peritoneal neutrophils in CD73−/− mice was
significantly increased at 4 h post-treatment, whereas other leukocyte populations were not different between these groups (Figure 7c). Importantly, the total numbers of other peritoneal
leukocyte populations in untreated WT and CD73−/− mice were comparable except for a modest decrease in T cells (CD5hi) in CD73−/− mice (Supplementary Figure 4f). Thus CD73 has an important
role in limiting neutrophil recruitment in the context of mild, self-limiting inflammation _in vivo_. This finding, together with our observation that CD73+ peritoneal macrophages express
relatively high levels of _Adora2a_ and _Adora2b_ under these conditions, supports a role for CD73 in generating adenosine necessary for the anti-inflammatory effects of A2 receptor
activation. DISCUSSION The precise role of apoptotic cell clearance by tissue macrophages in the resolution of tissue inflammation remains an open question.37, 38 Here, we show that the
5′-ecto-nucleotidase CD73 has a key role in regulating macrophage immune responses during efferocytosis by converting AMP released from apoptotic cells into adenosine to suppress multiple
pro-inflammatory cytokines. We specifically investigated the role of CD73 in regulating TNF during efferocytosis because suppression of TNF by apoptotic cells is one of the most widely
observed yet poorly understood consequences of efferocytosis on macrophages.9, 10 Although numerous studies have shown that A2a and A2b activation in macrophages can inhibit NFkB activity
and _Tnf_ transcription downstream of TLR4 signaling, to our knowledge our study is the first to show an essential role for CD73 in mediating adenosine receptor activation and immune
regulation of macrophages in response to dying cells.30, 31, 33 Interestingly, CD73 on either the macrophage or apoptotic cell is sufficient to mediate suppression of TNF during
efferocytosis. This is an important observation given that the expression of CD73 varies widely between different macrophage populations and that most macrophage populations in fact do not
express CD73.39 These findings suggest that in tissues where the resident macrophage populations are low or negative for CD73, it is possible that recruited CD73+ leukocytes (e.g.,
granulocytes, monocytes) could supply the nucleotidase activity necessary to convert apoptotic cell-derived AMP to adenosine and modulate macrophage inflammatory responses. Another important
finding from our study is that suboptimal TLR activation in macrophages potentiates the immunosuppressive effects of apoptotic cell-derived adenosine during efferocytosis. The
concentrations of AMP present in our apoptotic cell cultures (<1 _μ_M) were far below that required to cause significant inhibition of TNF in resting macrophages. However, we show that
under conditions where A2a levels were upregulated in macrophages, either due to LEC treatment or ectopic expression of A2a, the CD73-dependent conversion of AMP from apoptotic cells
effectively suppressed TNF production. Similarly, _Adora2a_ expression was significantly upregulated in lung and peritoneal macrophages 4–6 h after inducing mild inflammation _in vivo_.
Notably, _Adora2b_ levels were also increased in macrophages under these conditions, although A2b inhibition had only modest effects on TNF production by LEC-MФ in efferocytosis cultures.
Thus our mechanistic studies reveal that the capacity of apoptotic cell-derived AMP to suppress macrophages during efferocytosis requires both CD73 (to convert AMP→adenosine) and high levels
of A2a (to generate cAMP in response to low levels of apoptotic cell-derived adenosine) to suppress TNF production during efferocytosis. Because the upregulation of A2a in tissue
macrophages occurs in the first few hours of inflammation, it is possible that AMP released by apoptotic cells during inflammation could provide a key anti-inflammatory signal to macrophages
that promotes the transition from inflammation to resolution. In agreement with this idea, we observed significantly higher numbers of recruited neutrophils in the peritonea of
CD73-deficient mice compared with WT at 4 h after inducing mild peritonitis. Our findings also indicate that the outcomes of macrophage encounters with apoptotic cells are not uniformly
immunosuppressive, but instead are highly dependent on the physiologic state of the macrophage.4, 40 We found that the combination of apoptotic cells and high-dose LPS stimulation on
naive/resting peritoneal macrophages significantly increased the production of the 18 cytokines we measured compared with LPS stimulation alone, including many pro-inflammatory cytokines
like IL-1_β_, IL-6, IL-12 and TNF that have been shown to be suppressed by apoptotic cells in other studies.9, 10 By contrast, when macrophages were pre-conditioned with a suboptimal,
non-tolerizing dose of LPS, the pro-inflammatory effects of apoptotic cells were significantly blunted, suggesting that the early exposure of tissue-resident macrophages to low levels of
PAMPs and DAMPs present in the early stages of inflammation could have an important role in determining the response of macrophages to apoptotic cells later during inflammation. Our study
also shows that CD73 has an important role in this blunting effect for multiple pro-inflammatory cytokines, including TNF, MIP-1_β_, MCP-1, MCP-3, and CXCL10. On the basis of the data
presented here using a relatively simple macrophage conditioning model, we believe it is likely that the exposure of tissue macrophages to low levels of other PAMP or DAMP signals before
encountering apoptotic cells could have pronounced qualitative and quantitative effects on macrophage inflammatory responses. By developing advanced _in vitro_ efferocytosis models that
better reflect the complex immune environment of an inflamed tissue, it will be possible to more precisely define the molecular links and immunologic consequences of macrophage interactions
with apoptotic cells on the progression and resolution of inflammation _in vivo_. MATERIALS AND METHODS MICE Animal experiments were approved by the University of Rochester Animal Care and
Use Committee and mice were housed under specific-pathogen free conditions. C57BL/6J, BALB/cJ, 129S1 and CD73−/− (_Nt5e_−/−) mice were obtained from Jackson Labs (Bar Harbor, ME, USA);
CD39−/− (_Entpd1_−/−) mice have been previously described41 and were obtained from Amgen (Thousand Oaks, CA, USA). CD73−/− mice were maintained on a C57BL/6 background. Female mice between 8
and 12 weeks of age were used for experiments unless otherwise noted. REAGENTS AND ANTIBODIES Commercial reagents were obtained from the following vendors: heat-inactivated FBS (HI-FBS,
Atlanta Biologicals, Flowery Branch, GA, USA); cell culture media and additives (Cellgro, Manassas, VA, USA); SCH442416 (A2a antagonist), PSB603 (A2b antagonist) and POM-1 (CD39 inhibitor)
were purchased from Tocris Biosciences; bovine adenosine deaminase and puromycin (EMD Millipore, Billerica, MA, USA); apyrase (New England Biolabs, Ipswich, MA, USA); Ultrapure LPS (_E.
coli_ O111:B4, for _in vitro_ studies) and Z-VAD-FMK (Enzo Life Sciences, Farmingdale, NY, USA); adenosine, AMP, AMPCP (CD73 inhibitor) and bulk LPS (_E. coli_ O111:B4) and zymosan
(Sigma-Aldrich, St. Louis, MO, USA). Antibodies used in this study were CD16/32 (93), CD19 (eBio1D3) CD5 (53-7.3), CD39 (24DMS1), CD45 (30-F11), CD73 (eBioTY/11.8, eBioscience, San Diego,
CA, USA),Tim-4 (RMT4-54), Siglec-F (E50-2440, BD Biosciences, San Jose, CA, USA), hCD73 (AD2), F4/80 (BM8), CD11b (M1/70, Biolegend, San Diego, CA, USA). CELL ISOLATION AND CULTURE Resident
peritoneal macrophages were recovered from untreated mice via two serial lavages with 3 ml peritoneal lavage media (cation-free HBSS, 0.5% heat-inactivated FBS, 2 mM EDTA). Cells were washed
once in RFHP10 media (RPMI 1640, 10% HI-FBS, 10 mM HEPES, 1% penicillin–streptomycin–l-glutamine), resuspended in RFHP10 at a density of 2 × 105 cells per ml and plated at 5 × 104 cells per
well in a 48-well tissue culture plate. Elicited macrophages were harvested by peritoneal lavage from WT mice 4 days following i.p. injection with 1 ml 3% aged thioglycollate (BD, Spark,
MD, USA). For BMDM generation, bone marrow cells from C57BL/6J mice were collected and red blood cells lysed using ACK lysis buffer (Sigma-Aldrich). Bone marrow cells were washed and plated
at 1 × 106 cells per ml on non-treated petri dishes in RFHP10 containing 20 ng/ml M-CSF (eBioscience). Media was replaced on days 3 and 6, and mature BMDM used on day 7. Jurkat human T cells
(clone E6.1) and RAW 264.7 murine macrophage-like cells were grown in RFHP10. J774A.1 and BV-2 cells were cultured in DMEM containing 10% HI-FBS and 1% penicillin–streptomycin–l-glutamine.
Stable cell lines were grown in the absence of selection antibiotic for at least 18 h before use in experiments. FLOW CYTOMETRY Cells were treated with 1:100 anti-CD16/32 in cold FACS buffer
(cation-free PBS, 0.5% BSA, 0.05% NaN3) for 10 min on ice to block Fc receptors before antibody staining. Surface staining was performed for 30 min on ice, washed once with FACS buffer and
analyzed on a LSR II or FACScalibur cytometers (BD Bioscience). Flow cytometry data analysis was performed using FlowJo 9.6.3 software (FlowJo LLC, Ashland, OR, USA). Cell sorting was
performed at the University of Rochester Flow Cytometry Core facility on a FACSAria-II cell-sorter (BD Bioscience) or on a user-operated S3e cell-sorter (Bio-Rad, Hercules, CA, USA) at the
University of Rochester Center for Vaccine Biology and Immunology (CVBI). PLASMIDS AND TRANSFECTION The pA-puro-CD73 expression construct was generated by PCR cloning of human _NT5E_ cDNA
(Clone 6163101, Open Biosystems, Huntsville, AL, USA) into the _EcoR_I/_Sal_I sites of the pA-puro vector.42 Overall, 5 × 106 Jurkat cells were electroporated with 5 _μ_g _Not_I linearized
pA-puro vector using a BTX830 electroporator (Harvard Apparatus, Cambridge, MA, USA) and selection carried out using 400 ng/ml puromycin. Puromycin-resistant cells were stained with
anti-hCD73, and CD73+ cells FACS-sorted and single cell cloning carried out by limiting dilution. hA2a-YFP was generated by PCR coning of _ADORA2a_ from a human reference cDNA pool
(Clontech, Mountain View, CA, USA) into _EcoR_I/_BamH_I sites of eYFP-N1 (Clontech). RAW 264.7 cells were transfected with 2 _μ_g hA2a-eYFP or eYFP-N1 by Amaxa Nucleofection, and allowed to
grow for 24 h at which time cells were sorted to obtain >98% YFP+ cells and plated at 1 × 104 per well for experiments. Jurkat cells expressing hCD39 or site B mutant pannexin-1 have been
described elsewhere,25, 26 and were maintained in 1 _μ_g/ml puromycin. APOPTOSIS INDUCTION AND SUPERNATANT PREPARATION Jurkat cells were resuspended to 5 × 106/ml in RFHP2 and left
untreated (‘live’), treated with 250 ng/ml anti-Fas 2-3 hours (Clone CH11, EMD Millipore) or exposed to UV-C light for 5 min. Cells were cultured for 3 h at 37 °C/5% CO2 for apoptosis
induction. Cell-free supernatants were prepared by successive centrifugations: 5 min at 350 × _g_ and 5 min at 2000 × _g_. For whole cell preparations, contents of wells were collected and
centrifuged 5 min at 350 × _g_ and resuspended in RFHP2 before use in suppression assays. ATP levels in supernatants were determined from Jurkat cells cultured in RPMI 1640, 0.5% BSA, 10 mM
HEPES, 1% penicillin–streptomycin–l-glutamine and quantified as previously described.26 QUANTIFICATION OF ADENINE NUCLEOTIDES BY HPLC–MS Purine concentrations in conditioned media were
analyzed largely as previously described.43 Two hundred and fifty _μ_l of conditioned media were combined with an equivalent amount of 100% methanol (50% MeOH final). Samples were
subsequently spun down at full speed for 5 min at 4 °C and the remaining supernatant was transferred to HPLC sample vials. Ten _μ_l were analyzed with a reversed-phase liquid chromatography
(LC) method using tributylamine as an ion paring reagent. Chromatography was performed using a LC-20AD HPLC system (Shimadzu) and a Synergi Hydro-RP column (150 × 2 mm with a 5 _μ_m-particle
size; (Phenomenex) coupled to a mass spectrometer. The LC parameters were as follows: autosampler temperature, 4 °C; column temperature 40 °C; flow rate, 0.5 ml/min. The LC solvents were:
solvent A, 100% methanol; solvent B, 10 mM tributylamine and 15 mM acetic acid in 97 : 3 water : methanol. The gradient conditions were as follows: _t_=0, 85% B; _t_=4, 3% B; _t_=5, 3% B;
_t_=5.1, 85% B. Mass spectrometric analyses were performed on a TSQ Quantum Ultra triple-quadrupole mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA), running in negative mode
with the following selected-reaction-monitoring (SRM) transitions and collision energies: AMP 346 to 79 _m_/_z_ at 18 eV; ADP 426 to 159 _m_/_z_ at 25 eV; ATP 506 to 159 _m_/_z_ at 28 eV.
Peak heights for metabolite chromatograms were analyzed using the Excalibur software (Thermo Fisher Scientific). EFFEROCYTOSIS AND CYTOKINE SUPPRESSION ASSAYS RPMФ were plated at 5 × 104 per
well in 48-well tissue culture plates for 2 h at 37 °C/5%CO2. Media was then replaced with 250 _μ_l RFHP10 or priming media (RFHP10+1 ng/ml Ultrapure LPS) and cultured for 18 h. Media was
then replaced with stimulation media (RFHP2+100 ng/ml Ultrapure LPS). All drug treatments were added coincident with stimulation. At indicated times, cell-free supernatants were collected
and stored at −80 °C. ELISA AND LUMINEX ASSAYS ELISAs were performed using cell-free culture supernatants diluted 1 : 2–1 : 20 in assay diluent and analyzed for cytokine levels using ELISA
Ready-SET-Go kits from eBioscience according to manufacturer’s instructions. For Luminex assays, cell-free tissue culture supernatants were collected 8 h post-treatment and analyzed for
cytokine/chemokine levels using ProcartaPlex Mouse Cytokine & Chemokine Panel 1 (26 plex) from eBioscience according to manufacturer’s instructions. Sensitivities for all cytokines
tested were <3.43 pg/ml with the exception of IL-18 which has a sensitivity of 9.95 pg/ml. All data was collected on a Bio-Plex 200 (Bio-Rad) at the Human Immunology Core Facility
(University of Rochester, Rochester, NY, USA). REAL-TIME PCR RNA was isolated via DNase I-treated RNeasy columns (Qiagen, Hilden, Germany) and cDNA was synthesized from 50 to 500 ng of RNA
using iScript cDNA synthesis kit (Bio-Rad) and quantitative PCR analysis carried out on an ABI 7300 thermocycler (Applied Biosystems, Foster City, CA, USA) using TaqMan probes (Life
Technologies, Carlsbad, CA, USA). Owing to the low abundance of _Adora_ mRNA in unstimulated macrophages, some experiments included a 14-cycle pre-amplification step before qPCR analysis as
previously described.44 The following TaqMan probes were used: _Bact_ (Mm00607939_s1), _Adora2a_ (Mm00802075_m1), _Adora2b_ (Mm00839292_m1), _Adora3_ (Mm01296602_m1) _Hprt_ (Mm0046968_m1),
_Nos2_ (Mm00440502_m1), _Nr4a1_ (Mm01300401_m1), _TNF_ (Mm00443258_m1), _Tgfb1_ (Mm01178820_m1), _Timp1_ (Mm00441818_m1) and _Vegfa_ (Mm00437304_m1). STATISTICAL ANALYSIS Statistical
significance of data was analyzed by one-way ANOVA with Tukey’s multiple comparisons post-test using Prism 6 (GraphPad, La Jolla, CA, USA). Differences were considered significant if the
_P_-value was <0.05 (*_P_<0.05, **_P_<0.01, ***_P_<0.001 and ****_P_<0.0001 for all tests). REFERENCES * Soehnlein O, Lindbom L . Phagocyte partnership during the onset and
resolution of inflammation. _Nat Rev Immunol_ 2010; 10: 427–439. Article CAS Google Scholar * Ariel A, Serhan CN . New lives given by cell death: macrophage differentiation following
their encounter with apoptotic leukocytes during the resolution of inflammation. _Front Immunol_ 2012; 3: 4. PubMed PubMed Central Google Scholar * Gilroy D, De Maeyer R . New insights
into the resolution of inflammation. _Semin Immunol_ 2015; 27: 161–168. Article CAS Google Scholar * Krysko DV, Vandenabeele P . From regulation of dying cell engulfment to development of
anti-cancer therapy. _Cell Death Differ_ 2007; 15: 29–38. Article Google Scholar * Elliott MR, Ravichandran KS . Clearance of apoptotic cells: implications in health and disease. _J Cell
Biol_ 2010; 189: 1059–1070. Article CAS Google Scholar * Saas P, Kaminski S, Perruche S . Prospects of apoptotic cell-based therapies for transplantation and inflammatory diseases.
_Immunotherapy_ 2013; 5: 1055–1073. Article CAS Google Scholar * Nagata S, Hanayama R, Kawane K . Autoimmunity and the clearance of dead cells. _Cell_ 2010; 140: 619–630. Article CAS
Google Scholar * Muñoz LE, Berens C, Lauber K, Gaipl US, Herrmann M . Apoptotic cell clearance and its role in the origin and resolution of chronic inflammation. _Front Immunol_ 2015; 6:
139. PubMed PubMed Central Google Scholar * Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I . Immunosuppressive effects of apoptotic cells. _Nature_ 1997; 390: 350–351.
Article CAS Google Scholar * Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM . Macrophages that have ingested apoptotic cells _in vitro_ inhibit proinflammatory cytokine
production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. _J Clin Investig_ 1998; 101: 890–898. Article CAS Google Scholar * Lucas M, Stuart LM, Savill J,
Lacy-Hulbert A . Apoptotic cells and innate immune stimuli combine to regulate macrophage cytokine secretion. _J Immunol_ 2003; 171: 2610–2615. Article CAS Google Scholar * Ipseiz N,
Uderhardt S, Scholtysek C, Steffen M, Schabbauer G, Bozec A _et al_. The nuclear receptor Nr4a1 mediates anti-inflammatory effects of apoptotic cells. _J Immunol_ 2014; 192: 4852–4858.
Article CAS Google Scholar * Medina CB, Ravichandran KS . Do not let death do us part: ‘find-me’ signals in communication between dying cells and the phagocytes. _Cell Death Differ_ 2016;
23: 979–989. Article CAS Google Scholar * Yamaguchi H, Maruyama T, Urade Y, Nagata S . Immunosuppression via adenosine receptor activation by adenosine monophosphate released from
apoptotic cells. _Elife_ 2014; 3: e02172. Article Google Scholar * Koroskenyi K, Duro E, Pallai A, Sarang Z, Kloor D, Ucker DS _et al_. Involvement of adenosine A2A receptors in
engulfment-dependent apoptotic cell suppression of inflammation. _J Immunol_ 2011; 186: 7144–7155. Article CAS Google Scholar * Cekic C, Linden J . Purinergic regulation of the immune
system. _Nat Rev Immunol_ 2016; 16: 177–192. Article CAS Google Scholar * Zimmermann H, Zebisch M, Sträter N . Cellular function and molecular structure of ecto-nucleotidases. _Purinergic
Signal_ 2012; 8: 437–502. Article CAS Google Scholar * Okabe Y, Medzhitov R . Tissue-specific signals control reversible program of localization and functional polarization of
macrophages. _Cell_ 2014; 157: 832–844. Article CAS Google Scholar * Rosas M, Davies LC, Giles PJ, Liao C-T, Kharfan B, Stone TC _et al_. The transcription factor Gata6 links tissue
macrophage phenotype and proliferative renewal. _Science_ 2014; 344: 645–648. Article CAS Google Scholar * Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR . T regulatory and
primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5'-adenosine monophosphate to adenosine. _J Immunol_ 2006; 177: 6780–6786. Article CAS
Google Scholar * Hasko G, Csoka B, Koscso B, Chandra R, Pacher P, Thompson LF _et al_. Ecto-5'-nucleotidase (CD73) decreases mortality and organ injury in sepsis. _J Immunol_ 2011;
187: 4256–4267. Article CAS Google Scholar * Aeffner F, Woods PS, Davis IC . Ecto-5'-nucleotidase CD73 modulates the innate immune response to influenza infection but is not required
for development of influenza-induced acute lung injury. _Am J Physiol Lung Cell Mol Physiol_ 2015; 309: L1313–L1322. Article CAS Google Scholar * Biswas SK, López-Collazo E . Endotoxin
tolerance: new mechanisms, molecules and clinical significance. _Trends Immunol_ 2009; 30: 475–487. Article CAS Google Scholar * Lucas M, Stuart LM, Zhang A, Hodivala-Dilke K, Febbraio M,
Silverstein R _et al_. Requirements for apoptotic cell contact in regulation of macrophage responses. _J Immunol_ 2006; 177: 4047–4054. Article CAS Google Scholar * Elliott MR, Chekeni
FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF _et al_. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. _Nature_ 2009; 461: 282–286. Article
CAS Google Scholar * Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER _et al_. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability
during apoptosis. _Nature_ 2010; 467: 863–867. Article CAS Google Scholar * Qu Y, Misaghi S, Newton K, Gilmour LL, Louie S, Cupp JE _et al_. Pannexin-1 is required for ATP release during
apoptosis but not for inflammasome activation. _J Immunol_ 2011; 186: 6553–6561. Article CAS Google Scholar * Junger WG . Immune cell regulation by autocrine purinergic signalling. _Nat
Rev Immunol_ 2011; 11: 201–212. Article CAS Google Scholar * Idzko M, Ferrari D, Eltzschig HK . Nucleotide signalling during inflammation. _Nature_ 2014; 509: 310–317. Article CAS
Google Scholar * Hasko G, Kuhel DG, Chen JF, Schwarzschild MA, Deitch EA, Mabley JG _et al_. Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2a receptor-dependent and
independent mechanisms. _FASEB J_ 2000; 14: 2065–2074. Article CAS Google Scholar * Murphree LJ, Sullivan GW, Marshall MA, Linden J . Lipopolysaccharide rapidly modifies adenosine
receptor transcripts in murine and human macrophages: role of NF-κB in A2A adenosine receptor induction. _Biochem J_ 2005; 391: 575. Article CAS Google Scholar * Cohen HB, Briggs KT,
Marino JP, Ravid K, Robson SC, Mosser DM . TLR stimulation initiates a CD39-based autoregulatory mechanism that limits macrophage inflammatory responses. _Blood_ 2013; 122: 1935–1945.
Article CAS Google Scholar * Kreckler LM, Wan TC, Ge Z-D, Auchampach JA . Adenosine inhibits tumor necrosis factor-alpha release from mouse peritoneal macrophages via A2A and A2B but not
the A3 adenosine receptor. _J Pharmacol Exp Ther_ 2006; 317: 172–180. Article CAS Google Scholar * Ghosn EEB, Cassado AA, Govoni GR, Fukuhara T, Yang Y, Monack DM _et al_. Two physically,
functionally, and developmentally distinct peritoneal macrophage subsets. _Proc Natl Acad Sci USA_ 2010; 107: 2568–2573. Article CAS Google Scholar * Elson G, Eisenberg M, Garg C, Outram
S, Ferrante CJ, Hasko G _et al_. Induction of murine adenosine A2A receptor expression by LPS: analysis of the 5′ upstream promoter. _Genes Immun_ 2013; 14: 147–153. Article CAS Google
Scholar * Desrosiers MD, Cembrola KM, Fakir MJ, Stephens LA, Jama FM, Shameli A _et al_. Adenosine deamination sustains dendritic cell activation in inflammation. _J Immunol_ 2007; 179:
1884–1892. Article CAS Google Scholar * Savill J, Dransfield I, Gregory C, Haslett C . A blast from the past: clearance of apoptotic cells regulates immune responses. _Nat Rev Immunol_
2002; 2: 965–975. Article CAS Google Scholar * Serhan CN, Savill J . Resolution of inflammation: the beginning programs the end. _Nat Immunol_ 2005; 6: 1191–1197. Article CAS Google
Scholar * Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S _et al_. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of
mouse tissue macrophages. _Nat Immunol_ 2012; 13: 1118–1128. Article CAS Google Scholar * Torchinsky MB, Garaude J, Blander JM . Infection and apoptosis as a combined inflammatory
trigger. _Curr Opin Immunol_ 2010; 22: 55–62. Article CAS Google Scholar * Pinsky DJ, Broekman MJ, Peschon JJ, Stocking KL, Fujita T, Ramasamy R _et al_. Elucidation of the
thromboregulatory role of CD39/ectoapyrase in the ischemic brain. _J Clin Investig_ 2002; 109: 1031–1040. Article CAS Google Scholar * Takata M, Sabe H, Hata A, Inazu T, Homma Y, Nukada T
_et al_. Tyrosine kinases Lyn and Syk regulate B cell receptor-coupled Ca2+ mobilization through distinct pathways. _EMBO J_ 1994; 13: 1341–1349. Article CAS Google Scholar * DeVito SR,
Ortiz-Riaño E, Martínez-Sobrido L, Munger J . Cytomegalovirus-mediated activation of pyrimidine biosynthesis drives UDP-sugar synthesis to support viral protein glycosylation. _Proc Natl
Acad Sci USA_ 2014; 111: 18019–18024. Article CAS Google Scholar * Stevenson C, la Rosa de G, Anderson CS, Murphy PS, Capece T, Kim M _et al_. Essential role of elmo1 in dock2-dependent
lymphocyte migration. _J Immunol_ 2014; 192: 6062–6070. Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank Tim Mosmann, Jim Miller and Elliott lab members for
helpful suggestions. Also, we thank Maiken Nedergaard and Amgen for providing _Entpd1_−/− mice, and we thank Jane Malone, Jamie Strampe, Nate Laniewski, Xenia Schafer, Liz Albertorio-Saez
and the University of Rochester Flow Cytometry Facility for technical support. This work was supported by the Ellison Medical Foundation (MRE) and the National Institutes of Health: AI114554
(MRE), AI027767 (MRE), HL113495 (TWW). PSM was supported in part by National Institutes of Health Grant T32 GM007285. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of
Microbiology and Immunology, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA Patrick S Murphy, Joshua C Munger, Terry W Wright & Michael R Elliott * David H.
Smith Center for Vaccine Biology and Immunology, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA Patrick S Murphy & Michael R Elliott * Department of
Pediatrics, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA Jing Wang, Samir P Bhagwat & Terry W Wright * Department of Biochemistry and Biophysics,
University of Rochester School of Medicine and Dentistry, Rochester, NY, USA Joshua C Munger * Department of Medicine, National Jewish Health and University of Colorado Denver Anschutz
Campus, Denver, CO, USA William J Janssen Authors * Patrick S Murphy View author publications You can also search for this author inPubMed Google Scholar * Jing Wang View author publications
You can also search for this author inPubMed Google Scholar * Samir P Bhagwat View author publications You can also search for this author inPubMed Google Scholar * Joshua C Munger View
author publications You can also search for this author inPubMed Google Scholar * William J Janssen View author publications You can also search for this author inPubMed Google Scholar *
Terry W Wright View author publications You can also search for this author inPubMed Google Scholar * Michael R Elliott View author publications You can also search for this author inPubMed
Google Scholar CORRESPONDING AUTHOR Correspondence to Michael R Elliott. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no conflict of interest. ADDITIONAL INFORMATION Edited by
DR Green Supplementary Information accompanies this paper on _Cell Death and Differentiation_ website . SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURE 1 (PDF 881 KB) SUPPLEMENTARY FIGURE 2
(PDF 1079 KB) SUPPLEMENTARY FIGURE 3 (PDF 762 KB) SUPPLEMENTARY FIGURE 4 (PDF 1721 KB) RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Murphy, P., Wang,
J., Bhagwat, S. _et al._ CD73 regulates anti-inflammatory signaling between apoptotic cells and endotoxin-conditioned tissue macrophages. _Cell Death Differ_ 24, 559–570 (2017).
https://doi.org/10.1038/cdd.2016.159 Download citation * Received: 05 October 2016 * Revised: 07 December 2016 * Accepted: 12 December 2016 * Published: 06 January 2017 * Issue Date: March
2017 * DOI: https://doi.org/10.1038/cdd.2016.159 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is
not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative








