- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Sustained activation of nuclear factor-_κ_B (NF-_κ_B) in cancer cells has been shown to promote inflammation, expansion of cancer stem cell (CSC) population, and tumor development.
In contrast, recent studies reveal that CSCs exhibit increased inflammation due to constitutive NF-_κ_B activation; however, the underlying molecular mechanism remains unclear. In the
present study, the analysis of microarray data revealed upregulation of NF-_κ_B-regulated pro-inflammatory genes and downregulation of copper metabolism MURR1 domain-containing 1 (COMMD1)
during the enrichment for stemness in SAS head and neck squamous-cell carcinoma (HNSCC) cells. The 3′-UTR of COMMD1 mRNA contains microRNA (miR)-205 target site. Parallel studies with HNSCC
and NSCLC cells indicated that miR-205 is upregulated upon NF-_κ_B activation and suppresses COMMD1 expression in stemness-enriched cancer cells. COMMD1 negatively regulates the inflammatory
responses induced by TLR agonists, IL-1_β_, and TNF-_α_ by targeting RelA for degradation. The shRNA-mediated downregulation of COMMD1 in cancer cells enhanced inflammatory response,
generating favorable conditions for macrophage recruitment. In addition, genes associated with stemness were also upregulated in these cells, which exhibited increased potential for
anchorage-independent growth. Furthermore, COMMD1 downregulation promoted _in vivo_ tumorigenesis and tumor growth, and tumors derived from COMMD1-knockdown cells displayed elevated level of
NF-_κ_B activation, increased expression of inflammatory- and stemness-associated genes, and contain expanded population of tumor-associated leukocytes and stemness-enriched cancer cells.
These results suggest that COMMD1 downregulation by miR-205 promotes tumor development by modulating a positive feedback loop that amplifies inflammatory- and stemness-associated properties
of cancer cells. SIMILAR CONTENT BEING VIEWED BY OTHERS RNA-BINDING PROTEIN COMPLEX LIN28/MSI2 ENHANCES CANCER STEM CELL-LIKE PROPERTIES BY MODULATING HIPPO-YAP1 SIGNALING AND INDEPENDENTLY
OF LET-7 Article Open access 31 January 2022 IGF2BP2-MEIDATED M6A MODIFICATION OF CSF2 REPROGRAMS MSC TO PROMOTE GASTRIC CANCER PROGRESSION Article Open access 21 October 2023 MACC1 ABLATION
SUPPRESSES THE DEDIFFERENTIATION PROCESS OF NON-CSCS IN LUNG CANCER THROUGH STABILIZING KLF4 Article Open access 18 December 2024 MAIN Accumulating evidence supports the association between
inflammation and tumorigenesis. Persistent inflammatory response induced by extrinsic factors such as exposure to environmental agents, infection with a pathogen, genetic disease, or
metabolic disorder increases the predisposition of patients with subclinical conditions to cancer. Inflammatory conditions in the tumor microenvironment promote angiogenesis, tumor
progression, metastasis, and chemoresistance. Nuclear factor kappa B (NF-_κ_B) regulates the expression of multiple pro-inflammatory genes and is therefore a key mediator of acute and
chronic inflammatory responses.1, 2, 3, 4 Cancer stem cells (CSCs) possess the capacity for both self-renewal and differentiation; they promote tumor progression and metastasis and are
responsible for chemoresistance and cancer relapse.5, 6, 7, 8 Accumulating evidence indicates that NF-_κ_B activation supports the expansion of CSCs and promotes tumor development. The
activation of NF-_κ_B in cancer cells results in a number of downstream events, including induction of antiapoptotic genes and suppression of apoptosis. Moreover, induction of the regulators
of epithelial–mesenchymal transition (EMT) leads to EMT phenotype, which has been hypothesized to initiate metastasis as well as the de-differentiation of cancer cells into CSCs.9, 10, 11,
12, 13 In contrast, cancer cells enriched for stemness have been recently shown to exhibit inflammatory gene expression patterns due to constitutive NF-_κ_B activation;14, 15, 16, 17 this
could lead to a positive feedback loop resulting in the amplification of inflammatory- and stemness-associated properties in cancer cells, although the molecular mechanism remains unclear.
Toll-like receptors (TLRs) are a group of proteins expressed by cells of the innate immune system that recognize microbial pathogens and respond to endogenous molecules released from dying
cells during stress conditions such as chemotherapy.18, 19, 20 Interleukin (IL)-1 and tumor necrosis factor (TNF)-_α_ are potent pro-inflammatory cytokines; TLR agonists, IL-1_β_, and
TNF-_α_ function as major pro-inflammatory stimuli in the tumor microenvironment.21, 22 Activation of TLRs and IL-1 receptor (IL-1R) triggers the sequential recruitment of MyD88, IRAK, and
TRAF6 to form a complex; this, in turn, activates TAK, which leads to NF-_κ_B activation. The activation of NF-_κ_B downstream TNF-_α_ receptor (TNFR) is mediated by the signaling molecules
TRADD, RIP, and TRAF2. The molecular components involved in TLR/IL-1R and TNFR signaling pathways only partially overlap; nevertheless, the principle involved in the regulation of these
pathways is similar and involves the recruitment of adaptor molecules, with protein levels and protein–protein interactions regulated by ubiquitination and deubiquitination. Several E3
ubiquitin-protein ligases, deubiquitinases, and co-factors involved in the ubiquitination system have been shown to regulate inflammatory properties of cancer cells through regulation of
NF-_κ_B activation.23, 24, 25, 26 MicroRNAs (or miRs) are a group of small (18–24 nt) non-coding RNAs that regulate target gene expression by binding target sites on the 3′ untranslated
regions (UTRs) of messenger RNAs (mRNAs), leading to their degradation or translational inhibition.27, 28 Copper Metabolism MURR1 Domain-containing 1 (COMMD1) is the primary member of the
COMM domain family that functions as an interface for protein–protein interactions and promotes ubiquitin-mediated degradation of the interaction partners.29, 30 The current study
investigates the key molecules governing NF-_κ_B activity and inflammatory properties in stemness-enriched cancer cells through analysis of microarray-derived expression profiles of negative
regulators of the TLR, IL-1R, and TNFR signaling pathways in the course of stemness enrichment in head and neck squamous-cell carcinoma (HNSCC) cells. Parallel studies with HNSCC cells and
non-small-cell lung cancer (NSCLC) cells indicated that upregulation of NF-_κ_B-driven miR-205 in stemness-enriched cancer cells leads to COMMD1 downregulation; this results in amplification
of inflammatory- and stemness-associated properties of cancer cells and also promotes _in vivo_ tumorigenesis and tumor growth. RESULTS ELEVATED NF-_Κ_B ACTIVATION AND INFLAMMATION IN
STEMNESS-ENRICHED CANCER CELLS The changes in NF-_κ_B activation and inflammation during the enrichment of stemness in cancer cells were investigated by analyzing microarray data derived
from SAS HNSCC cells subject to enrichment for stemness in defined serum-free (DSF) selection medium for 2, 3, 5, or 9 weeks; this microarray data set was previously published31 and
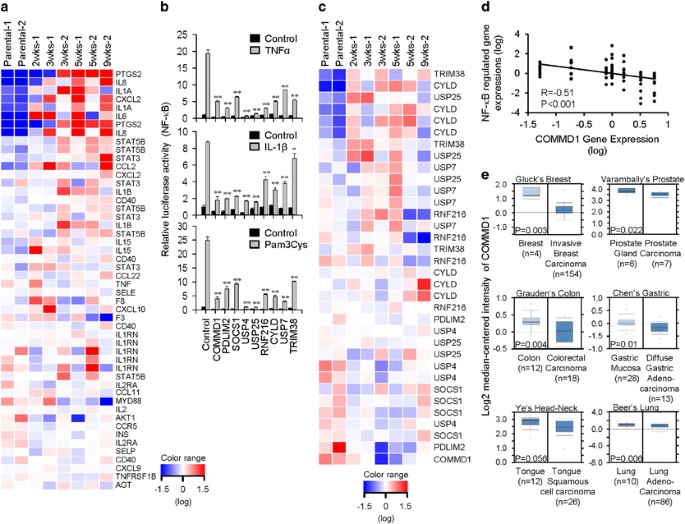
submitted to Gene Expression Omnibus (GEO) database (Accession No. GSE35603). Multiple NF-_κ_B-regulated cytokines and chemokines were upregulated during the enrichment of stemness of SAS
cells (Figure 1a), suggesting the elevation of NF-_κ_B activity and inflammation during enrichment for stemness. IDENTIFICATION OF KEY REGULATORS OF NF-_Κ_B ACTIVATION DURING ENRICHMENT FOR
STEMNESS We hypothesized that downregulating the negative regulators of NF-_κ_B activation in the TLR, IL-1R, and TNFR signaling pathways could lead to elevated NF-_κ_B activation and
inflammation in the stemness-enriched cancer cells. In order to identify the key regulator, a set of known negative regulators of NF-_κ_B activation associated with these pathways were
shortlisted for further analysis; these function through regulation of ubiquitination and deubiquitination processes. The inhibitory activities of these regulators on NF-_κ_B activation
induced by the TLR2 agonist Pam3Cys, IL-1_β_, and TNF-_α_ were first confirmed by NF-_κ_B luciferase reporter assay using TLR2-overexpressing or parental HEK 293 cells (Figure 1b). The
expression profile of these regulators was also analyzed using the microarray data set obtained from stemness-enriched SAS cells in Figure 1a. Heat map generated from the microarray data
revealed downregulation of a few of these negative regulators during the enrichment of stemness in SAS cells (Figure 1c). Of these, COMMD1 was the most downregulated gene with a good reverse
correlation (_R_=−0.51) between its downregulation and the upregulated, NF-_κ_B-controlled genes, including PTGS2, IL8, IL1A, CXCL2, IL-6, STAT5B, STAT3, CCL2, IL1B, CD40, and IL-15
(Figures 1a, c, and d). In addition, a search of the the Oncomine database revealed reduced expression of COMMD1 in different types of tumors from patients (Figure 1e). Thus, the functions
of COMMD1 in the regulation of inflammatory- and stemness-associated properties of cancer cells were further investigated in this study. DOWNREGULATION OF COMMD1 IN STEMNESS-ENRICHED HNSCC
CELLS AND NSCLC CELLS The downregulation of COMMD1 observed from microarray data was further confirmed using SAS cells and also the human H460 and mouse D121 NSCLC cell lines. The cells were
cultured in DSF medium to allow enrichment of sphere-forming cells exhibiting stemness and spheres were microscopically observed after 2 weeks (Figures 2a–c; top left panels). Real-time
quantitative PCR (RT-qPCR) analysis revealed that COMMD1 expression was downregulated in the stemness-enriched SAS cells (Figure 2a; top right panel), which is consistent with microarray
analysis (Figure 1c); similar observations were made in stemness-enriched H460 and D121 cells (Figures 2b and c; top right panels). RT-qPCR was employed for analyzing the expression of genes
associated with stemness. Although the induction patterns of these genes differed between the three cell types, increased expression of these genes was observed in spheres compared with
parental cells (Figures 2a–c; bottom panels), indicating the enrichment of stemness in cells of the sphere. 3′-UTR OF COMMD1 MRNA CONTAINS MIR-205 TARGET SITE MicroRNAs have been implicated
in the regulation of stemness in cancer cells through downregulation of their target genes.32, 33 To investigate the molecular mechanism underlying the downregulation of COMMD1, the 3′-UTR
of COMMD1 mRNA was examined for microRNA target sites using the mirSVR scoring method.34, 35 Three target sites corresponding to miR-205, miR-491-5p, and miR-7, respectively, were predicted
(Supplementary Figure S1a); of these, the miR-205 target site exhibited good mirSVR score of −1.2126 (Supplementary Figure S1b), which falls within the top 7% of mirSVR scores.35 Therefore,
the regulation of COMMD1 expression by miR-205 was further investigated. DOWNREGULATION OF COMMD1 BY NF-_Κ_B-REGULATED MIR-205 DURING ENRICHMENT FOR STEMNESS To investigate whether miR-205
is responsible for COMMD1 downregulation in cancer cells enriched for stemness, the expression of miR-205 and COMMD1 were compared between parental cells and spheres cultured in DSF medium
for 4 weeks through RT-qPCR; increased expression of miR-205 and downregulation of COMMD1 was observed in spheres derived from SAS, H460, and D121 cells (Figure 3a). Subsequent experiments
aimed to investigate whether COMMD1 is a bona fide target of miR-205. Reporter constructs expressing luciferase regulated by the wild-type (wt) 3′-UTR of COMMD1 or a mutant (mut) containing
mutated miR-205 target site were generated (Supplementary Figure S2a). These reporter constructs were co-transfected with miR-control or miR-205 mimic into HEK 293 cells stably expressing
the control-sponge or miR-205-sponge designed to inhibit miR-205 (Supplementary Figure S2b). The experiment revealed that miR-205 destabilized the luciferase activity regulated by COMMD1
3′UTR-wt; this effect of miR-205 was antagonized by miR-205-sponge (Figure 3b, left panel). In contrast, miR-205 failed to exert any effect on luciferase activity regulated by COMMD1
3′-UTR-mut containing the mutant miR-205 target site (Figure 3b, right panel). Further studies revealed that overexpression of miR-205 resulting in COMMD1 downregulation at both the mRNA and
protein levels in SAS, H460, and D121 cells (Figures 3c and d). A previous study reported the presence of NF-_κ_B-binding site upstream to the _miR-205_ gene locus.36 Upregulation of
miR-205 through NF-_κ_B activation in NSCLC cells such as H460 has been reported to result in increased tumor growth.36 Thus, the induction of miR-205 in SAS and D121 cells was further
investigated in the present study. MiR-205 was induced by IL-1_β_ and TLR2 ligands, and the induction was reversed upon treatment of cells with BMS-345541, an IKK inhibitor37 (Figure 3e).
Taken together, these results suggest that COMMD1 expression is downregulated by miR-205, which is upregulated by NF-_κ_B activation during enrichment for stemness. COMMD1 DOWNREGULATION IN
CANCER CELLS PROMOTES INFLAMMATION The function of COMMD1 in regulating the inflammatory properties of cancer cells was investigated. To this end, shRNA viral vectors for the knockdown of
COMMD1 mRNA (shCOMMD1) and luciferase mRNA (shLuc) were generated (Supplementary Figure 3). The control and COMMD1-knockdown cells were stimulated with TNF-_α_, IL-1, and various TLR
ligands, and TNF-_α_ expression was analyzed using RT-qPCR. COMMD1 knockdown was found to increase the responsiveness of SAS, H460, and D121 cells to inflammatory stimuli (Figures 4a–c). In
RAW264.7 cells, a mouse monocytic cell line containing multiple TLRs, COMMD1 overexpression reversed NF-_κ_B activation induced by TNF-_α_, IL-1, and various TLR ligands (Figure 4d),
indicating that COMMD1 is a negative regulator of inflammatory responses induced by TNF-_α_, IL-1, and TLR ligands in monocytic and cancer cells. The molecular target that mediates the
regulation of inflammation by COMMD1 was investigated. The overexpression of various signaling molecules downstream TNFR, IL-1R, and TLR could cause NF-_κ_B activation.38 COMMD1 was found to
reverse NF-_κ_B activation induced by these signaling molecules (Figures 4e and f), suggesting that the molecular target of COMMD1 is likely to be present further downstream in these
pathways. Therefore, expression vectors for three downstream signaling molecules in the NF-_κ_B activation pathways, namely, TAK1, IKK_β_, and RelA, were co-transfected into HEK 293 cells in
the presence or absence of COMMD1-expression vector; analysis of the expression of these three signaling molecules by immunoblot analysis revealed downregulation of RelA but not IKK_β_ and
TAK1 upon COMMD1 co-expression (Figure 4g). Further, elevated expression levels of endogenous RelA were detected when COMMD1 was knocked down in SAS, H460, and D121 cells (Figure 4h), and
increased phosphorylation levels of RelA (phospho-RelA), an indicator of NF-_κ_B activation,39, 40 were measured by flow cytometric analysis in COMMD1-knockdown D121 cells (Supplementary
Figure S4). These results suggest that RelA is the molecular target of COMMD1 in NF-_κ_B activation pathways. DOWNREGULATION OF COMMD1 IN CANCER CELLS PROMOTES MACROPHAGE RECRUITMENT
Subsequent investigation focused on the role of COMMD1 in regulating inflammation in the tumor microenvironment to modulate macrophage recruitment. Control and COMMD1-downregulated SAS and
D121 cells were stimulated with TNF-_α_ and the production of various cytokines and chemokines was analyzed by RT-qPCR using gene-specific primers (Supplementary Tables S2 and S3). Elevated
levels of various cytokines and chemokines were observed in COMMD1-downregulated cells irrespective of TNF-_α_ treatment (Figure 5a), suggesting that COMMD1 regulates both intrinsic and
induced inflammatory responses in cancer cells. The role of COMMD1 in regulating the crosstalk between cancer cells and macrophages was further investigated through _in vitro_ macrophage
recruitment assay. Consistent with the production of various cytokines and chemokines by COMMD1-knockdown cells, the conditioned medium obtained from these cells proved more effective in
macrophage recruitment (Figure 5b). COMMD1 DOWNREGULATION ENHANCES STEMNESS IN CANCER CELLS The function of COMMD1 in regulating stemness was investigated. Analysis of the expression of
stemness-associated genes using RT-qPCR revealed elevated expression of POU5F1, KLF4, NANOG, CD117, CD133, ALDH, and ABCG2 to varying extents in COMMD1-knockdown SAS and D121 cells compared
with the respective controls (Figures 6a and b, left panels). The sphere-forming ability of COMMD1-knockdown cells was investigated in DSF medium. Decreased COMMD1 expression in cells
correlated with increased number of spheres (Figures 6a and b, middle and right panels). Whether the function of COMMD1 was involved with NF-_κ_B activation was further investigated and
compared with the function of miR-205. In these experiments, different cell lines derived from H460 cells were treated with BMS-345541. Results indicated that knockdown of COMMD1 and
overexpression of miR-205 increased the expression of stemness-associated genes and sphere formation, and these effects were blocked by the inhibition of NF-_κ_B activation with BMS-345541
(Figures 6c and d). These results suggest that COMMD1 is a negative regulator and miR-205 is a positive regulator of stemness enrichment in cancer cells and that their function is mediated
by NF-_κ_B activation. COMMD1 DOWNREGULATION ENHANCES THE TRANSFORMING ABILITY OF CANCER CELLS The role of COMMD1 knockdown in regulating the transforming ability of cancer cells was
investigated in parallel with the function of miR-205 overexpression using anchorage-independent growth assay. The control and COMMD1-knockdown cells of D121 (serial diluted from 2 × 104),
H460 and SAS (1 × 104), and the control and miR-205-overexpressing cells of D121 and H460 (1 × 104) were cultured in soft agar for 2–3 weeks and colonies were enumerated. Significantly
higher colony numbers were observed with COMMD1-knockdown cells and miR-205-overexpressing cells compared with the respective controls (Figures 7a and e). Thus, COMMD1 downregulation and
miR-205 overexpression in cancer cells enhances their transforming ability, which is in agreement with the enhanced stemness associated with COMMD1 downregulation and miR-205 overexpression.
COMMD1 DOWNREGULATION PROMOTES _IN VIVO_ TUMORIGENICITY AND TUMOR GROWTH The _in vivo_ role of COMMD1 in regulation of tumorigenicity and tumor growth were investigated. C57BL/6J (B6) mice
were inoculated with varying numbers of control or COMMD1-knockdown D121 cells. A higher tumor development rate was observed in mice injected with COMMD1-knockdown cells than in mice
injected with control cells (Figure 8a). Tumor growth was investigated by inoculating (1 x 105) cells per mouse of COMMD1 knockdown, miR-205 overexpressing, and their respective control D121
cells; faster growth rates were observed in tumors derived from COMMD1-knockdown and miR-205-overexpressing cells relative to their control cells (Figures 8b and c). These observations
suggest that downregulation of COMMD1 by miR-205 in cancer cells can promote tumorigenicity and tumor growth. The properties associated with inflammation and stemness were investigated in
the tumors derived from COMMD1-knockdown and control cells. The expression of genes associated with inflammation and stemness was investigated in these tumors by RT-qPCR; higher expression
of inflammatory cytokines and chemokines (Figure 8d) as well as stemness-associated genes (Figure 8e) was observed in tumors derived from COMMD1-knockdown cells. H&E staining of tumor
sections revealed higher leukocyte infiltration in the tumors (Figure 8f). Moreover, flow cytometric analysis revealed an elevated level of phospho-RelA in whole tumor cells, Cd11b+
tumor-associated leukocytes, and Cd117+ stemness-enriched tumor cells in tumors derived from COMMD1-knockdown cells relative to their respective cells in tumors derived from control cells
(Figures 8g and i). Flow cytometric analysis also showed expanded populations of Cd11b+ leukocytes and Cd117+ stemness-enriched cells in the tumors derived from COMMD1-knockdown cells
(Figures 8h and i). The Cd117+ cells and Cd117− were isolated from the tumors grown from COMMD1-knockdown cells and reinjected into mice to access their capacity for tumorigenesis. Results
showed more potency for the Cd117+ cells to develop tumors than the Cd117− cells (Figure 8j), confirming that the Cd117+ stemness-enriched cells were more aggressive cancer cells. Taken
together, these observations suggest that COMMD1 regulates tumorigenicity and tumor growth by modulating inflammatory- and stemness-associated properties of cancer cells. DISCUSSION
Tumor-promoting inflammation has been recognized as a hallmark of cancer.1 NF-_κ_B is key mediator of inflammation in cancer cells.9, 10, 11 CSCs are implicated in tumor recurrence,
metastasis, and higher mortality rates.7, 8 Recent studies have supported a role for NF-_κ_B in modulation of stemness in cancer cells and in inflammatory signaling, which is important for
CSC self-renewal and maintenance.12, 13 Moreover, pro-inflammatory cytokines and TLR agonists in inflammatory tumor microenvironment support the survival and tumorigenic activities of CSCs,
whereas NF-_κ_B inhibitors have been shown to inhibit CSC growth.12, 13, 41, 42, 43, 44 These observations suggest that the mechanism underlying the promotion of malignancy by inflammation
could involve enhancement of stemness in cancer cells resulting in increased CSC population. Apart from these, the present and several previous studies14, 15, 16, 17 revealed increased
expression of inflammatory genes in CSCs due to constitutive NF-_κ_B activation; this can further promote the release of pro-inflammatory mediators into the tumor microenvironment, thereby
forming a positive feedback loop for increasing stemness in cancer cells and amplifying inflammation in the cancer cells, CSCs, and the tumor microenvironment. This positive feedback loop is
likely to be an important mechanism for malignancy, despite the lack of clarity regarding the underlying molecular controls. TLRs, IL-1R, and TNFR initiate potent pro-inflammatory signaling
pathways resulting in NF-_κ_B activation in cancer cells, thereby promoting cancer cell growth, CSC expansion, and tumor development.21, 22 These signaling pathways are regulated by
multiple negative regulators that mediate ubiquitination and deubiquitination of signaling molecules to modulate protein–protein interactions and proteasomal degradation.23, 24, 25, 26 These
negative regulators not only modulate the magnitude of inflammatory responses to stimuli but also maintain the balance of NF-_κ_B activity within cells. Thus, downregulation of these
negative regulators is likely to cause the elevated NF-_κ_B activation observed during enrichment of stemness in cancer cells and in the CSCs. In this study, our results reveal that
miR-205-mediated downregulation of COMMD1 is responsible for the increased inflammatory and stemness features in stemness-enriched cancer cells and promotes tumorigenesis and tumor growth.
Both antitumor and protumor functions of miR-205 have been reported.45, 46 The antitumor activity of miR-205 is attributable to its targeting various oncogenes. For example, expression of
miR-205 suppresses cell proliferation by suppressing E2F1 expression in melanoma cells, and by inhibition of Src-mediated oncogenic pathways for its growth-inhibitory effects in renal
carcinoma.47, 48 In breast cancer cells, knockdown of miR-205 enhances stem cell traits and promotes development of mammary lesions in cancer animal model.49 In addition, miR-205 is
downregulated in gastric cancer and miR-205 inhibition promotes the proliferation of gastric cancer cells.50 On the other hand, accumulating evidence indicates that miR-205 promotes
tumorigenesis, tumor progression, and chemoresistance. In mouse mammary epithelial cells, miR-205 expression is enhanced in stem-like cells and suppresses phosphatase and tensin homolog
(PTEN), leading to the expansion of progenitor cell population.51 In human breast cancer cells, miR-205 overexpression increases the enrichment of stem-like cells exhibiting self-renewal
ability.52 Elevated expression of miR-205 is associated with poor prognosis in endometrial tumors, with miR-205 promoting the proliferation of endometrial cancer cells.53 In addition,
miR-205 expression correlates with the initiation of nasopharyngeal carcinoma and contributes to its radioresistance.54 In ovarian cancer cells, miR-205 is upregulated upon VEGF-A treatment
and targets Ezrin and Lamin A/C to promote invasion and proliferation.55 Furthermore, several studies have shown that upregulation of miR-205 in NSCLC cells promotes growth, invasion, and
chemoresistance of the cancer cells.36, 56, 57 These observations suggest that the pro- or antitumor functions of miR-205 are determined by its target genes, the context of cancer, and the
tumor microenvironment. In contrast, COMMD1 is shown to have an antitumor function. Previous studies suggested that COMMD1 inhibits NF-_κ_B activation by functioning as a scaffold protein in
conjugation with ECSSOCS1, a multimeric E3 ubiquitin ligase complex, to promote ubiquitination and proteasomal degradation of the NF-_κ_B subunit RelA.58 The conditional knockdown of COMMD1
in myeloid cells rendered mice hyper-responsive to LPS stimulation as well as more sensitive to dextran sodium sulfate-induced colitis and colitis-associated cancer.59 In concordance with
these results, reduced COMMD1 expression was observed in several human cancers and low expression of COMMD1 correlated with a reduced survival rate among patients with endometrial cancer.60
The level of COMMD1 protein is negatively regulated by secretory clusterin (sCLU), a stress-induced small heat-shock protein, in prostate cancer cells. The expression of sCLU showed inverse
correlation with that of COMMD1 but positive correlation with NF-_κ_B-regulated genes.61 Consistent with this observation, the present study showed that COMMD1 downregulation promotes
tumorigenesis and tumor growth. Downregulation of COMMD1 during the enrichment of stemness in cancer cells resulted in upregulation of genes associated with inflammation and stemness.
Furthermore, the present study shows that COMMD1 downregulation can be regulated at the level of mRNA stability by the NF-_κ_B-regulated miR-205, in addition to the previous observed
regulation at protein level by sCLU.61 In summary, this study has shown that during the enrichment of stemness in HNSCC cells and NSCLC cells, COMMD1 is downregulated by miR-205, while
miR-205 is upregulated upon NF-_κ_B activation. The downregulation of COMMD1 promotes the expression of stemness-associated genes and enhances inflammation-induced as well as sustained
intrinsic NF-_κ_B activation in cancer cells. This, in turn, promotes the release of pro-inflammatory mediators into the tumor microenvironment and further enhances miR-205 expression in
cancer cells. Thus, COMMD1 plays a role in the regulation of tumorigenesis and tumor growth _via_ regulation of a positive feedback loop that amplifies inflammatory- and stemness-associated
properties in cancer cells. MATERIALS AND METHODS REAGENTS AND ANTIBODIES The TLR agonists Pam3Cys, polyI:C, LPS, flagellin, and R848 were purchased from Invivogen (San Diego, CA, USA);
CpG-2006 and CpG-1826 from Invitrogen (Carlsbad, CA, USA) or Genomics Biosci & Tech (New Taipei, Taiwan); BMS-345541, collagenase II, collagenase IV, deoxyribonuclease, and anti-Flag M2
antibody from Sigma-Aldrich Co. (St. Louis, MO, USA); and HA.11, PE-anti-human CD117, PE- anti-mouse CD117, and PE-rat IgG2b _κ_ isotype control antibody from BioLegend (San Diego, CA, USA);
PE-anti-human CD133 antibody from Miltenyi Biotec (Bergisch Gladbach, Germany); PE-anti-mouse CD11b antibody from eBioscience (San Diego, CA, USA); anti-phospho-RelA (Alexa Fluor 647
conjugate) and rabbit IgG isotype control antibody from Cell Signaling (Beverley, MA, USA). The miR-205 mimics and control were synthesized by GeneDirex (Gueishan Township, Taiwan).
Antibodies against human and mouse COMMD1 were purchased from Abnova (Taipei, Taiwan) and Proteintech (Chicago, IL, USA), respectively; human recombinant TNF-_α_, IL-1_β_, epidermal growth
factor (EGF), and basic fibroblast growth factor (bFGF) from Peprotech (Rocky Hill, NJ, USA); and reagents for luciferase assay, from Promega (Medison, WI, USA). CELL CULTURE AND ENRICHMENT
OF SPHERE-FORMING CANCER CELLS Human SAS HNSCC, HEK 293, murine D121 NSCLC, and RAW264.7 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal
bovine serum (FBS). Human H460 NSCLC and THP-1 cells were grown in RPMI supplemented with 10% FBS. For enrichment of sphere-forming cells, SAS, H460, and D121 cells were cultured at a
density of 10 000 cells/ml in six-well plates coated with poly(2-hydroxyethyl methacrylate) (polyHEMA; Sigma-Aldrich Co.) in sphere medium consisting of serum-free DMEM/F12-K medium, N2
supplement (GIBCO, Carlsbad, CA, USA), 20 ng/ml human recombinant EGF, and 20 ng/ml human recombinant bFGF. PolyHEMA-coated plates were prepared by dissolving 10 mg/ml polyHEMA in 95%
ethanol; 1 ml of the solution was added onto the six-well plates and placed overnight in a laminar flow hood at room temperature for air drying. BIOINFORMATICS ANALYSIS OF MICROARRAY DATA,
GENE EXPRESSION IN TUMOR, AND IDENTIFICATION OF MIRNA TARGET SITES Microarray data obtained at various time points during the enrichment of stemness for SAS HNSCC cells (GEO data sets,
GSE35603) were analyzed for expression profiles of NF-_κ_B-regulated inflammatory genes as well as negative regulators of the TLR, IL-1R, and TNFR signaling pathways. Oncomine database
(https://www.oncomine.org/) was searched for the expression profiles of COMMD1 in normal tissues and tumors from patients. MicroRNA target sites in the 3′-UTR of COMMD1 mRNA were analyzed
using the algorithm miRanda (http://www.microrna.org/). PLASMID CONSTRUCTION The expression constructs for human COMMD1, PDLIM2, SOCS1, CYLD, USP4, USP7, USP25, and TRIM38 were generated
through PCR amplification of the corresponding protein-coding regions from the first-strand cDNA library derived from human spleen cells generated as previous reported.62 The amplified DNA
fragments were cloned into pRK5 vector for protein expression. The 3′-UTR reporter constructs were generated through PCR amplification of the 3′-UTR of COMMD1 from human genomic DNA
purchased from Clontech Laboratories, Inc. (Mountain View, CA, USA), followed by cloning into pMIR-REPORT luciferase vector. LENTIVIRAL SHRNA, MIR-205-SPONGE AND PRECURSOR MIR-205
CONSTRUCTION AND INFECTION The shRNA constructs were generated by synthesizing and annealing sense and antisense shRNA oligonucleotides with 5′GATC and 5′AATT overhangs, respectively. The
double-stranded shRNA was cloned into the _Bam_HI/_Eco_RI sites of pGreenpuro plasmid. The miR-205-sponge was constructed by annealing sense and antisense oligonucleotides containing six
tandem repeats of the bulge-containing miR-205-binding motif and cloning into _Xba_I/_Bam_HI sites of pCDH plasmid. Precursor miR-205 construct was generated through PCR amplification of the
precursor miR-205 from human genomic DNA, followed by cloning into _Eco_RI/_Bam_HI sites of pCDH plasmid. Lentivirus was produced by harvesting culture supernatants obtained upon
transfecting 293 T cells with the generated constructs along with three packaging plasmids. HEK 293 and cancer cell lines were spin-infected by plating cells in 12-well plates in the
presence of lentiviral supernatants and 8 _μ_g/ml polybrene (Sigma-Aldrich Co.), followed by centrifugation at 1100 × _g_ for 30 min. The cells were subject to selection with puromycin (3
ng/ml) to obtain stable cell lines. The sequences of oligonucleotides employed for generating shRNA, miR-205, and miR-205-sponge constructs, and precursor miR-205 are listed (Supplementary
Tables S1). LUCIFERASE REPORTER ASSAYS For NF-_κ_B activation assay, cells were plated on 24-well plates, allowed to adhere overnight, and co-transfected with NF-_κ_B-driven luciferase
reporter, _β_-galactosidase, and the indicated expression plasmids using polyethylenimine (Sigma-Aldrich Co.). The following day, the cells were subject to 10-h treatment with TNF-_α_ (10
ng/ml), IL-1_β_ (10 ng/ml), or Pam3Cys (200 ng/ml), followed by lysis and determination of luciferase activity. Relative luciferase activities were calculated as fold induction relative to
unstimulated control. For 3′-UTR reporter assay, HEK 293 cells stably transfected with control or miR-205-sponge were co-transfected for 16 h with COMMD1 3′-UTR luciferase reporter plasmid,
_β_-galactosidase plasmid, and the indicated microRNA precursors, followed by cell lysis and determination of luciferase activities. SDS-PAGE AND IMMUNOBLOT ANALYSIS Cells were lysed with
lysis buffer (100 mM NaCl, 50 mM Tris-Cl pH 7.5, 0.5 mM EDTA, 1% NP-40) containing complete protease inhibitor cocktail (Roche Life Science, Indianapolis, IN, USA). Cell lysates were
separated by SDS-PAGE and transferred onto PVDF membranes. The membranes were blocked for 30 min with 5% fat-free milk in Tris-buffered saline containing Tween-20 (TBST; 50 mM Tris-Cl pH
7.5, 150 mM NaCl, 0.1% Tween-20) and incubated for 2 h with the indicated antibody in TBST containing 2% fat-free milk. The membranes were subsequently washed in TBST and incubated for 1 h
with HRP-conjugated secondary antibody. The immunoreactive bands were visualized using chemiluminescent HRP substrate (Immobilon Western; Millipore, Temecula, CA, USA) and the UVP
BioSpectrum Imaging System. RT-QPCR ANALYSIS OF GENE AND MICRORNA EXPRESSION Total RNA including small RNA species was purified using TRIzol (Invitrogen), according to the manufacturer’s
instructions. Reverse transcription (RT) was performed using SuperScript III first-strand synthesis system (Invitrogen) and oligo-dT for first-strand cDNA synthesis or corresponding RT
primers for miRNA synthesis. RT-qPCR was carried out using ABI PRISM 7900_HT_ Sequence Detection System and KAPA SYBR fast qPCR kit (KK4605) for gene expression analysis or FastStart
Universal Probe Master and Universal ProbeLibrary Probe #21 (Roche Life Science) for microRNA detection. Data were analyzed using the 2−ΔΔCt method described in the ABI user manual. The
expression of mRNA and microRNA were normalized to _β_-actin and RNU6, respectively. The primer sequences are listed (Supplementary Table S1–S3). ANCHORAGE-INDEPENDENT GROWTH Bottom
agar–medium mixture (DMEM, 10% FBS, 0.8% agarose) was added to each well of six-well tissue culture plates and allowed to solidify. Top agar–medium mixture (DMEM, 10% FBS, 0.4% agarose)
containing (1 × 104) cells was added, followed by the addition of growth medium. The plates were incubated at 37 °C for 2–3 weeks and colony formation was monitored. The plates were fixed
with 4% formaldehyde and stained with 0.005% crystal violet in PBS for 1 h. The colonies were photographed and counted. The total number of colonies with a diameter of ≥100 _μ_m were
quantified over four fields per well for a total of 12 fields in triplicate samples. MACROPHAGE RECRUITMENT ASSAY Conditioned medium (CM) was collected from cancer cells treated with TNF-_α_
or control for 24 h and added to the lower chamber of transwell plates containing polyethylene terephthalate (PET) membrane insert of 5-_μ_m pore size (Corning Life Sciences, Manassas, VA,
USA). THP-1 (1 × 105) or RAW264.7 (2 × 104) cells were plated onto the upper chamber and incubated for 8 h at 37 °C. Cells that failed to migrate through the pores were removed using a
cotton swab, while the migratory cells were fixed, stained with 0.05% crystal violet, and counted in five randomly selected fields. FLOW CYTOMETRIC ANALYSIS For flow cytometric analysis,
cells were suspended in PBS containing 2% FCS and incubated with PE-conjugated Abs as indicated at 4 °C for 30 min. After washing, cells were analyzed on a FACSCalibur flow cytometer with
CellQuest software (Becton Dickinson, San Jose, CA, USA). For intracellular staining of phospho-RelA, cell line and tumor mass cells were first fixed and permeabilized by BD Cytofix/Cytoperm
Kit (BD Bioscience, San Diego, CA, USA) for 20 min. After fixation and permeabilization, cells were washed and resuspensed in 1 × BD Perm/wash buffer and then incubated with
anti-phospho-RelA for 30 min. ANIMAL CARE, PREPARATION OF TUMOR SINGLE-CELL SUSPENSION, TUMORIGENESIS, AND ANALYSIS OF TUMOR GROWTH Animal experiments were approved by the Institutional
Animal Care and Use Committee (IACUC) of the National Health Research Institutes, Taiwan. C57BL/6J mice were maintained and handled in accordance with the stated guidelines. D121 cells
stably expressing shLuc and shCOMMD1 were harvested, resuspended in PBS, and injected subcutaneously into C57BL/6 mice of 6–8 weeks of age. Tumor single-cell suspension was prepared by
mincing and digestion of tumor mass in PBS containing 0.5% BSA, 0.25% collagenase II, 0.25% collagenase IV, and 0.05% deoxyribonuclease for 30 min. The reaction was stopped with DMEM
containing 10% FBS. The mixture was then strained through a 70- _μ_ M strainer and lysis of red blood cells with RBC lysis buffer (eBioscience). Cd117+ tumor cells were separated using MACS
from tumor single-cell suspension with Cd117 MicroBeads and MACS column (Miltenyi Biotec) following the manufacturer’s instruction. Tumor volume (TV) was calculated using the following
formula: TV (mm3)=(length × width2)/2. HISTOLOGICAL ANALYSIS Tissue samples were collected, immersed in 10% formalin, embedded in paraffin wax, and sectioned. The sections were stained with
H&E and microscopically visualized. STATISTICAL ANALYSIS Data are expressed as mean±S.D. Statistical analysis was performed on data derived from three or more independent experiments
using Student’s _t_-test. A _P-_value of <0.05 was considered to represent statistically significant differences between the experimental groups. ABBREVIATIONS * COMMD1: copper metabolism
MURR1 domain-containing 1 * NF-_κ_B: nuclear factor-_κ_B * TLR: toll-like receptor * IL-1: interleukin 1 * TNF-_α_: tumor necrosis factor-_α_ * CSC: cancer stem cells * HNSCC: head and neck
squamous-cell carcinoma * NSCLC: non-small cell lung cancer REFERENCES * Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. _Cell_ 2011; 144: 646–674. Article CAS PubMed
Google Scholar * Mantovani A, Allavena P, Sica A, Balkwill F . Cancer-related inflammation. _Nature_ 2008; 454: 436–444. Article CAS PubMed Google Scholar * Grivennikov SI, Greten FR,
Karin M . Immunity, inflammation, and cancer. _Cell_ 2010; 140: 883–899. Article CAS PubMed PubMed Central Google Scholar * Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA .
Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. _Nat Rev Cancer_ 2013; 13: 759–771. Article CAS PubMed Google Scholar * Reya T, Morrison SJ,
Clarke MF, Weissman IL . Stem cells, cancer, and cancer stem cells. _Nature_ 2001; 414: 105–111. Article CAS PubMed Google Scholar * Visvader JE, Lindeman GJ . Cancer stem cells: current
status and evolving complexities. _Cell Stem Cell_ 2012; 10: 717–728. Article CAS PubMed Google Scholar * Mertins SD . Cancer stem cells: a systems biology view of their role in
prognosis and therapy. _Anticancer Drugs_ 2014; 25: 353–367. Article CAS PubMed PubMed Central Google Scholar * Colak S, Medema JP . Cancer stem cells – important players in tumor
therapy resistance. _FEBS J_ 2014; 281: 4779–4791. Article CAS PubMed Google Scholar * Gasparini C, Feldmann M . NF-kappaB as a target for modulating inflammatory responses. _Curr Pharm
Des_ 2012; 18: 5735–5745. Article CAS PubMed Google Scholar * Sarkar FH, Li Y, Wang Z, Kong D . NF-kappaB signaling pathway and its therapeutic implications in human diseases. _Int Rev
Immunol_ 2008; 27: 293–319. Article CAS PubMed Google Scholar * DiDonato JA, Mercurio F, Karin M . NF-kappaB and the link between inflammation and cancer. _Immunol Rev_ 2012; 246:
379–400. Article PubMed Google Scholar * Zhou C, Liu J, Tang Y, Liang X . Inflammation linking EMT and cancer stem cells. _Oral Oncol_ 2012; 48: 1068–1075. Article CAS PubMed Google
Scholar * Shigdar S, Li Y, Bhattacharya S, O'Connor M, Pu C, Lin J _et al_. Inflammation and cancer stem cells. _Cancer Lett_ 2014; 345: 271–278. Article CAS PubMed Google Scholar
* Rajasekhar VK, Studer L, Gerald W, Socci ND, Scher HI . Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-kappaB signalling. _Nat Commun_ 2011; 2: 162.
Article PubMed Google Scholar * Garner JM, Fan M, Yang CH, Du Z, Sims M, Davidoff AM _et al_. Constitutive activation of signal transducer and activator of transcription 3 (STAT3) and
nuclear factor kappaB signaling in glioblastoma cancer stem cells regulates the Notch pathway. _J Biol Chem_ 2013; 288: 26167–26176. Article CAS PubMed PubMed Central Google Scholar *
Yamashina T, Baghdadi M, Yoneda A, Kinoshita I, Suzu S, Dosaka-Akita H _et al_. Cancer stem-like cells derived from chemoresistant tumors have a unique capacity to prime tumorigenic myeloid
cells. _Cancer Res_ 2014; 74: 2698–2709. Article CAS PubMed Google Scholar * Jinushi M . Role of cancer stem cell-associated inflammation in creating pro-inflammatory tumorigenic
microenvironments. _Oncoimmunology_ 2014; 3: e28862. Article PubMed PubMed Central Google Scholar * Hoebe K, Jiang Z, Georgel P, Tabeta K, Janssen E, Du X _et al_. TLR signaling
pathways: opportunities for activation and blockade in pursuit of therapy. _Curr Pharm Des_ 2006; 12: 4123–4134. Article CAS PubMed Google Scholar * Chen K, Huang J, Gong W, Iribarren P,
Dunlop NM, Wang JM . Toll-like receptors in inflammation, infection and cancer. _Int Immunopharmacol_ 2007; 7: 1271–1285. Article CAS PubMed Google Scholar * Piccinini AM, Midwood KS .
DAMPening inflammation by modulating TLR signalling. _Mediators Inflamm_ 2010; 2010 PII: 672395. Article PubMed PubMed Central Google Scholar * Lawrence T . The nuclear factor NF-kappaB
pathway in inflammation. _Cold Spring Harb Perspect Biol_ 2009; 1: a001651. Article PubMed PubMed Central Google Scholar * Hoesel B, Schmid JA . The complexity of NF-kappaB signaling in
inflammation and cancer. _Mol Cancer_ 2013; 12: 86. Article CAS PubMed PubMed Central Google Scholar * Verstrepen L, Bekaert T, Chau TL, Tavernier J, Chariot A, Beyaert R . TLR-4, IL-1R
and TNF-R signaling to NF-kappaB: variations on a common theme. _Cell Mol Life Sci_ 2008; 65: 2964–2978. Article CAS PubMed Google Scholar * Bibeau-Poirier A, Servant MJ . Roles of
ubiquitination in pattern-recognition receptors and type I interferon receptor signaling. _Cytokine_ 2008; 43: 359–367. Article CAS PubMed Google Scholar * Wertz IE, Dixit VM . Signaling
to NF-kappaB: regulation by ubiquitination. _Cold Spring Harb Perspect Biol_ 2010; 2: a003350. Article PubMed PubMed Central Google Scholar * Zinngrebe J, Montinaro A, Peltzer N,
Walczak H . Ubiquitin in the immune system. _EMBO Rep_ 2014; 15: 28–45. Article CAS PubMed Google Scholar * Bushati N, Cohen SM . microRNA functions. _Annu Rev Cell Dev Biol_ 2007; 23:
175–205. Article CAS PubMed Google Scholar * Huntzinger E, Izaurralde E . Gene silencing by microRNAs: contributions of translational repression and mRNA decay. _Nat Rev Genet_ 2011; 12:
99–110. Article CAS PubMed Google Scholar * Maine GN, Burstein E . COMMD proteins and the control of the NF kappa B pathway. _Cell Cycle_ 2007; 6: 672–676. Article CAS PubMed Google
Scholar * Bartuzi P, Hofker MH, van de Sluis B . Tuning NF-kappaB activity: a touch of COMMD proteins. _Biochim Biophys Acta_ 2013; 1832: 2315–2321. Article CAS PubMed Google Scholar *
Lo JF, Yu CC, Chiou SH, Huang CY, Jan CI, Lin SC _et al_. The epithelial-mesenchymal transition mediator S100A4 maintains cancer-initiating cells in head and neck cancers. _Cancer Res_ 2011;
71: 1912–1923. Article CAS PubMed Google Scholar * Iorio MV, Croce CM . microRNA involvement in human cancer. _Carcinogenesis_ 2012; 33: 1126–1133. Article CAS PubMed PubMed Central
Google Scholar * Garofalo M, Croce CM . Role of microRNAs in maintaining cancer stem cells. _Adv Drug Deliv Rev_ 2015; 81: 53–61. Article CAS PubMed Google Scholar * Betel D, Wilson
M, Gabow A, Marks DS, Sander C . The microRNA.org resource: targets and expression. _Nucleic Acids Res_ 2008; 36: D149–D153. Article CAS PubMed Google Scholar * Betel D, Koppal A, Agius
P, Sander C, Leslie C . Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. _Genome Biol_ 2010; 11: R90. Article PubMed PubMed Central
Google Scholar * Cai J, Fang L, Huang Y, Li R, Yuan J, Yang Y _et al_. miR-205 targets PTEN and PHLPP2 to augment AKT signaling and drive malignant phenotypes in non-small cell lung cancer.
_Cancer Res_ 2013; 73: 5402–5415. Article CAS PubMed Google Scholar * McIntyre KW, Shuster DJ, Gillooly KM, Dambach DM, Pattoli MA, Lu P _et al_. A highly selective inhibitor of I kappa
B kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in mice. _Arthritis Rheum_ 2003; 48: 2652–2659. Article CAS PubMed Google Scholar * Xu
C, Liu J, Hsu LC, Luo Y, Xiang R, Chuang TH . Functional interaction of heat shock protein 90 and Beclin 1 modulates Toll-like receptor-mediated autophagy. _FASEB J_ 2011; 25: 2700–2710.
Article CAS PubMed PubMed Central Google Scholar * Park GY, Christman JW . Nuclear factor kappa B is a promising therapeutic target in inflammatory lung disease. _Curr Drug Targets_
2006; 7: 661–668. Article CAS PubMed Google Scholar * Vallabhapurapu S, Karin M . Regulation and function of NF-kappaB transcription factors in the immune system. _Annu Rev Immunol_
2009; 27: 693–733. Article CAS PubMed Google Scholar * Chefetz I, Alvero AB, Holmberg JC, Lebowitz N, Craveiro V, Yang-Hartwich Y _et al_. TLR2 enhances ovarian cancer stem cell
self-renewal and promotes tumor repair and recurrence. _Cell Cycle_ 2013; 12: 511–521. Article CAS PubMed PubMed Central Google Scholar * Chen K, Huang YH, Chen JL . Understanding and
targeting cancer stem cells: therapeutic implications and challenges. _Acta Pharmacol Sin_ 2013; 34: 732–740. Article CAS PubMed PubMed Central Google Scholar * Liu WT, Jing YY, Yu GF,
Han ZP, Yu DD, Fan QM _et al_. Toll like receptor 4 facilitates invasion and migration as a cancer stem cell marker in hepatocellular carcinoma. _Cancer Lett_ 2015; 358: 136–143. Article
CAS PubMed Google Scholar * Pistollato F, Giampieri F, Battino M . The use of plant-derived bioactive compounds to target cancer stem cells and modulate tumor microenvironment. _Food Chem
Toxicol_ 2015; 75: 58–70. Article CAS PubMed Google Scholar * Qin AY, Zhang XW, Liu L, Yu JP, Li H, Wang SZ _et al_. MiR-205 in cancer: an angel or a devil? _Eur J Cell Biol_ 2013; 92:
54–60. Article CAS PubMed Google Scholar * Orang AV, Safaralizadeh R, Hosseinpour Feizi MA . Insights into the diverse roles of miR-205 in human cancers. _Asian Pac J Cancer Prev_ 2014;
15: 577–583. Article PubMed Google Scholar * Dar AA, Majid S, de Semir D, Nosrati M, Bezrookove V, Kashani-Sabet M . miRNA-205 suppresses melanoma cell proliferation and induces
senescence via regulation of E2F1 protein. _J Biol Chem_ 2011; 286: 16606–16614. Article CAS PubMed PubMed Central Google Scholar * Majid S, Saini S, Dar AA, Hirata H, Shahryari V,
Tanaka Y _et al_. MicroRNA-205 inhibits Src-mediated oncogenic pathways in renal cancer. _Cancer Res_ 2011; 71: 2611–2621. Article CAS PubMed PubMed Central Google Scholar * Chao CH,
Chang CC, Wu MJ, Ko HW, Wang D, Hung MC _et al_. MicroRNA-205 signaling regulates mammary stem cell fate and tumorigenesis. _J Clin Invest_ 2014; 124: 3093–3106. Article CAS PubMed PubMed
Central Google Scholar * Yin WZ, Li F, Zhang L, Ren XP, Zhang N, Wen JF . Down-regulation of microRNA-205 promotes gastric cancer cell proliferation. _Eur Rev Med Pharmacol Sci_ 2014; 18:
1027–1032. PubMed Google Scholar * Greene SB, Gunaratne PH, Hammond SM, Rosen JM . A putative role for microRNA-205 in mammary epithelial cell progenitors. _J Cell Sci_ 2010; 123:
606–618. Article CAS PubMed PubMed Central Google Scholar * Vilquin P, Donini CF, Villedieu M, Grisard E, Corbo L, Bachelot T _et al_. MicroRNA-125b upregulation confers aromatase
inhibitor resistance and is a novel marker of poor prognosis in breast cancer. _Breast Cancer Res_ 2015; 17: 13. Article PubMed PubMed Central Google Scholar * Karaayvaz M, Zhang C,
Liang S, Shroyer KR, Ju J . Prognostic significance of miR-205 in endometrial cancer. _PLoS One_ 2012; 7: e35158. Article CAS PubMed PubMed Central Google Scholar * Qu C, Liang Z, Huang
J, Zhao R, Su C, Wang S _et al_. MiR-205 determines the radioresistance of human nasopharyngeal carcinoma by directly targeting PTEN. _Cell Cycle_ 2012; 11: 785–796. Article CAS PubMed
PubMed Central Google Scholar * Li J, Li L, Li Z, Gong G, Chen P, Liu H _et al_. The role of miR-205 in the VEGF-mediated promotion of human ovarian cancer cell invasion. _Gynecol Oncol_
2015; 137: 125–133. Article CAS PubMed Google Scholar * Lei L, Huang Y, Gong W . miR-205 promotes the growth, metastasis and chemoresistance of NSCLC cells by targeting PTEN. _Oncol Rep_
2013; 30: 2897–2902. Article CAS PubMed Google Scholar * Larzabal L, de Aberasturi AL, Redrado M, Rueda P, Rodriguez MJ, Bodegas ME _et al_. TMPRSS4 regulates levels of integrin alpha5
in NSCLC through miR-205 activity to promote metastasis. _Br J Cancer_ 2014; 110: 764–774. Article CAS PubMed PubMed Central Google Scholar * Maine GN, Mao X, Komarck CM, Burstein E .
COMMD1 promotes the ubiquitination of NF-kappaB subunits through a cullin-containing ubiquitin ligase. _EMBO J_ 2007; 26: 436–447. Article CAS PubMed Google Scholar * Li H, Chan L,
Bartuzi P, Melton SD, Weber A, Ben-Shlomo S _et al_. Copper metabolism domain-containing 1 represses genes that promote inflammation and protects mice from colitis and colitis-associated
cancer. _Gastroenterology_ 2014; 147: e3. Article Google Scholar * van de Sluis B, Mao X, Zhai Y, Groot AJ, Vermeulen JF, van der Wall E _et al_. COMMD1 disrupts HIF-1alpha/beta
dimerization and inhibits human tumor cell invasion. _J Clin Invest_ 2010; 120: 2119–2130. Article CAS PubMed PubMed Central Google Scholar * Zoubeidi A, Ettinger S, Beraldi E,
Hadaschik B, Zardan A, Klomp LW _et al_. Clusterin facilitates COMMD1 and I-kappaB degradation to enhance NF-kappaB activity in prostate cancer cells. _Mol Cancer Res_ 2010; 8: 119–130.
Article CAS PubMed PubMed Central Google Scholar * Liu J, Xu C, Hsu LC, Luo Y, Xiang R, Chuang TH . A five-amino-acid motif in the undefined region of the TLR8 ectodomain is required
for species-specific ligand recognition. _Mol Immunol_ 2010; 47: 1083–1090. Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We thank the Laboratory Animal Center
of the National Health Research Institutes, Taiwan, for assistance with animal work. This work was supported in part by the National Health Research Institutes, Taiwan (grant IM-103-PP-02)
and Ministry of Science and Technology of Taiwan (grants MOST102-2320-B-400-009-MY3). D-WY carried out thesis research under the auspices of the Graduate Program of Biotechnology in
Medicine, National Tsing-Hua University, and National Health Research Institutes. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Immunology Research Center, National Health Research
Institutes, Miaoli, Taiwan D-W Yeh, C-Y Lai, Y-L Liu, C-H Lu & T-H Chuang * Institute of Molecular Medicine, National Tsing-Hua University, Hsinchu, Taiwan D-W Yeh & L Chen *
Institute of Oral Biology, National Yang-Ming University, Taipei, Taiwan Y-S Chen & J-F Lo * Institute of Molecular Medicine, National Taiwan University, Taipei, Taiwan L-C Hsu *
Department of Immunology, Institute of Basic Medical Science, Chinese Academy of Medical Science and Peking Union Medical College, Beijing, China Y Luo * School of Medicine, University of
Nankai, Tianjin, PR China R Xiang * Program in Environmental and Occupational Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan T-H Chuang * Research and Development Center for
Immunology, China Medical University, Taichung, Taiwan T-H Chuang Authors * D-W Yeh View author publications You can also search for this author inPubMed Google Scholar * Y-S Chen View
author publications You can also search for this author inPubMed Google Scholar * C-Y Lai View author publications You can also search for this author inPubMed Google Scholar * Y-L Liu View
author publications You can also search for this author inPubMed Google Scholar * C-H Lu View author publications You can also search for this author inPubMed Google Scholar * J-F Lo View
author publications You can also search for this author inPubMed Google Scholar * L Chen View author publications You can also search for this author inPubMed Google Scholar * L-C Hsu View
author publications You can also search for this author inPubMed Google Scholar * Y Luo View author publications You can also search for this author inPubMed Google Scholar * R Xiang View
author publications You can also search for this author inPubMed Google Scholar * T-H Chuang View author publications You can also search for this author inPubMed Google Scholar
CORRESPONDING AUTHOR Correspondence to T-H Chuang. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no conflict of interest. ADDITIONAL INFORMATION Edited by R De Maria
Supplementary Information accompanies this paper on Cell Death and Differentiation website SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATIONS (PDF 264 KB) RIGHTS AND PERMISSIONS This work
is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s
Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the
license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Yeh, DW., Chen, YS., Lai, CY. _et al._ Downregulation of COMMD1 by miR-205 promotes a positive feedback loop for amplifying inflammatory- and stemness-associated properties of cancer
cells. _Cell Death Differ_ 23, 841–852 (2016). https://doi.org/10.1038/cdd.2015.147 Download citation * Received: 27 April 2015 * Revised: 22 September 2015 * Accepted: 02 October 2015 *
Published: 20 November 2015 * Issue Date: May 2016 * DOI: https://doi.org/10.1038/cdd.2015.147 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content:
Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative







