- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
From the beginning of research on receptors of the tumor necrosis factor (TNF) receptor superfamily (TNFRSF), agonistic antibodies have been used to stimulate TNFRSF receptors in vitro and
in vivo. Indeed, CD95, one of the first cloned TNFRSF receptors, was solely identified as the target of cell death-inducing antibodies. Early on, it became evident from in vitro studies that
valency and Fcγ receptor (FcγR) binding of antibodies targeting TNFRSF receptors can be of crucial relevance for agonistic activity. TNFRSF receptor-specific antibodies of the IgM subclass
and secondary cross-linked or aggregation prone dimeric antibodies typically display superior agonistic activity compared with dimeric antibodies. Likewise, anchoring of antibodies to cell
surface-expressed FcγRs potentiate their ability to trigger TNFRSF receptor signaling. However, only recently has the relevance of oligomerization and FcγR binding for the in vivo activity
of antibody-induced TNFRSF receptor activation been straightforwardly demonstrated in vivo. This review discusses the crucial role of oligomerization and/or FcγR binding for
antibody-mediated TNFRSF receptor stimulation in light of current models of TNFRSF receptor activation and especially the overwhelming relevance of these issues for the rational development
of therapeutic TNFRSF receptor-targeting antibodies.
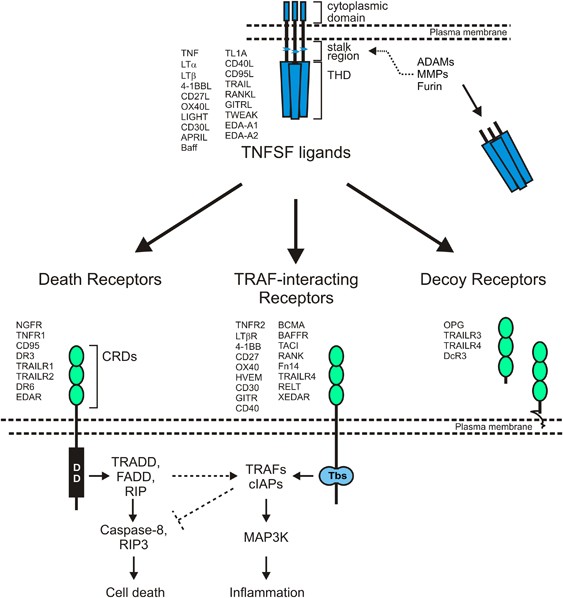
Ligands of the TNF superfamily (TNFSF) occur as trimeric transmembrane proteins but also as soluble trimeric molecules.
A subgroup of the TNF receptor superfamily (TNFRSF) is not or only slightly activated by soluble TNFSF ligands.
Oligomerization and cell surface-anchoring of soluble TNFSF ligands provide these molecules with membrane TNFSF ligand-like activities.
Dimeric TNFRSF receptor-specific antibodies have typically no or only a moderate agonistic activity.
Oligomerization and Fcγ receptor-binding frequently converts dimeric TNFRSF receptor-specific antibodies into strong agonists.
What is the molecular basis of the different responsiveness of TNFRSF receptors toward binding of soluble TNFSF ligands?
How one can generate antibody-based TNFRSF receptor agonists with oligomerization- and FcγR binding-independent activity?
What are the mechanisms underlying the FcγR binding-independent agonistic activity of TNFRSF receptor-specific human IgG2 isoform B antibodies?
Receptors of the tumor necrosis factor (TNF) receptor superfamily (TNFRSF) are naturally activated by ligands of the TNF superfamily.1, 2 Cytokines are assigned to the TNF superfamily
(TNFSF) based on a conserved carboxy-terminal homology domain called the TNF homology domain (THD) (Figure 1).1, 2 The THD promotes the assembly of homotrimeric molecules, or in rare cases
the formation of dimeric (murine GITRL)3, 4 or heterotrimeric (LTαβ2)5 ligands, and is essential for interaction with receptors of the TNFRSF. With exception of LTα, TNFSF ligands are
expressed as trimeric type II transmembrane proteins in which the THD is separated from the transmembrane domain by a stalk region of variable length (Figure 1). Due to proteolytic
processing in the stalk region or by alternative splicing, TNFSF ligands can also be found in the form of soluble trimeric molecules (Figure 1). Soluble TNFSF ligands still contain the THD
and thus retain the ability to interact with TNFRSF receptors.1, 2 X-ray crystallographic studies of various soluble TNFSF ligands, alone or in complex with TNFRSF receptor ectodomains
(Table 1), not only confirmed the trimeric organization of TNFSF ligands deduced from biochemical assays but also revealed that each of the three protomer–protomer interfaces of a TNFSF
ligand trimer binds a single TNFRSF receptor molecule.
Ligands of the TNF superfamily (TNFSF) stimulate receptors of the TNF receptor superfamily (TNFRSF). The TNFSF comprises 19 human ligands, which are defined by a conserved C-terminal
trimerization domain, designated as TNF homology domain (THD), and include TNF, CD40L, CD95L and TWEAK. LTα is a secreted ligand while the other TNFSF ligands are single spanning
transmembrane proteins. In many cases, however, soluble ligand molecules can be released from the membrane-bound proteins by proteolytic cleavage in the stalk region by proteases of the
furin, matrix metalloproteinase (MMP) and a disintegrin and metalloproteinase89 family. TNFSF ligands exert their activity by stimulation of TNFRSF receptors. The latter are characterized by
having one or more cysteine-rich domains (CRDs) in their extracellular parts and can be classified into three groups according to functional and structural similarities: (i) death receptors
that have a cytoplasmic protein–protein interaction domain called death domain that enables some death receptors to trigger cell death pathways, (ii) TRAF-interacting receptors that contain
one to three binding motifs for adapter proteins of the TNF receptor-associated factor (TRAF) family that link these receptors to proinflammatory signaling pathways, and (iii) decoy
receptors without own signaling capabilities that control the activity of other TNFRSF receptors. With regard to function the classification of the signaling competent TNFRSF receptors into
cell death-inducing death receptors and proinflammatory TRAF-interacting receptors is an oversimplification. Death receptors are also able to trigger proinflammatory pathways and
TRAF-interacting receptors via versa can boast apoptotic responses by blocking TRAF-dependent survival activities
In view of the structural organization of TNFSF ligand/TNFRSF receptor complexes, a sequential model of TNFRSF receptor activation was initially assumed. According to this model, a single
TNFRSF receptor molecule initially interacts with a TNFSF trimer and the resulting cell surface-associated TNFSF ligand3–TNFRSF receptor complex then recruits in two further steps two
additional monomeric TNFRSF receptor molecules to form an active TNFSF ligand3–TNFRSF receptor3 complex (Figure 2a). This early model of TNFRSF receptor activation, however, is incompatible
with some fundamental observations. First, ligand binding studies gave no evidence for a sequential assembly of TNFSF ligand–TNFRSF receptor complexes and consistently argued for a single
binding site interaction between TNFSF ligands and TNFRSF receptors. Second, the affinity of a single soluble TNFRSF receptor ectodomain for its ligand is usually rather low (>1 μM).6, 7
Indeed, efficient functional neutralization of TNFSF ligands with soluble TNFRSF receptor variants requires the assembly of two or more receptor molecules, for example, by genetic fusion
with dimerizing or trimerizing protein domains (e.g., Holler et al.8). Third, the sequential TNFRSF receptor activation model cannot explain why some mutants of the TNFRSF receptors CD95 and
TACI, which are defective in ligand binding, nevertheless act in a dominant-negative manner and cause autoimmune lymphoproliferative syndrome (ALPS)9 and common variable immunodeficiency
(CVID).10
PLAD-assisted oligomerization model of TNFRSF receptor activation. This model is based on the fundamental observation that at least some TNFRSF receptors pre-assembles in the absence of
ligand. The self-affinity of TNFRSF receptors would not only allow to explain TNFSF ligand binding by formation of high affinity dimeric or trimeric TNFRSF complexes but may also drive
secondary interaction of TNFSF ligand3–TNFRSF receptor3 complexes. The initially formed TNFSF ligand3–TNFRSF receptor3 complexes may already allow the recruitment of TNFRSF
receptor-associated signaling molecules but do not ensure full activation of these molecules by transactivation. Please note, the capacity of soluble TNFSF ligand-induced TNFSF
ligand3–TNFRSF receptor3 complexes to secondary aggregate spontaneously into fully active receptor clusters may vary considerably between TNFRSF receptors. In some cases (right, upper part)
the self-affinity of TNFRSF receptors is maybe too low to trigger spontaneous clustering of soluble TNFSF ligand-induced receptor complexes while in other cases (right, lower part) the
self-affinity is high enough to trigger this
The limitations of the sequential TNFRSF receptor activation model were solved by the discovery of a protein domain within several TNFRSF receptors that mediates self-assembly in the absence
of ligand molecules.9, 11, 12, 13 The interaction of two (or three) receptor molecules by this so-called 'pre-ligand assembly domain’ (PLAD) may create single high affinity binding sites
for TNFSF ligand trimers. This not only explains the single binding site interaction typically found for TNFSF ligands and cell bound TNFRSF receptors but also delivers a rationale for the
dominant-negative activity of ligand binding-defective CD95/TACI mutants. If such mutants still contain a functional PLAD, then this results in the trapping of wild-type receptor molecules
in complexes with mutant receptor molecules. The latter do not contribute to ligand binding, thus in this case dimerization of receptor molecules does not result in a relevant increase in
apparent affinity. It is noteworthy that the affinity of the PLAD–PLAD interaction is rather low and almost in the mM range.14 This corresponds to the observation that soluble TNFRSF
receptor ectodomains are typically very poor TNFSF ligand agonists unless they are fused with multimerizing scaffolds. In view of the weak PLAD-PLAD affinity an unclear aspect of the
PLAD-based TNFRSF receptor activation model concerns the equilibrium between monomeric and PLAD-assembled TNFRSF receptors. At one extreme, the PLAD-PLAD affinity, despite its weakness, is
possibly sufficient to drive the huge majority of receptors in the PLAD-assembled state due to the spatial pre-orientation and immobilization of the receptor molecules in the plasma membrane
(Figure 2b). However, at the other extreme, the equilibrium point favors monomeric TNFRSF receptors and suggests that there are only a few receptors in the PLAD-assembled state at any given
moment (Figure 2b). In this second scenario, the binding of a TNFSF ligand trimer to the rare PLAD-assembled receptor species would result in the stabilization of the few assembled
receptors and their removal from the equilibrium with the monomeric receptor species. According to the principle of LeChatelier, the pool of ligand-free PLAD-assembled TNFRSF receptors is
then recovered at the expense of the pool of the monomeric receptor species. Thus, with time almost the complete pool of TNFRSF receptor molecules would become accessible for ligand binding
via the ligand-free PLAD-assembled TNFRSF receptors despite the rare occurrence of this receptor species. Currently, it is not possible to differentiate between the two extremes and there
are certainly TNFRSF receptor type-dependent quantitative differences in the PLAD–PLAD interaction that may considerably affect the dynamic equilibrium between monomeric and PLAD-assembled
TNFRSF receptors.
The PLAD-based model for the formation of TNFSF ligand3–TNFRSF receptor3 complexes alone, however, does not adequately explain one fundamental observation of overwhelming functional
importance namely why a significant fraction of TNFRSF receptors bind soluble TNFSF ligands with high affinity but nevertheless fail to efficiently activate receptor-associated signaling
pathways. While interaction with a membrane-bound TNFSF ligand in any case results in strong receptor activation, TNFRSF receptors differ in their response to binding of soluble ligand
trimers. Some TNFRSF receptors strongly stimulate intracellular signaling pathways in response to soluble TNFSF ligands whereas another group of TNFRSF receptors binds soluble ligand
molecules with a limited effect on signal transduction (Table 2). The limited responsiveness to soluble TNFSF ligands of this second type of TNFRSF receptors reflects an intrinsic quality of
the TNFRSF receptor type and not an insufficiency of the soluble ligand. For example, soluble TNF efficiently stimulates TNFR1 signaling but fails to properly activate TNFR2 despite
efficient binding.15, 16 Similarly, soluble APRIL interacts with the TNFRSF receptors TACI and Baff receptor-3 (BR3) but only activates the latter.17, 18 TNFRSF receptors that fail to signal
properly in response to binding of soluble ligand trimers, typically respond quite well when the ligand molecules become secondarily oligomerized (Table 2). The latter can be achieved for
example by antibodies recognizing a tag attached to the cytokine molecules or by genetic fusion with protein domains triggering the assembly of two or more ligand trimers in a single
molecule (Table 3). Because oligomerization has no major effect on the apparent affinity of TNFSF ligand–TNFRSF receptor interaction.19, 20 This indicates that secondary interaction of two
or more TNFSF ligand3–TNFRSF receptor3 complexes is a key event in stimulation of TNFRSF receptor-associated signaling pathways.
There is, however, initial evidence that different types of TNFRSF receptor-associated signaling pathways differ in the need for secondary interaction of two or more TNFSF ligand3–TNFRSF
receptor3 complexes for activation. The need for clustering of TNFSF ligand3–TNFRSF receptor3 complexes for receptor activation has been typically observed in experiments where apoptosis
induction or activation of the classical NFκB pathway has been investigated (see Table 2). Recent studies indicated that soluble CD95L, at low concentrations where it typically fails to
trigger apoptosis without crosslinking, induces cell migration and proliferation (for review, see Wajant21). Soluble TWEAK ((TNF)-like weak inducer of apoptosis) furthermore stimulates
strong and efficient activation of the alternative NFκB pathway but activates the classical NFκB pathway only weakly whereas both NFκB pathways were strongly activated by membrane TWEAK and
oligomerized soluble TWEAK.22 The different oligomerization requirement for CD95L-induced apoptosis and CD95L-induced cell migration as well as the different need of oligomerization for
soluble TWEAK-triggered classical and alternative NFκB signaling correspond in both cases to different mechanisms how these pathways are activated. Interestingly, form studies comparing
ligand- and antibody-induced activation of CD40 and Fn14, there is also evidence for pathway-specific activation requirements of TNFRSF receptors. For example, it has been reported that
antibody production and IL6 secretion in B cells are induced after CD40 stimulation with membrane-bound CD40L while an agonistic CD40-specific antibody triggered antibody but not IL6
production.23 Fn14 targeting antibodies, furthermore, can stimulate the alternative NFκB pathway without a significant effect on the classical NFκB pathway.24
Fn14-mediated activation of the classical NFκB pathway requires the recruitment of the adapter protein TRAF2 and the TRAF2-interacting E3 ligases cIAP1 and cIAP2.25, 26 TRAF2 forms
homotrimeric molecules that binds tightly to a probably monomeric and thus inactive cIAP1 or cIAP2 E3 ligase molecule.27, 28, 29, 30 Dimerization of two cIAPs results in an active
conformation with E3 activity and the capacity to promote signaling via the classical NFκB pathway.27, 31 Thus, in view of the data discussed above soluble TWEAK seems to induce the
formation of complexes that only contain a single cIAP1/2 molecule (TWEAK3-Fn143-TRAF23-cIAP1/2) and which are still unable to trigger the classical NFκB pathway but are competent to do this
upon cIAP1/2 transactivation-enabling crosslinking. In contrast, the formation of TWEAK-Fn14 complexes containing only one TRAF2 trimer and a single cIAP1/2 molecule is already sufficient
to activate the alternative NFκB pathway, because in this case, it is sufficient to withdraw TRAF2–cIAP1/2 complexes from the cytosol32, 33 where they are involved in triggering the
destruction of the alternative NFκB inducing kinase NIK. In the case of CD95-induced apoptosis, there is crystallographic evidence that a pentameric/oligomeric complex of the CD95-recruited
death domain-containing adapter protein FADD has to be formed to trigger efficient dimerization and activation of caspase-8 in oligomeric structures.34, 35, 36, 37, 38 In contrast, soluble
CD95L-induced CD95-mediated cell migration and proliferation are independent from FADD and occur by help of tyrosine kinases that directly interact with CD95.39 In this case, signaling
pathway activation could already emerge from CD95L3–CD953 complexes. In sum, the evidence for oligomerization-independent selective activation of only certain receptor-associated signaling
pathways by soluble TWEAK and soluble CD95L favors a two-step model of TNFRSF receptor activation. In a first step, there is ligand induced formation of signaling competent TNFSF
ligand3–TNFRSF receptor3 complexes, which might already trigger certain signaling pathways. In a second step, there is then oligomerization of TNFSF ligand3–TNFRSF receptor3 complexes that
eventually enables activation of signaling pathways requiring transactivation/oligomerization of TNFSF ligand3–TNFRSF receptor3 complex-associated signaling intermediates (Figure 2b).
The capacity of membrane-bound TNFSF ligands to trigger TNFRSF receptor clustering has not been extensively investigated. The finding that membrane-bound CD95L but not soluble CD95L induces
the formation of durable supramolecular ligand-receptor clusters, however, is in good accordance with this idea.40 In accordance with the evidence discussed above that activation of only a
subset of CD95-induced signaling pathways, including apoptosis induction, requires oligomerization of CD95L3–CD953 complexes and thus membrane-bound CD95L, O’Reilly et al. reported that mice
expressing only soluble CD95L have defective CD95-induced apoptosis but also obtained evidence for soluble CD95L-mediated non-apoptotic activities.41 It is furthermore worth mentioning that
artificially anchoring soluble TNFSF ligands to the cell surface is all that is required to equip these molecules with the activity of the corresponding membrane-bound cytokine. For
example, soluble TNFSF ligand fusion proteins with interaction domains recognizing a cell surface exposed molecular structure/protein acquire membrane ligand-like activity after target
binding.42, 43 Similarly, soluble CD95L gain high apoptotic activity after fibronectin binding and APRIL stimulates Baff-R when trapped by the extracelluar matrix via a heparan sulfate
proteoglycan binding motif in the stalk region.18, 44, 45 Moreover, it has been observed that the enhanced TNFR2-stimulating activity of a cell surface-anchored fusion protein of soluble TNF
is accompanied by clustering of TNFR2 complexes.46
Ligand binding and self-assembly occur via different parts of the ectodomain of TNFRSF receptors.9, 11 TNFRSF receptors have therefore the ability to interact with each other also when
complexed by their ligand suggesting a model of TNFRSF receptor activation in which PLAD–PLAD interactions not only facilitate the binding of TNFSF ligands to TNFRSF receptors to form
signaling competent TNFSF ligands3–TNFRSF receptors3 complexes but also promote secondarily their clustering into supramolecular aggregates where transactivation of TNFRSF
receptor3-associated signaling complexes become possible (Figure 2b).
The two-step model of TNFRSF receptor activation is based on data of the subgroup of TNFRSF receptors that do not or only poorly activate apoptosis and classical NFκB signaling in response
to binding of soluble TNFSF ligands. An obvious question that has not been addressed so far is how TNFRSF receptors that are readily activated by soluble TNFSF ligands, such as TNFR1, fit in
the two-step model of TNFRSF activation. One possibility is that the PLAD-dependent self-affinity of these TNFRSF receptors is simply high enough to drive secondary clustering of initially
formed TNFSF ligand3–TNFRSF receptor3 complexes. However, it cannot be ruled out that this TNFRSF receptor type uses still unknown mechanisms/factors enabling these receptors to promote
oligomerization of TNFRSF-associated adapter proteins without oligomerization of TNFSF ligand3–TNFRSF receptor3 complexes.
Agonistic receptor-specific antibodies were important tools for studying functions of TNFRSF receptors as long as their corresponding TNFSF ligands were unknown and are accordingly still of
special relevance for the analysis of the orphan TNFRSF receptors DR6, TROY and RELT. Agonistic antibodies are also a great help for research on TNFRSF receptors that share a common TNFSF
ligand, as for example the TNF-related apoptosis inducing ligand (TRAIL) receptors. Above all, however, agonistic antibodies are still the means of choice in scenarios where activation of
TNFRSF receptors is needed. Indeed, antibodies have superior pharmacokinetics compared with recombinant TNFSF ligands that have quite low serum half-life of around 10–30 min47, 48, 49 and
therefore require elaborate clinical treatment regimes, such as infusion. Moreover, there is broad experience in the development, production and approval of antibodies. Accordingly, there
are various agonistic TNFRSF receptor-specific antibodies that are currently under consideration in clinical trials (Table 4). Typically, TNFRSF receptor-specific antibodies are used with
the intention to activate TNFRSF receptors on tumor cells to trigger cell death (TRAILR1, TRAILR2) or to activate costimulatory receptors on immune cells to promote antitumor immunity
(4-1BB, GITR, CD27, OX40 CD40). In some cases (CD30, Fn14), the tumor-associated expression pattern of certain TNFRSF receptors is exploited to target tumor cells with ADCC-inducing
antibodies or antibody immunotoxins.
Soon after the description of the first TNFRSF receptor-specific agonistic antibodies, it turned out that the valency of antibodies, thus the antigen binding sites per molecule, is of
crucial relevance for the agonistic activity. In a panel of 17 human TNFR1-specific IgG2a and IgG2b antibodies, Engelmann et al. identified only two antibodies that moderately mimicked the
cytotoxic activity of TNF while all of the these antibodies showed strong TNFR1-mediated killing upon cross-linking with secondary antibodies.50 Likewise, it was found that cross-linking
converts the antagonistic TNFR1-specific IgG2a antibody H398 into a potent TNFR1 agonist.51 Another study characterized the in vitro activities of two IgG1 antibodies and an IgM specific for
TNFR1 and reported superior agonistic activity for the pentameric IgM variant.52 Related data have been reported for CD95-specific antibodies. The highly agonistic CD95-specific antibody
APO-1 is an IgG3 and has thus a considerable tendency to self-aggregate. In contrast, IgG1, IgG2a, IgG2b and IgA variants of APO-1, that have no or only a low capacity to aggregate, elicit
no or less efficient CD95 activation in vitro.53 Cross-linking with protein A or secondary antibodies, however, restored the high agonistic activity of these APO-1 variants.53 In line with
this, various other CD95-specific mAbs of the IgG1 and IgG2a/b subclass have been described that only display strong agonistic activity after cross-linking while the pentameric CD95-specific
IgM CH-11, but not Fab2 fragments derived of this antibody, has high, aggregation-independent agonistic activity.54, 55, 56 The potentiating, or even uncovering, effect of cross-linking on
the agonistic activity of dimeric antibodies has also been broadly documented for other TNFRSF receptors including CD40,57, 58 CD27,59 TRAILR1/DR4,60 TRAILR2/DR561, 62, 63, 64, 65 and
Fn14.24, 66, 67, 68 The relevance of cross-linking for the agonistic activity of dimeric TNFRSF receptor-specific antibodies is also reflected by the fact that antibodies recognizing
non-overlapping epitopes synergistically induce receptor activation.58 In a variation of this theme, it has been recently demonstrated that the therapeutic agonistic activity of the rat
IgG2a murine 4-1BB-specific antibody 3H3 in mouse models of experimental autoimmune encephalomyelitis and allergic asthma is based on the expression of galectin-9 which binds to 4-1BB
without affecting antibody binding.69 Thus, the endogenously present galectin-9 molecule may act as a natural crosslinker here. Although antibody-specific factors, such as affinity and
epitope localization in the targeted TNFRSF receptor, certainly play a role for agonistic activity, the data discussed, in sum suggest that the valency of TNFRSF receptor-specific antibodies
and antibody preparations is the dominant factor that determines their receptor-stimulatory capacity. In particular in view of the importance of clustering of trimeric ligand–receptor
comple
xes for the activation of TNFRSF receptor-associated signaling pathways, it seems natural that interaction of two or more receptor2–antibody complexes is required to form active
[receptor2–antibody]n aggregates (Figure 3a).
TNFRSF receptor activation by oligomerized and FcγR-bound dimeric antibodies. The binding of two TNFRSF molecules by a bivalent antibody may lead, to some extent, to the recruitment of
TNFRSF-associated proteins but with lower efficiency than in the case of stimulation by trimeric ligand. There is, however, no transactivation of TNFRSF receptor3-associated signaling
complexes. Optimal recruitment of adapter proteins as well as transactivation of receptor-bound effector molecules, thus full receptor activation, only occurs after secondary crosslinking of
antibody–TNFRSF receptor2 complexes by protein A or G or secondary antibodies (a) or can be promoted by the self-affinity of the TNFRSF receptors when there is assistance by the spatial and
mobility constraints given by binding to plasma membrane localized FcγRs (b)
The need for secondary interaction of initially formed trimeric ligand–receptor complexes for full TNFRSF receptor activation is nicely reflected by the ability of some per se non-agonistic
TNFRSF receptor-specific antibodies to synergistically stimulate receptor signaling in concert with soluble TNFSF ligands. Already in the 1990s, we described the TNFR2-specific monoclonal
antibody 80M2 that allowed robust TNFR2 activation by soluble TNF which alone is an inefficient stimulator of TNFR2 signaling.15 Likewise, it has been found that poorly active, soluble CD95L
trimers synergistically induce cell death with non-apoptotic CD95-specific antibodies and that some CD40-specific antibodies enhance soluble CD40L activity.58, 70 Of course, a
straightforward explanation of these observations is that these TNFRSF receptor antibodies bring together individually assembled trimeric ligand–receptor complexes.
The typically quite limited agonistic potential of bivalent TNFRSF receptor-specific antibodies may further suggest that monomeric receptors are the dominant receptor species in the
equilibrium of monomeric receptors and PLAD-assembled receptors. In the case of a significant fraction of PLAD-assembled receptors, one would predict the formation of flexible ‘chains’ or
clusters formed due to the bivalency of the antibodies and the two or three epitopes present in dimeric (or trimeric) PLAD-assembled receptors. It is not so obvious why further cross-linking
should have here the huge functional relevance that has been observed experimentally. In the case of a low degree of PLAD-driven complex formation, however, cross-linking of dimeric
antibodies would have an almost obligate impact on the secondary interaction of receptor2–antibody complexes and thus on receptor2–antibody chain/cluster formation.
The overwhelming importance of the intrinsically limited activity of soluble TNFSF ligand trimers and dimeric anti-TNFRSF receptor antibodies for the development of TNFRSF receptor-targeting
therapeutic concepts becomes particularly apparent in the development of TRAIL death receptor-targeting drugs. TRAIL has been initially identified due to its homologies to TNF. TRAIL binds
to five different receptor types that all belong to the TNFRSF receptor family: TRAILR1 to TRAILR4 and osteoprotegerin (OPG). While TRAILR3, TRAILR4 and OPG act as membrane-associated or
soluble decoy receptors, TRAILR1 and TRAILR2 are typical representatives of the death receptor type of TNFRSF receptors.71 Early on, it has been observed that TRAIL triggers apoptosis in a
variety of transformed cell lines but not or only rarely in non-transformed cell types. Accordingly, there were/are considerable efforts of a variety of research groups and companies to
develop TRAIL death receptor-targeting therapeutics for tumor treatment.71 Indeed, recombinant soluble TRAIL (Dulanermin) and several TRAIL death receptor-specific antibodies have been
subjected to clinical trials (Table 4). As monotherapy but also in combination with other anticancer drugs, all these TRAIL death receptor-targeting therapeutics have found to be well
tolerated to date.71 Unfortunately, however, there was also no or quite limited clinical efficacy. From the beginning a variety of in vitro studies demonstrated that oligomerization
potentiates the activity of soluble TRAIL (e.g., Schneider et al.72 and Wiley et al.73) and TRAILR1/2 targeting antibodies (see above). Thus, the TRAIL death receptor-targeting reagents
tested so far in the clinic obviously failed to unleash the full apoptotic activity of the two TRAIL death receptors and the poor therapeutic activity, but also the excellent tolerability,
is therefore perhaps no real surprise. It is noteworthy that in accordance with the already discussed fact that poorly active soluble TNFSF ligand trimers can co-operate with barely active
TNFRSF receptor-specific antibodies to trigger maximal receptor activation, it has been recently shown in vitro and in vivo that co-treatment with soluble TRAIL and the TRAILR2-specific
antibody AMG655 (Conatumumab) results in enhanced apoptosis induction and improved antitumor responses.74, 75 Soluble TRAIL and the murine TRAILR2-specific antibody MD5-1 also
synergistically induce cell death in vitro in various murine cell lines.74 More importantly, the combined treatment with these reagents showed superior antitumor activity and good
tolerability in vivo.74 This suggests that it is possible to target at least TRAILR2 with highly active agonists without paying with detrimental off-target effects.
TNFRSF receptor-specific bivalent antibodies not only resemble soluble TNFSF ligands with respect to the agonistic activity-potentiating effect of oligomerization but also mirror the
differential ability of soluble and membrane-bound TNFSF ligands to activate certain types of TNFRSF receptors. Similar to soluble TNFSF ligand fusion proteins that functionally mimic
membrane TNFSF ligands upon anchoring to cell surface-exposed molecules (Figure 3b), antigen-bound antibodies naturally anchor to certain cell types in an antigen-independent manner by
interaction with Fc receptors recognizing the constant parts of antibodies. For the clinically most important IgG isotypes, there are five human and four murine Fc receptors, the so-called
Fcγ receptors (FcγR; Table 5) that are expressed to a varying extent on B cells and myeloid cell types.76, 77 After binding of antigen–antibody complexes the activatory Fcγ receptors (human:
FcγRI, FcγRIIA, FcγRIIC, FcγRIIIA, FcγRIIIB; murine: FcγRI, FcγRIII, FcγRIV) trigger immune effector functions, such as cytokine release, phagocytosis, antibody-dependent cellular
cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). The activity of these activatory Fcγ receptors is antagonized by the inhibitory FcγRIIB.76, 77 There is now broad in vitro
and in vivo evidence that Fcγ receptor-bound antibodies display strongly enhanced agonistic activity. Crystallographic studies showed that a single IgG molecule interacts with a single FcγR
molecule78, 79, 80, 81, 82 arguing against activation of TNFRSF receptors by sole FcγR-mediated cross-linking of receptor2–antibody complexes as discussed above for protein A and secondary
antibodies. Instead, it is tempting to speculate that in analogy to membrane-bound TNFSF ligands and cell surface anchored fusion proteins of soluble TNFSF ligands, the plasma
membrane-associated spatial and mobility constraints of FcγR-bound antibodies assist TNFRSF receptor self-affinity driven clustering of receptor2–antibody complexes (Figure 3b).
The potential relevance of FcγR binding for TNFRSF antibody activity in vivo became already indirectly obvious in the early studies with antibody class switch variants of the CD95 targeting
APO-1 antibody. While it turned out that the IgG2b isoform of APO-1 is inactive in vitro, it nevertheless displayed significant antitumor activity in vivo.53 Although, it was not clarified
in an early report to which extent antibody-dependent effector functions, such as ADCC and CDC, and FcγR binding-dependent agonistic activity of APO-1 IgG2b contributed to the antitumoral
effect, in vitro studies performed with the hamster IgG2 anti-mouse CD95 mAb Jo2 revealed later strong FcγR binding-dependent agonistic activity.83 Most importantly, however, in vivo studies
with Jo2 and various mice strains with defective expression of one or more FcγRs revealed a crucial role of the inhibitory FcγRII receptor in Jo2-induced hepatotoxicity, the deadly hallmark
of systemic CD95 activation.84, 85 This straightforwardly showed for the first time that the FcγR binding-dependent agonistic activity of a TNFRSF receptor-specific IgG antibody, and thus
receptor activation, is decisive for the observed in vivo effects.
Some important factors that determine the FcγR binding-dependent agonistic activity of TNFRSF receptor-specific antibodies have been revealed in recent years in preclinical studies by
investigating the mode of action of CD40- and TRAILR2-specific antibodies by help of FcγR-deficient mice and FcγR discriminating antibody panels. In a vaccination model where the mouse
CD40-reactive rat anti-CD40 IgG2a mAb 1C10 has been used as an adjuvant, Li and Ravetch86 observed abrogation of CD40-dependent T-cell expansion/activation and antitumor activity in mice
without the common Fc receptor γ (FcRγ) chain. As all three activating FcγRs in mice require the common FcRγ chain for expression and signaling, this observation pointed to a crucial role of
the remaining inhibitory FcγRII for the adjuvant activity of 1C10 and ruled out a major role of ADCC. In line with the idea of a FcγRII-dependent mode of CD40 activation, it turned out
furthermore that 1C10-derived Fab2 preparations and a deglycosylated form of 1C10, thus 1C10 variants that fail to interact with Fcγ receptors, elicit no adjuvant activity in this model,
too.86 Similar findings were made with 3/23, another murine CD40-specific rat IgG2a. A chimeric murine IgG1 variant of 3/23, which significantly binds to FcγRII and the activating FcγRIII,
showed in vitro and in vivo strong stimulatory effects on antigen-presenting cells (B cells, dendritic cells) that are indicative for CD40 activation.87 In contrast, a chimeric murine IgG2a
variant of 3/23 displaying strong binding to the murine activating Fcγ receptors but only poor binding to FcγRII showed no or only marginal immune stimulatory activities.87 Analogous results
were also revealed in studies with the murine TRAILR2/DR5-specific hamster IgG2 antibody MD5-1 and the human TRAILR2/DR5-specific human IgG1 Drozitumab.88, 89 Again, the activating FcγRs
were found to be dispensable for agonistic antibody activity in vivo. A murine IgG1 variant of Drozitumab, which does not interact with FcγRIV, retained antitumoral activity in FcγRI/FcγRIII
double deficient mice.89 Similarly, the well-documented mouse strain-specific hepatotoxicity and tumoricidial activity of MD5-190, 91 was completely abrogated in FcγRII mice.88 Moreover, Fc
domain mutants of MD5-1 and Drozitumab devoid of FcγR binding lost in vivo activity and a variant of MD5-1 with enhanced binding to human FcγRIIB showed improved activity in FcγRII KO mice
with a human FcγRIIB transgene.88
It is worth note that upon immobilization on plastic the aforementioned murine 3/23 chimeras were highly effective with respect to triggering CD40 activation irrespective of their FcγR
preferences.87 In vitro studies with cells expressing a cytoplasmic deletion mutant of FcγRII indicated furthermore that triggering of intracellular signaling pathways is dispensable for
FcγRII to unleash the agonistic activity of 3/23.87 Last but not least, it has been shown that all the activating FcγRs also promote CD40 activation by anti-CD40 IgGs and TRAILR2 activation
by Drozitumab in vitro and a similar FcγR type-independent enhanced activity of FcγR-bound IgGs have also been reported for Fn14-specific antibodies.24, 68, 87, 89 At the first glance, in
sum these data suggest that the sole binding of dimeric antibodies to cell surface-expressed molecules or a plastic surface is sufficient to enable these molecules to activate TNFRSF
receptors. However, this simple view is challenged by the observation that inhibitors of the actin cytoskeleton strongly inhibit the receptor-stimulating activity of CD95- and DR5-specific
IgG antibodies without affecting their binding to FcγRs.83, 89
Against the background that binding to all FcγR types is sufficient to confer strong agonistic activity to TNFRSF receptor-specific antibodies in vitro, it is tempting to speculate that the
observed dominant role of the inhibitory FcγRII in vivo reflects its better bioavailability compared with the activating FcγRs. In further accordance with the idea that the available number
of Fcγ receptors is important for the in vivo activity of dimeric anti-TNFRSF receptor antibodies, Li and Ravetch92 reported that the agonistic in vivo activities of the CD40-specific 1C10
and the TRAILR2-specific mAb MD5-1 are abrogated not only in FcγRII KO mice but also in heterozygous FcγRII animals.
Taken together, FcγR-bound bivalent antibodies display high, membrane-bound TNFSF ligand mimicking TNFRSF receptor-stimulating activity and resemble in this regard extracellular matrix-bound
soluble TNFSF ligands and soluble TNFSF ligand fusion proteins that have been anchored to a cell surface-expressed molecular target. Of course, this does not mean that ‘conventional’ Fc
effector activities of antibodies, such as ADCC or CDC, are unimportant for the in vivo effects of TNFRSF receptor-specific antibodies. Indeed, the antitumoral activity of IgGs targeting the
costimulatory TNFRSF receptors GITR and OX40 have been found to be dominated by ADCC of tumor-associated regulatory T cells.93, 94
The knowledge accumulated in recent years on the relevance of valency, oligomerization and FcγR binding for the agonistic activity of TNFRSF receptor-targeted antibodies will certainly
improve the rational design of antibody-derived TNFRSF receptor agonists but will also help to avoid pitfalls. The agonism-generating effects of oligomerization and FcγR binding are also of
obvious relevance for the development of antagonistic ligand binding-blocking TNFRSF antibodies. Corresponding efforts have not only to avoid the use of antibody variants that bind FcγRs but
must also ensure lack of immunogenicity to prevent the development of cross-linking secondary antibodies.
The recognition of the overwhelming importance of FcγRII/FcγRIIB binding for the agonistic activity of most TNFRSF receptor-specific IgGs may revitalize/enhance efforts to target the TRAIL
death receptors in cancer therapy with antibody variants with FcγRIIB-binding properties superior to the antibodies used so far. In cases where FcγRIIB anchoring has its limitations, for
example, due to poor bioavailability of FcγRIIB expressing cells, artificial oliogmerization of TNFRSF receptor-specific antibodies or antibodies fragments may deliver an alternative
solution to overcome the poor agonistic activity of conventional IgGs. Indeed, high, secondary oligomerization-independent activity has been described for trimeric, tetrameric and pentameric
TRAILR2/DR5-specific nanobody/scFv variants.95, 96 A first clinical trial with the tetravalent nanobody TAS266 revealed reversible hepatoxicity.97 Thus, multivalent highly active
TRAILR2-targeting antibody constructs may offer the promise of increased antitumoral activity but there is also a need to reconsider the possible side effects of systemic TRAILR2 activation
when potent agonists are used in vivo.
The relevance of oligomerization and FcγRIIB anchoring for the agonistic activity of bivalent TNFRSF receptor-specific antibodies has been clearly recognized yet and corresponds very well
with current concepts of TNFRSF receptor activation by secondary interaction of TNFSF ligand3–TNFRSF receptor3 complexes. Oligomerization and FcγRIIB anchoring of bivalent antibodies,
however, are presumably not the only factors that determine agonistic activity of TNFRSF-specific IgGs. There are at least two basal observations that cannot be straightforwardly integrated
in a TNFRSF receptor activation model where oligomerized and cell surface-anchored IgGs promote the clustering of TNFSF ligand3–TNFRSF receptor3 complexes. First, only just, an unexpected,
clinically potentially relevant, FcγR binding-independent agonistic activity has been observed for CD40-targeting human IgG2 isoform B antibodies.98 Here, future studies must show whether
this type of bivalent antibody indeed activates TNFRSF receptor-associated pathways without TNFRSF receptor clustering or have to clarify how this antibody type triggers TNFRSF receptor
clustering without an obvious capacity to auto-aggregate and without evidence for antigen-independent cell surface binding. Second, it is currently not understood why the agonistic activity
of FcγR-bound CD95- and TRAILR2/DR5-specific IgG antibodies is abrogated by pretreatment of the FcγR-expressing cells with actin inhibitors although this do not interfere with antibody
binding.83, 89
This work was supported by research grants from Deutsche Forschungsgemeinschaft (DFG, grant WA 1025/24-1) and from Deutsche Krebshilfe (111703).
Division of Molecular Internal Medicine, Department of Internal Medicine II, University Hospital Würzburg, Würzburg, Germany
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the
article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission
from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
Anyone you share the following link with will be able to read this content:






