- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT A primary failsafe program against unrestrained proliferation and oncogenesis is provided by the p53 tumor suppressor protein, inactivation of which is considered as a hallmark of
cancer. Intriguingly, mutations of the _TP53_ gene are rarely encountered in neuroblastoma tumors, suggesting that alternative p53-inactivating lesions account for escape from p53 control in
this childhood malignancy. Several recent studies have shed light on the mechanisms by which neuroblastoma cells circumvent the p53-driven antitumor barrier. We review here these mechanisms
for evasion of p53-mediated growth control and conclude that deregulation of the p14ARF-MDM2-p53 axis seems to be the principal mode of p53 inactivation in neuroblastoma, opening new
perspectives for targeted therapeutic intervention. SIMILAR CONTENT BEING VIEWED BY OTHERS TARGETING P53 PATHWAYS: MECHANISMS, STRUCTURES AND ADVANCES IN THERAPY Article Open access 01 March
2023 TRANSLATING P53-BASED THERAPIES FOR CANCER INTO THE CLINIC Article 29 January 2024 _MCL-1_ GAINS OCCUR WITH HIGH FREQUENCY IN LUNG ADENOCARCINOMA AND CAN BE TARGETED THERAPEUTICALLY
Article Open access 10 September 2020 MAIN The p53 tumor surveillance network constitutes the core defense mechanism of the cell against loss of genomic integrity and malignant
transformation. Evasion of p53 activity is, therefore, a prerequisite for tumor cells to survive and thrive, and this is attainable either through mutation of the _TP53_ gene or through
defects in the molecular components that govern or execute the various aspects of the p53 response. Elucidation of the mechanisms by which tumor cells override the p53-orchestrated failsafe
program is not only important to gain insight into the ontogenesis of a tumor, but may also point to preferable modes of therapeutic intervention. A striking feature of the childhood cancer
neuroblastoma is the low frequency (<2%) of _TP53_ mutations at diagnosis.1 There is considerable evidence that _TP53_ mutations may be acquired during chemotherapy and malignant
progression of neuroblastoma.1, 2, 3, 4 Accordingly, an increased frequency of _TP53_ mutations is observed in multidrug-resistant neuroblastoma cell lines and in neuroblastoma cell lines
established at relapse, but even in this context, the majority of cell lines remain characterized by a wild-type _TP53_ gene.5, 6 Furthermore, many studies indicate that the p53 signal
transduction pathway is intrinsically intact in neuroblastoma,1, 4, 7, 8, 9 suggesting that circumvention of the p53 barrier in this tumor entity relies primarily on an inappropriately
increased activity of inhibitors of p53 signaling or, alternatively, on a loss of positive regulators of p53 activity. This review summarizes our current understanding of the mechanisms by
which neuroblastoma cells escape from p53-mediated tumor surveillance and positions deregulation of the p14ARF-MDM2-p53 axis as a central switch for p53 inactivation in neuroblastoma. THE
P14ARF-MDM2-P53 AXIS AND LESIONS AT THE _MDM2_ AND _CDKN2A_ (_P16__INK4A__/P14__ARF_) LOCI IN NEUROBLASTOMA The MDM2 oncoprotein, a human homolog of the ‘mouse double minute 2’ gene product
that was originally identified in a spontaneously transformed mouse cell line with double minute chromosomes,10 is a critical negative regulator of p53 stability and activity. It has been
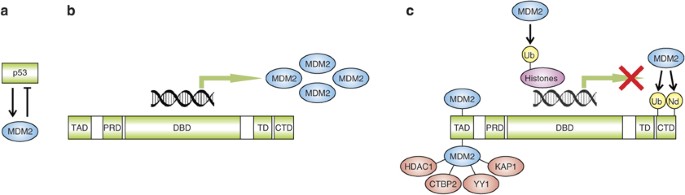
well established that p53 and MDM2 mutually control their cellular levels and form a tight autoregulatory feedback loop (Figure 1a). Under normal physiological conditions, p53 protein levels
are very low because of MDM2-dependent proteasomal degradation.11 Exposure of cells to harmful stimuli, such as DNA damage, hypoxia, telomere erosion, ribonucleotide depletion, or oncogene
activation, results in a number of modifications on the p53 protein (e.g. phosphorylation and acetylation), which suppress the binding of p53 to MDM2 and which lead to accumulation and
increased transcriptional activity of p53.12 In addition to inducing expression of target genes involved in cell-cycle arrest, DNA damage repair, senescence, and apoptosis, p53 also
transactivates the _MDM2_ gene (Figure 1b). The resulting increase in _MDM2_ expression limits the duration and intensity of a non-lethal stress response. There are several mechanisms by
which MDM2 is capable of counteracting p53 activity and stability (Figure 1c). First, MDM2 binds to the transactivation domain of p53 and, therefore, directly interferes with recruitment of
the basal transcriptional machinery and transcriptional coactivators.13, 14, 15 Second, MDM2 acts as an E3 ubiquitin ligase for p53 in a dosage-dependent manner. Low levels of MDM2 promote
p53 monoubiquitination, which may both stimulate nucleocytoplasmic shuttling of p53 because of unmasking of a nuclear export signal and decrease p53 transactivation capacity owing to
unavailability of the ubiquitinated lysine residues for acetylation. At higher levels, the activity of MDM2 results in polyubiquitination and subsequent proteasomal degradation of p53.11,
16, 17 Third, MDM2 also induces monoubiquitination of histone proteins in the vicinity of p53-responsive promoters, resulting in transcriptional repression.18 Fourth, MDM2 has been reported
to inhibit p53 transcriptional activity by promoting conjugation of the ubiquitin-like protein NEDD8 to p53.19 Fifth, MDM2 may also contribute to p53 inactivation by recruiting several
corepressor proteins, such as HDAC1,20 CTBP2,21 YY1,22 and KAP1.23 A central negative regulator of MDM2 is the tumor suppressor protein p14ARF, which is an alternate reading frame product of
the _CDKN2A_ locus on chromosome 9p21. This locus encodes two structurally unrelated growth-inhibitory proteins, p16INK4a and p14ARF, that govern the activities of the pRb and p53 tumor
suppressor pathways, respectively.24 The p14ARF protein serves as a key sensor of hyperproliferative signals generated by activated oncogenes and engages both p53-dependent and
p53-independent pathways to protect cells from malignant transformation.25 The importance of p14ARF-mediated signaling of oncogene activity in the p53 tumor surveillance network is
underscored by observations in mice models that the cancer-protective activity of p53 is abolished in the absence of the murine homolog p19ARF.26, 27 The physical interaction between p14ARF
and MDM2 is in large part responsible for the ability of p14ARF to stabilize and activate p53. p14ARF prevents MDM2 from targeting p53 for degradation by inhibiting the E3 ubiquitin ligase
activity of MDM228 and by blocking nuclear export of MDM2 and p53.29, 30 It has also been firmly established that p14ARF, which is predominantly a nucleolar protein, is capable of mobilizing
MDM2 into the nucleolus, and it has, therefore, been proposed that p14ARF releases nucleoplasmic p53 from the inhibitory grip of MDM2 by inducing nucleolar sequestration of MDM2.30, 31
Although MDM2 redistribution to nucleoli may contribute to p14ARF-induced p53 activity, several reports indicate that neither localization of p14ARF in the nucleolus nor nucleolar
sequestration of MDM2 is essential for stabilization and activation of p53 by p14ARF.32, 33, 34, 35 In this regard, it has been suggested that p14ARF is stored within the nucleolus in
complexes with nucleophosmin, regulating ribosome biogenesis, and displaced to the nucleoplasm by stress-induced nucleolar perturbation, in which it can efficiently counteract MDM2 and
activate the p53 pathway.34, 35, 36 In addition, p14ARF may also enhance p53 function by MDM2-independent mechanisms, for example by inhibiting the activity of another E3 ubiquitin ligase
involved in p53 degradation, ARF-BP1/Mule,37 and by neutralizing the p53-antagonizing NF-_κ_B pathway.38 The mechanisms by which p14ARF promotes p53 stability and activity are shown in
Figure 2. Not surprisingly, many forms of cancer develop defects in MDM2 or p14ARF to escape from p53 control. Genetic aberrations of the _MDM2_ locus as well as genetic or epigenetic
disruption of the _CDKN2A_ (_p16__INK4a__/p14__ARF_) locus may account for inactivation of the p53 pathway in a subset of neuroblastoma tumors, mainly at relapse. Amplification of chromosome
12q–derived sequences encompassing the _MDM2_ gene has been described almost exclusively in neuroblastoma tumors and cell lines that simultaneously have amplification of the _MYCN_ oncogene
on chromosome 2p24, and is associated with attenuated p53 transcriptional function and multidrug resistance.5, 39, 40, 41, 42, 43 The _CDKN2A_ (_p16__INK4a__/p14__ARF_) locus at 9p21 is the
most frequent target of homozygous deletion in both neuroblastoma cell lines44 and primary tumors,45 and has been found to be silenced by methylation in several neuroblastoma cell lines.46,
47 It has been estimated that approximately half of all neuroblastoma cell lines established at relapse are subject to a genetic or epigenetic lesion of the _MDM2_ or _CDKN2A_
(_p16__INK4a__/p14__ARF_) locus,6 but these findings await confirmation in a study that takes also neuroblastoma tumor samples into account. A recent line of evidence supporting a role for
MDM2 activity in the development and malignant behavior of neuroblastoma stems from epidemiological studies of a T>G single nucleotide polymorphism in the _MDM2_ promoter (SNP309;
rs2279744). The presence of this polymorphism increases the affinity of the _MDM2_ promoter for a transcriptional activator, Sp1. This results in enhanced transcription of _MDM2_,
overexpression of the MDM2 protein, attenuation of the p53 pathway, and may eventually lead to accelerated tumor formation.48 Both individuals homozygous for SNP309 (G/G) and subjects
heterozygous for SNP309 (T/G) have an increased risk for the development of neuroblastoma, and neuroblastoma patients carrying the SNP309 variant (G/G or T/G) present with a more advanced
clinical stage and have a shorter 5-year overall survival than patients homozygous for the wild-type allele (T/T).49 A survival study in children with stage 4 neuroblastoma yielded similar
results, with patients homozygous for SNP309 (G/G) having a worse overall survival and a worse survival after relapse than those homozygous for the wild-type allele (T/T), and with
heterozygous SNP309 variant carriers (T/G) showing intermediate survival rates.50 These findings suggest that an increased activity of MDM2 because of the presence of SNP309 has an adverse
effect on the development, aggressiveness, and outcome of neuroblastoma, and provide a direct incentive for the development of novel therapeutic strategies aimed at MDM2 inhibition.
TRANSACTIVATION OF _MDM2_ EXPRESSION BY MYCN Amplification of the _MYCN_ oncogene plays a central role in the pathophysiology and clinical behavior of high-risk neuroblastoma. This genetic
aberration is found in approximately 22% of all neuroblastoma tumors51 and is highly correlated with advanced stages of disease, rapid progression, treatment failure, and fatal outcome.52,
53 _MYCN_ amplification results in overexpression of the MYCN protein, which is a bHLH transcription factor that operates in a heterodimeric complex with Max family proteins to promote cell
growth and proliferation.54 The oncogenic effects of _MYCN_ overexpression have been convincingly established in a variety of model systems. Enhanced expression of _MYCN_ elicits neoplastic
transformation of mammalian cells,55, 56 induces autocrine growth factor activity and increases proliferative potential,57 accelerates cell-cycle progression,58 enhances tumor cell motility
and invasiveness,59 evokes genomic instability through disruption of the regulation of centrosome replication,60, 61 diminishes expression of angiogenesis inhibitors,62, 63 and promotes
immune escape in neuroblastoma by inhibiting the chemoattraction of natural killer T cells.64 Direct evidence for a causative role of _MYCN_ amplification in the pathogenesis of
neuroblastoma is derived from the observation that transgenic mice with targeted expression of _MYCN_ in normal neuroblasts develop tumors with a phenotype very similar to human
neuroblastoma.65 However, aberrant _MYCN_ expression also potently sensitizes neuroblastoma cells to drug- and stress-induced apoptosis,66, 67, 68, 69 and, therefore, needs to be accompanied
by a collateral impairment of the cell death program to provide a selective advantage for the tumor. This counterbalance to the intrinsic apoptosis-sensitizing effect of MYCN may be
delivered by an increased activity of MDM2. A ChIP cloning approach combined with oligonucleotide pull-down and luciferase reporter assays has identified _MDM2_ as a direct transcriptional
target of MYCN in neuroblastoma cells.70 In the same study, endogenous _MDM2_ mRNA and MDM2 protein levels were rapidly upregulated on induction of MYCN in _MYCN_-conditional neuroblastoma
cell lines, whereas targeted inhibition of MYCN in _MYCN_-amplified neuroblastoma cells resulted in reduced MDM2 levels with stabilization of p53 and induction of apoptosis. These data
suggest that MYCN-driven expression of _MDM2_ may constitutively debilitate the p53 pathway in _MYCN_-amplified neuroblastoma cells, providing both a possible mechanism for evasion of
MYCN-primed apoptosis and an explanation for the low frequency of _TP53_ mutations in these cells. This view is further strengthened by evidence that the closely related MYC (c-MYC)
oncoprotein also relies on MDM2 to restrain p53-mediated apoptosis, as Myc-induced lymphomagenesis in mice is profoundly suppressed by haploinsufficiency of _Mdm2_ because of a drastic
increase in p53-dependent apoptosis.71 SUPPRESSION OF P14ARF AND P53 BY TWIST1 Another excellent candidate to explain escape from MYCN-dependent apoptotic sensitization is TWIST1. Just like
MYCN, TWIST1 is a bHLH transcription factor with a fundamental role in embryonic and fetal development. This evolutionary conserved protein is involved in mesoderm formation and
diversification, myogenesis, neurogenesis, and neural crest cell migration and differentiation.72 Loss-of-function mutations in the _TWIST1_ gene have been identified as the main cause of
the Saethre–Chotzen syndrome, an autosomal dominant disorder of craniosynostosis with craniofacial and limb abnormalities.73, 74 In addition to its developmental function in mesodermal and
neural crest cell populations, TWIST1 also acts as an oncoprotein in several cancer types. Neuroblastoma tumors with _MYCN_ amplification consistently exhibit MYCN-driven overexpression of
_TWIST1_, resulting in an oncogenic cooperation that protects neuroblastoma cells from the proapoptotic effect of MYCN and that increases tumorigenicity _in vivo_.75 It could be shown that
the protective effect conferred by TWIST1 was due to suppression of the p53 response and that the dampened p53 function was at least partially attributable to impaired p14ARF activity.75
These findings are in agreement with an earlier study that pointed to downregulation of p19ARF expression by Twist1 as a mechanism for compensating the apoptosis-priming properties of Myc.76
Several other mechanisms may also contribute to the p53-inhibitory activity of TWIST1, including inhibition of acetyltransferases that serve as transcriptional coactivators for p53,77
modulation of the activity of a transactivator of the _TP53_ promoter,78 prevention of p53 phosphorylation,78 and direct suppression of the DNA-binding activity of p53.79 Of note, the TWIST1
and TWIST2 proteins have also recently been shown to prevent oncogene-induced premature senescence with concomitant abrogation of p16INK4a and p21WAF1/CIP1 activation, and to induce, in
cooperation with activated mitogenic oncoproteins, epithelial-mesenchymal transition, suggesting a role as general inhibitors of multiple oncogene-induced safeguard programs.80 INACTIVATION
OF THE P14ARF-P53 PATHWAY BY BMI1 The Polycomb-group transcriptional repressor BMI1 has been proposed as another roadblock to MYCN-induced apoptosis by suppressing the p14ARF-p53 signaling
pathway.81, 82 BMI1 is a component of the Polycomb repressive complex 1, which mediates transcriptional silencing through chromatin modifications and which is involved in embryonic and adult
stem cell maintenance and in the development of several cancer types.83 It has been convincingly shown that Bmi1 is indispensable for the self-renewal capacity and postnatal maintenance of
hematopoietic and neural stem cells in mice by repressing the _Cdkn2a_ (_p16__INK4a__/p19__ARF_) locus.84, 85, 86, 87, 88 Notably, Bmi1 also collaborates strongly with Myc in murine
lymphomagenesis,89, 90, 91, 92 and the molecular basis of this oncogenic cooperation is the ability of Bmi1 to prohibit Myc-induced apoptosis by downregulating _Cdkn2a_
(_p16__INK4a__/p19__ARF_) expression.93 Similar to TWIST1, the BMI1 oncoprotein inhibits oncogene-induced premature senescence and cooperates with activated H-Ras to induce neoplastic
transformation and epithelial-mesenchymal transition.84, 94 Thus, both the TWIST1 and BMI1 transcriptional regulators may overcome several oncogene-induced failsafe barriers and may serve as
examples of corrupt exploitation of normal developmental programs by tumor cells. _BMI1_ is strongly expressed in neuroblastoma cell lines and tumors,81, 82 and has been shown to be
essential for the tumorigenicity of neuroblastoma cells.82 BMI1 negatively regulates p53 expression in neuroblastoma cells, potently inhibits the apoptotic activity of MYCN, and functions as
an oncogenic partner of MYCN in the transformation of normal neural crest cells and in the malignant progression of neuroblastoma cells.82 These findings have been attributed to the ability
of BMI1 to repress the _CDKN2A_ (_p16__INK4a__/p14__ARF_) locus, although it cannot be excluded that CDKN2A-independent pathways may also play a role. Interestingly, the collaborative
activity between MYCN and BMI1 may be switched on by a single initiating event, as deregulated E2F1 activity, which is a characteristic lesion in highly proliferative neuroblastoma tumors,95
is capable of directly driving the expression of both oncogenes.81, 96 The role of BMI1 in neuroblastoma pathogenesis seems not to be limited to _MYCN_-amplified tumors, as _BMI1_ is also
expressed and required for tumorigenicity in neuroblastoma cells with a normal copy number of _MYCN_.82 In line with the requirement of BMI1 activity in self-renewal of neural stem cells, it
has been argued that BMI1 may be of critical importance for the maintenance of neuroblastoma stem cells by regulating clonogenic self-renewal and multilineage differentiation, offering an
attractive target for therapeutic intervention.97 REPRESSION OF P14ARF-P53 SIGNALING BY LOSS OF CHD5 Escape from p53 surveillance in neuroblastoma cells may also be accomplished by the loss
of another chromatin-remodeling protein involved in transcriptional control of the _CDKN2A_ (_p16__INK4a__/p14__ARF_) locus. One of the most characteristic genomic lesions in neuroblastoma
is deletion of the short arm of chromosome 1, which is found in 25 to 35% of primary neuroblastoma tumors and 80 to 90% of neuroblastoma cell lines.98 The actual target of this deletion has
remained elusive for a long time, but detailed analysis of the different genes located in the smallest region of deletion at 1p36.31 has recently identified _CHD5_ as the strongest candidate
tumor suppressor gene.99, 100 _CHD5_ encodes a protein with _chr_omatin-_o_rganizing _mo_dulator (chromo), helicase, and DNA-binding motifs that is preferentially expressed in the nervous
system and the adrenal gland.101 Expression of _CHD5_ is very low or absent in neuroblastoma cell lines and is inversely correlated with 1p deletion, _MYCN_ amplification, advanced clinical
stage, unfavorable histology, and poor event-free and overall survival in neuroblastoma tumors.99, 100, 101 Homozygous deletion and mutational inactivation of _CHD5_ are infrequent events,99
but it has been shown that the remaining _CHD5_ allele in neuroblastoma cells with heterozygous 1p deletion may be transcriptionally silenced by promoter methylation.100 Reintroduction of
_CHD5_ in such neuroblastoma cells with 1p deletion and epigenetic _CHD5_ silencing significantly reduced clonogenicity and _in vivo_ tumorigenicity, validating _CHD5_ as a _bona fide_ tumor
suppressor gene.100 Of note, an independent study that used chromosome engineering to produce mouse strains with deletions or duplications of a region corresponding to human 1p36 identified
_Chd5_ as a potent tumor suppressor that controls proliferation, senescence, and apoptosis through the p19ARF-p53 pathway.102 Silencing of _Chd5_ by short hairpin RNA in MEFs severely
compromised p53 function and promoted tumorigenesis, and these effects were associated with a substantial reduction in the basal and oncogene-induced expression levels of p16INK4a and
p19ARF. Knockdown of p19ARF, but not p16INK4a, was capable of bypassing the proliferation defect of MEFs that harbored an engineered duplication of the 1p36-syntenic region, indicating that
Chd5, which could be shown to be responsible for the proliferation-suppressive properties of the 1p36-syntenic region, exerts its antiproliferative activity by facilitating expression of
p19ARF. Altogether, the findings of this study support a model in which the chromatin-remodeling activity of Chd5 is required for proper transcriptional activation of the _Cdkn2a_
(_p16__INK4a__/p19__ARF_) locus. Although a direct link between CHD5 and the p14ARF-p53 network remains to be established in the context of human neuroblastoma, it is tempting to speculate
that loss of _CHD5_ by 1p deletion and epigenetic silencing may promote the pathogenesis of neuroblastoma by crippling the p14ARF-p53 signaling pathway. DEREGULATION OF THE P14ARF-MDM2-P53
AXIS BY PPM1D (WIP1) The most frequent and the prognostically most unfavorable genomic alteration in neuroblastoma is gain or amplification of genetic material from the long arm of
chromosome 17.103, 104 A gene at 17q23.2 encoding a key negative regulator of p53, _PPM1D_, has been put forward as the most likely target of 17q gain/amplification, based on its location in
the minimal common region of gain/amplification, its consistent pattern of overexpression in neuroblastoma cell lines with 17q gain/amplification, its growth-promoting and antiapoptotic
activity in neuroblastoma cells, and the adverse impact of its expression level on the prognosis of primary neuroblastoma.105 Similarly, _PPM1D_ has been blamed as the culprit oncogene
behind gain or amplification of 17q23 in breast cancer,106, 107, 108 ovarian clear cell adenocarcinoma,109 and medulloblastoma.110, 111 The protein encoded by _PPM1D_ is a serine/threonine
phosphatase that is transcriptionally induced by wild-type p53 in response to DNA-damaging stimuli such as ionizing radiation, and it has, therefore, been given the name Wip1 (_w_ild-type
p53–_i_nduced _p_hosphatase 1).112 The p53-dependent expression of Wip1 creates a negative feedback loop that helps to turn off p53 at the end of a stress response, as Wip1 suppresses p53
activity and stability through multiple mechanisms (Figures 3a–c). First, Wip1 dephosphorylates and inactivates several kinases that mediate p53 stabilization and activation after genotoxic
stress, for example p38 MAPK,113 Chk1,114 Chk2,115, 116, 117 ATM,118, 119 and probably ATR.120 In addition, Wip1 dephosphorylates p53 itself at serine 15, thereby probably promoting both p53
degradation and inactivation.114 The most important block provided by Wip1 on p53 function is mediated through stabilization and enhanced p53 binding of MDM2, which result from Wip1-induced
dephosphorylation of MDM2 at serine 395 and which argue for a role of Wip1 as a molecular gatekeeper in the p53-MDM2 autoregulatory feedback loop.121 Finally, studies using _Ppm1d_-null
MEFs have shown that Wip1 is also capable of suppressing p19ARF levels through a p38 MAPK–dependent mechanism, which seems to involve transcriptional repression of the _Cdkn2a_
(_p16__INK4a__/p19__ARF_) locus, thus offering an additional explanation of how Wip1 may keep p53 in check.122 The potent inhibitory activity of Wip1 on p53 provides, in principle, an
appealing opportunity for tumor cells to escape from p53 control. Indeed, as discussed above, copy number gain/amplification and overexpression of _PPM1D_ are observed in a variety of human
tumors including neuroblastoma, which then typically retain wild-type p53 and often carry a poor prognosis.105, 107, 108, 109, 110 Overexpression experiments have shown that Wip1 induces
malignant transformation in collaboration with other oncogenes, protects against oncogene-induced premature senescence and apoptosis, and accelerates tumorigenesis _in vivo_.106, 107, 123,
124 Conversely, _Ppm1d_-null MEFs and mice are resistant to oncogene-induced transformation and to spontaneous and oncogene-driven tumorigenesis, respectively.119, 122, 123 Although most of
the oncogenic properties of Wip1 are ascribed to its ability to suppress the p53 pathway and the DNA damage response, concomitant inhibition of pRb tumor suppressor activity because of
transcriptional repression of p16INK4a expression may also play a role.122 Of note in the context of neuroblastoma is the observation that homozygous or heterozygous deficiency of _Ppm1d_ in
mice confers protection against Myc-induced lymphomagenesis in a p53- and ATM-dependent manner, indicative of a strict requirement for Wip1 activity in the suppression of Myc-triggered
apoptosis.119 This is reminiscent of the critical role of Mdm2 in evasion of Myc-primed apoptosis in murine lymphomagenesis,71 which is a concept directly transferable to human neuroblastoma
and MYCN.70 It could, therefore, be speculated that increased dosage of _PPM1D_ provides another mechanism for escape from MYCN-stimulated apoptosis and a molecular explanation for the
strong association between 17q gain and _MYCN_ amplification in neuroblastoma cells.105 CYTOPLASMIC SEQUESTRATION OF P53 Aberrant cytoplasmic localization of wild-type p53 has been proposed
as another mechanism for p53 inactivation in neuroblastoma cells. Although controversy exists on the frequency and functional relevance of this phenomenon, it has been extensively documented
that cytoplasmic p53 sequestration does occur in at least some cases of neuroblastoma. Interestingly, as will be discussed below, cumulating evidence indicates that an increased activity of
MDM2 or a dysfunction of its functional counterpart HAUSP, a principal p53-deubiquitinating enzyme, lies at the molecular basis of cytoplasmic p53 retention in neuroblastoma, further
underscoring the importance of MDM2 deregulation as a means to escape from p53 control. An initial study found cytoplasmic p53 sequestration in 96% of undifferentiated neuroblastoma tumors,
whereas this phenotype was absent in differentiated neuroblastoma tumors.125 However, other studies have reported a predominant nuclear localization of p53 in undifferentiated neuroblastoma
tumors, and both cytoplasmic and nuclear p53 in differentiating neuroblastoma.9, 43, 126 Conflicting results also exist for neuroblastoma cell lines, as the subcellular localization of p53
has been reported to be exclusively cytoplasmic (e.g. in IMR-32 and SK-N-SH cells),127 primarily cytoplasmic and weakly nuclear (e.g. in IMR-32 and SK-N-SH cells),126, 128 equally
cytoplasmic and nuclear (e.g. in SK-N-SH cells),7 predominantly nuclear (e.g. in IMR-32 cells),43 and completely nuclear (e.g. in IMR-32 and SK-N-SH cells).129 Some of the discrepancies may
be explained by cross-reactivity of the antibodies used to detect p53 and by different methods of tissue fixation and cell preparation.9, 43, 126 Nonetheless, it is generally accepted that
some cytoplasmic p53 does exist in neuroblastoma, although the prevalence and importance of cytoplasmic p53 sequestration remain a subject of debate.9, 130 It has been reported that abnormal
cytoplasmic p53 localization may attenuate the DNA damage–induced G1 checkpoint function127 and the apoptotic activity131, 132 of wild-type p53 in some neuroblastoma cells. On the contrary,
many studies have shown that the DNA-binding and transactivation capacity of p53 and the p53 signal transduction pathway are intact in neuroblastoma cells with wild-type p53,1, 4, 7, 8, 9
indicating that cytoplasmic retention of wild-type p53 is either an infrequent anomaly or a relative block on p53 that can be overcome by appropriate p53-inducing stimuli. Proposed
mechanisms for abnormal p53 accumulation in the cytoplasm of neuroblastoma cells include hyperactive nuclear export of p53, cytoplasmic tethering of p53, resistance of p53 to proteasomal
degradation, and possibly impaired nuclear re-import of p53. A unifying theme common to these diverse mechanisms may be the involvement of a disrupted MDM2/HAUSP regulation of p53. It has
been firmly established that cytoplasmic p53 sequestration in neuroblastoma cells is at least in part caused by enhanced nuclear export133 and that MDM2 plays an important role in this
nuclear exclusion of p53.131, 134 Once transported to the cytoplasm, p53 may be held in this compartment by a cytoplasmic anchor protein, such as Parc.128 Neuroblastoma cells express high
levels of Parc, which have been shown to prevent nuclear localization of p53 and a normal apoptotic response to the genotoxic drug etoposide.128 A comparable cytoplasmic anchoring function
may be exerted by the large T antigen from human polyomavirus BK,135, 136 by the glucocorticoid receptor,137 and by the MDM2-related protein MDM4 (also known as MDMX).138 It has also been
shown that p53 in neuroblastoma cells is aberrantly ubiquitinated because of an impaired interaction between p53 and the deubiquitinating enzyme HAUSP, and that this hyperubiquitination
contributes to cytoplasmic p53 sequestration.130 As both Parc and HAUSP interact with the carboxy terminus of p53, competition between Parc and HAUSP for p53 binding has been postulated to
underly the impaired p53–HAUSP interaction in neuroblastoma cells, although this remains to be formally proven.130 The defective deubiquitination of p53 results in the appearance of
(multi)monoubiquitinated p53 species, which are relatively resistant to proteasomal degradation139 and which are subject to increased nuclear export and possibly to diminished re-import,
thus yielding a phenotype of cytoplasmic p53 sequestration.130 In keeping with the deregulation of MDM2/HAUSP, interference with p53 hyperubiquitination by targeted inhibition of the
p53–MDM2 interaction in neuroblastoma cells has been shown to relocate p53 from the cytoplasm to the nucleus and to restore the transcriptional and apoptotic activities of p53.130
CONCLUSIONS The rarity of _TP53_ mutations in neuroblastoma has been a puzzling issue to many investigators given the potent antitumor capacity of wild-type p53 protein. A substantial number
of alternative p53-inactivating lesions have been identified in neuroblastoma during the past few years, many of which interfere with proper functioning of the p14ARF-MDM2-p53 axis (Figure
4). A recent mouse study underscores the importance of direct inhibition of p53 by MDM2 and suppression of p19ARF in the pathogenesis of neuroblastoma.140 However, it should be kept in mind
that cellular decisions of growth, survival, and death result from the integration of a complex network of intertwined signaling cascades and, therefore, that also pathways that do not
impinge directly on the core p53 machinery may still provide a means to oppose or neutralize p53 activity. Full characterization of the nature and relative importance of the different blocks
on the p53 pathway in neuroblastoma cells awaits genome-wide experimental approaches in well-controlled model systems. It may not be that surprising, after all, that turning off the
p14ARF-MDM2-p53 axis is a preferential mode of p53 inactivation in neuroblastoma cells. It has been convincingly shown that deregulated _MYC_ expression is a potent trigger for induction of
the p14ARF protein, and it is very likely that the same holds true for _MYCN_.25, 141 In addition, aggressive neuroblastoma tumors typically express high levels of the E2F1 transcription
factor,95 which is capable of inducing p14ARF expression through binding to an E2F-responsive element in the _p14__ARF_ promoter.142, 143 Hence, acquisition of defects that inactivate p14ARF
or that uncouple p14ARF from its p53-dependent effector pathway (i.e. through uncontrolled MDM2 activity) may provide the most effective route to non-mutational p53 inactivation in
neuroblastoma cells by directly dismantling the molecular circuitry that signals the malicious identity of these cells to the p53 guardian. In conclusion, it has become increasingly clear in
recent years that inappropriately increased activity of MDM2 is the primary culprit for p53 inactivation in neuroblastoma cells. Preclinical work from our laboratory and others has shown
that small-molecule MDM2 inhibitors are capable of eliciting potent antitumor effects against neuroblastoma by selectively and non-genotoxically reactivating p53.8, 130, 144, 145 These
findings may provide a new therapeutic avenue for the treatment of children with high-risk neuroblastoma. ABBREVIATIONS * bHLH: basic helix-loop-helix * CHD5: chromodomain helicase
DNA-binding protein 5 * ChIP: chromatin immunoprecipitation * chromo: chromatin-organizing modulator * MDM2: mouse double minute 2 homolog * MEFs: mouse embryonic fibroblasts * p14ARF: human
alternate reading frame protein of 14 kDa * p19ARF: murine alternate reading frame protein of 19 kDa * Parc: p53-associated parkin-like cytoplasmic protein * pRb: retinoblastoma protein *
SNP309: single nucleotide polymorphism at position 309 (T>G) in _MDM2_ * Wip1: wild-type p53–induced phosphatase 1 REFERENCES * Tweddle DA, Pearson AD, Haber M, Norris MD, Xue C, Flemming
C _et al_. The p53 pathway and its inactivation in neuroblastoma. _Cancer Lett_ 2003; 197: 93–98. Article CAS PubMed Google Scholar * Tweddle DA, Malcolm AJ, Bown N, Pearson AD, Lunec J
. Evidence for the development of _p53_ mutations after cytotoxic therapy in a neuroblastoma cell line. _Cancer Res_ 2001; 61: 8–13. CAS PubMed Google Scholar * Kotchetkov R, Driever PH,
Cinatl J, Michaelis M, Karaskova J, Blaheta R _et al_. Increased malignant behavior in neuroblastoma cells with acquired multi-drug resistance does not depend on P-gp expression. _Int J
Oncol_ 2005; 27: 1029–1037. CAS PubMed Google Scholar * Xue C, Haber M, Flemming C, Marshall GM, Lock RB, MacKenzie KL _et al_. p53 determines multidrug sensitivity of childhood
neuroblastoma. _Cancer Res_ 2007; 67: 10351–10360. Article CAS PubMed Google Scholar * Keshelava N, Zuo JJ, Chen P, Waidyaratne SN, Luna MC, Gomer CJ _et al_. Loss of p53 function
confers high-level multidrug resistance in neuroblastoma cell lines. _Cancer Res_ 2001; 61: 6185–6193. CAS PubMed Google Scholar * Carr J, Bell E, Pearson AD, Kees UR, Beris H, Lunec J
_et al_. Increased frequency of aberrations in the p53/MDM2/p14_ARF_ pathway in neuroblastoma cell lines established at relapse. _Cancer Res_ 2006; 66: 2138–2145. Article CAS PubMed
Google Scholar * Goldman SC, Chen CY, Lansing TJ, Gilmer TM, Kastan MB . The p53 signal transduction pathway is intact in human neuroblastoma despite cytoplasmic localization. _Am J Pathol_
1996; 148: 1381–1385. CAS PubMed PubMed Central Google Scholar * Van Maerken T, Speleman F, Vermeulen J, Lambertz I, De Clercq S, De Smet E _et al_. Small-molecule MDM2 antagonists as a
new therapy concept for neuroblastoma. _Cancer Res_ 2006; 66: 9646–9655. Article CAS PubMed Google Scholar * Chen L, Malcolm AJ, Wood KM, Cole M, Variend S, Cullinane C _et al_. p53 is
nuclear and functional in both undifferentiated and differentiated neuroblastoma. _Cell Cycle_ 2007; 6: 2685–2696. Article CAS PubMed Google Scholar * Cahilly-Snyder L, Yang-Feng T,
Francke U, George DL . Molecular analysis and chromosomal mapping of amplified genes isolated from a transformed mouse 3T3 cell line. _Somat Cell Mol Genet_ 1987; 13: 235–244. Article CAS
PubMed Google Scholar * Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G . Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. _Cell Death Differ_ 2006;
13: 927–934. Article CAS PubMed Google Scholar * Vousden KH, Lu X . Live or let die: the cell's response to p53. _Nat Rev Cancer_ 2002; 2: 594–604. Article CAS PubMed Google
Scholar * Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B . Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. _Nature_ 1993; 362:
857–860. Article CAS PubMed Google Scholar * Thut CJ, Goodrich JA, Tjian R . Repression of p53-mediated transcription by MDM2: a dual mechanism. _Genes Dev_ 1997; 11: 1974–1986. Article
CAS PubMed PubMed Central Google Scholar * Wadgaonkar R, Collins T . Murine double minute (MDM2) blocks p53-coactivator interaction, a new mechanism for inhibition of p53-dependent
gene expression. _J Biol Chem_ 1999; 274: 13760–13767. Article CAS PubMed Google Scholar * Kohn KW, Pommier Y . Molecular interaction map of the p53 and Mdm2 logic elements, which
control the Off On switch of p53 in response to DNA damage. _Biochem Biophys Res Commun_ 2005; 331: 816–827. Article CAS PubMed Google Scholar * Brooks CL, Gu W . p53 ubiquitination:
Mdm2 and beyond. _Mol Cell_ 2006; 21: 307–315. Article CAS PubMed PubMed Central Google Scholar * Minsky N, Oren M . The RING domain of Mdm2 mediates histone ubiquitylation and
transcriptional repression. _Mol Cell_ 2004; 16: 631–639. Article CAS PubMed Google Scholar * Xirodimas DP, Saville MK, Bourdon JC, Hay RT, Lane DP . Mdm2-mediated NEDD8 conjugation of
p53 inhibits its transcriptional activity. _Cell_ 2004; 118: 83–97. Article CAS PubMed Google Scholar * Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E _et al_.
MDM2–HDAC1-mediated deacetylation of p53 is required for its degradation. _EMBO J_ 2002; 21: 6236–6245. Article CAS PubMed PubMed Central Google Scholar * Mirnezami AH, Campbell SJ,
Darley M, Primrose JN, Johnson PW, Blaydes JP . Hdm2 recruits a hypoxia-sensitive corepressor to negatively regulate p53-dependent transcription. _Curr Biol_ 2003; 13: 1234–1239. Article
CAS PubMed Google Scholar * Sui G, Affar el B, Shi Y, Brignone C, Wall NR, Yin P _et al_. Yin Yang 1 is a negative regulator of p53. _Cell_ 2004; 117: 859–872. Article CAS PubMed
Google Scholar * Wang C, Ivanov A, Chen L, Fredericks WJ, Seto E, Rauscher 3rd FJ _et al_. MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. _EMBO J_ 2005; 24:
3279–3290. Article CAS PubMed PubMed Central Google Scholar * Lowe SW, Sherr CJ . Tumor suppression by _Ink4a–Arf_: progress and puzzles. _Curr Opin Genet Dev_ 2003; 13: 77–83. Article
CAS PubMed Google Scholar * Sherr CJ . Divorcing ARF and p53: an unsettled case. _Nat Rev Cancer_ 2006; 6: 663–673. Article CAS PubMed Google Scholar * Efeyan A, Garcia-Cao I,
Herranz D, Velasco-Miguel S, Serrano M . Tumour biology: policing of oncogene activity by p53. _Nature_ 2006; 443: 159. Article CAS PubMed Google Scholar * Christophorou MA, Ringshausen
I, Finch AJ, Swigart LB, Evan GI . The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. _Nature_ 2006; 443: 214–217. Article CAS PubMed Google
Scholar * Honda R, Yasuda H . Association of p19ARF with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. _EMBO J_ 1999; 18: 22–27. Article CAS PubMed PubMed
Central Google Scholar * Zhang Y, Xiong Y . Mutations in human _ARF_ exon 2 disrupt its nucleolar localization and impair its ability to block nuclear export of MDM2 and p53. _Mol Cell_
1999; 3: 579–591. Article CAS PubMed Google Scholar * Tao W, Levine AJ . P19ARF stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. _Proc Natl Acad Sci USA_ 1999; 96:
6937–6941. Article CAS PubMed PubMed Central Google Scholar * Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D . Nucleolar Arf sequesters Mdm2 and activates p53. _Nat Cell Biol_
1999; 1: 20–26. Article CAS PubMed Google Scholar * Midgley CA, Desterro JM, Saville MK, Howard S, Sparks A, Hay RT _et al_. An N-terminal p14ARF peptide blocks Mdm2-dependent
ubiquitination _in vitro_ and can activate p53 _in vivo_. _Oncogene_ 2000; 19: 2312–2323. Article CAS PubMed Google Scholar * Lin AW, Lowe SW . Oncogenic _ras_ activates the ARF-p53
pathway to suppress epithelial cell transformation. _Proc Natl Acad Sci USA_ 2001; 98: 5025–5030. Article CAS PubMed PubMed Central Google Scholar * Llanos S, Clark PA, Rowe J, Peters G
. Stabilization of p53 by p14ARF without relocation of MDM2 to the nucleolus. _Nat Cell Biol_ 2001; 3: 445–452. Article CAS PubMed Google Scholar * Korgaonkar C, Hagen J, Tompkins V,
Frazier AA, Allamargot C, Quelle FW _et al_. Nucleophosmin (B23) targets ARF to nucleoli and inhibits its function. _Mol Cell Biol_ 2005; 25: 1258–1271. Article CAS PubMed PubMed Central
Google Scholar * Gjerset RA, Bandyopadhyay K . Regulation of p14ARF through subnuclear compartmentalization. _Cell Cycle_ 2006; 5: 686–690. Article CAS PubMed Google Scholar * Chen D,
Kon N, Li M, Zhang W, Qin J, Gu W . ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. _Cell_ 2005; 121: 1071–1083. Article CAS PubMed Google Scholar * Rocha S, Campbell
KJ, Perkins ND . p53- and Mdm2-independent repression of NF-_κ_B transactivation by the ARF tumor suppressor. _Mol Cell_ 2003; 12: 15–25. Article CAS PubMed Google Scholar * Corvi R,
Savelyeva L, Amler L, Handgretinger R, Schwab M . Cytogenetic evolution of _MYCN_ and _MDM2_ amplification in the neuroblastoma LS tumour and its cell line. _Eur J Cancer_ 1995; 31A:
520–523. Article CAS PubMed Google Scholar * Corvi R, Savelyeva L, Breit S, Wenzel A, Handgretinger R, Barak J _et al_. Non-syntenic amplification of _MDM2_ and _MYCN_ in human
neuroblastoma. _Oncogene_ 1995; 10: 1081–1086. CAS PubMed Google Scholar * Van Roy N, Forus A, Myklebost O, Cheng NC, Versteeg R, Speleman F . Identification of two distinct chromosome
12-derived amplification units in neuroblastoma cell line NGP. _Cancer Genet Cytogenet_ 1995; 82: 151–154. Article CAS PubMed Google Scholar * McKenzie PP, Guichard SM, Middlemas DS,
Ashmun RA, Danks MK, Harris LC . Wild-type p53 can induce p21 and apoptosis in neuroblastoma cells but the DNA damage-induced G1 checkpoint function is attenuated. _Clin Cancer Res_ 1999; 5:
4199–4207. CAS PubMed Google Scholar * Tweddle DA, Malcolm AJ, Cole M, Pearson AD, Lunec J . p53 cellular localization and function in neuroblastoma: evidence for defective G1 arrest
despite WAF1 induction in _MYCN_-amplified cells. _Am J Pathol_ 2001; 158: 2067–2077. Article CAS PubMed PubMed Central Google Scholar * Thompson PM, Maris JM, Hogarty MD, Seeger RC,
Reynolds CP, Brodeur GM _et al_. Homozygous deletion of _CDKN2A_ (_p16__INK4a__/p14__ARF_) but not within 1p36 or at other tumor suppressor loci in neuroblastoma. _Cancer Res_ 2001; 61:
679–686. CAS PubMed Google Scholar * Caren H, Erichsen J, Olsson L, Enerback C, Sjoberg RM, Abrahamsson J _et al_. High-resolution array copy number analyses for detection of deletion,
gain, amplification and copy-neutral LOH in primary neuroblastoma tumors: Four cases of homozygous deletions of the CDKN2A gene. _BMC Genomics_ 2008; 9: 353. Article CAS PubMed PubMed
Central Google Scholar * Takita J, Hayashi Y, Kohno T, Yamaguchi N, Hanada R, Yamamoto K _et al_. Deletion map of chromosome 9 and _p16_ (_CDKN2A_) gene alterations in neuroblastoma.
_Cancer Res_ 1997; 57: 907–912. CAS PubMed Google Scholar * Takita J, Hayashi Y, Nakajima T, Adachi J, Tanaka T, Yamaguchi N _et al_. The _p16 (CDKN2A)_ gene is involved in the growth of
neuroblastoma cells and its expression is associated with prognosis of neuroblastoma patients. _Oncogene_ 1998; 17: 3137–3143. Article CAS PubMed Google Scholar * Bond GL, Hu W, Bond EE,
Robins H, Lutzker SG, Arva NC _et al_. A single nucleotide polymorphism in the _MDM2_ promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. _Cell_
2004; 119: 591–602. Article CAS PubMed Google Scholar * Cattelani S, Defferrari R, Marsilio S, Bussolari R, Candini O, Corradini F _et al_. Impact of a single nucleotide polymorphism in
the _MDM2_ gene on neuroblastoma development and aggressiveness: results of a pilot study on 239 patients. _Clin Cancer Res_ 2008; 14: 3248–3253. Article CAS PubMed Google Scholar *
Perfumo C, Parodi S, Mazzocco K, Defferrari R, Inga A, Haupt R _et al_. Impact of _MDM2_ SNP309 genotype on progression and survival of stage 4 neuroblastoma. _Eur J Cancer_ 2008; 44:
2634–2639. Article CAS PubMed Google Scholar * Brodeur GM . Neuroblastoma: biological insights into a clinical enigma. _Nat Rev Cancer_ 2003; 3: 203–216. Article CAS PubMed Google
Scholar * Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM . Amplification of N-_myc_ in untreated human neuroblastomas correlates with advanced disease stage. _Science_ 1984; 224:
1121–1124. Article CAS PubMed Google Scholar * Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY _et al_. Association of multiple copies of the N-_myc_ oncogene with rapid
progression of neuroblastomas. _N Engl J Med_ 1985; 313: 1111–1116. Article CAS PubMed Google Scholar * Wenzel A, Schwab M . The mycN/max protein complex in neuroblastoma. Short review.
_Eur J Cancer_ 1995; 31A: 516–519. Article CAS PubMed Google Scholar * Schwab M, Varmus HE, Bishop JM . Human N-_myc_ gene contributes to neoplastic transformation of mammalian cells in
culture. _Nature_ 1985; 316: 160–162. Article CAS PubMed Google Scholar * Small MB, Hay N, Schwab M, Bishop JM . Neoplastic transformation by the human gene N-_myc_. _Mol Cell Biol_
1987; 7: 1638–1645. Article CAS PubMed PubMed Central Google Scholar * Schweigerer L, Breit S, Wenzel A, Tsunamoto K, Ludwig R, Schwab M . Augmented _MYCN_ expression advances the
malignant phenotype of human neuroblastoma cells: evidence for induction of autocrine growth factor activity. _Cancer Res_ 1990; 50: 4411–4416. CAS PubMed Google Scholar * Lutz W, Stohr
M, Schurmann J, Wenzel A, Lohr A, Schwab M . Conditional expression of N-_myc_ in human neuroblastoma cells increases expression of _α_-prothymosin and ornithine decarboxylase and
accelerates progression into S-phase early after mitogenic stimulation of quiescent cells. _Oncogene_ 1996; 13: 803–812. CAS PubMed Google Scholar * Goodman LA, Liu BC, Thiele CJ, Schmidt
ML, Cohn SL, Yamashiro JM _et al_. Modulation of N-_myc_ expression alters the invasiveness of neuroblastoma. _Clin Exp Metastasis_ 1997; 15: 130–139. Article CAS PubMed Google Scholar
* Sugihara E, Kanai M, Matsui A, Onodera M, Schwab M, Miwa M . Enhanced expression of _MYCN_ leads to centrosome hyperamplification after DNA damage in neuroblastoma cells. _Oncogene_ 2004;
23: 1005–1009. Article CAS PubMed Google Scholar * Slack AD, Chen Z, Ludwig AD, Hicks J, Shohet JM . MYCN-directed centrosome amplification requires MDM2-mediated suppression of p53
activity in neuroblastoma cells. _Cancer Res_ 2007; 67: 2448–2455. Article CAS PubMed Google Scholar * Fotsis T, Breit S, Lutz W, Rossler J, Hatzi E, Schwab M _et al_. Down-regulation of
endothelial cell growth inhibitors by enhanced MYCN oncogene expression in human neuroblastoma cells. _Eur J Biochem_ 1999; 263: 757–764. Article CAS PubMed Google Scholar * Hatzi E,
Breit S, Zoephel A, Ashman K, Tontsch U, Ahorn H _et al_. MYCN oncogene and angiogenesis: down-regulation of endothelial growth inhibitors in human neuroblastoma cells. Purification,
structural, and functional characterization. _Adv Exp Med Biol_ 2000; 476: 239–248. Article CAS PubMed Google Scholar * Song L, Ara T, Wu HW, Woo CW, Reynolds CP, Seeger RC _et al_.
Oncogene MYCN regulates localization of NKT cells to the site of disease in neuroblastoma. _J Clin Invest_ 2007; 117: 2702–2712. Article CAS PubMed PubMed Central Google Scholar * Weiss
WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM . Targeted expression of _MYCN_ causes neuroblastoma in transgenic mice. _EMBO J_ 1997; 16: 2985–2995. Article CAS PubMed PubMed
Central Google Scholar * Lutz W, Fulda S, Jeremias I, Debatin KM, Schwab M . MycN and IFN_γ_ cooperate in apoptosis of human neuroblastoma cells. _Oncogene_ 1998; 17: 339–346. Article CAS
PubMed Google Scholar * Fulda S, Lutz W, Schwab M, Debatin KM . MycN sensitizes neuroblastoma cells for drug-induced apoptosis. _Oncogene_ 1999; 18: 1479–1486. Article CAS PubMed
Google Scholar * Fulda S, Lutz W, Schwab M, Debatin KM . MycN sensitizes neuroblastoma cells for drug-triggered apoptosis. _Med Pediatr Oncol_ 2000; 35: 582–584. Article CAS PubMed
Google Scholar * van Golen CM, Soules ME, Grauman AR, Feldman EL . N-Myc overexpression leads to decreased _β_1 integrin expression and increased apoptosis in human neuroblastoma cells.
_Oncogene_ 2003; 22: 2664–2673. Article CAS PubMed Google Scholar * Slack A, Chen Z, Tonelli R, Pule M, Hunt L, Pession A _et al_. The p53 regulatory gene _MDM2_ is a direct
transcriptional target of MYCN in neuroblastoma. _Proc Natl Acad Sci USA_ 2005; 102: 731–736. Article CAS PubMed PubMed Central Google Scholar * Alt JR, Greiner TC, Cleveland JL,
Eischen CM . Mdm2 haplo-insufficiency profoundly inhibits Myc-induced lymphomagenesis. _EMBO J_ 2003; 22: 1442–1450. Article CAS PubMed PubMed Central Google Scholar * Firulli AB,
Conway SJ . Phosphoregulation of Twist1 provides a mechanism of cell fate control. _Curr Med Chem_ 2008; 15: 2641–2647. Article CAS PubMed PubMed Central Google Scholar * Howard TD,
Paznekas WA, Green ED, Chiang LC, Ma N, Ortiz de Luna RI _et al_. Mutations in _TWIST_, a basic helix–loop–helix transcription factor, in Saethre-Chotzen syndrome. _Nat Genet_ 1997; 15:
36–41. Article PubMed Google Scholar * el Ghouzzi V, Le Merrer M, Perrin-Schmitt F, Lajeunie E, Benit P, Renier D _et al_. Mutations of the _TWIST_ gene in the Saethre-Chotzen syndrome.
_Nat Genet_ 1997; 15: 42–46. Article CAS PubMed Google Scholar * Valsesia-Wittmann S, Magdeleine M, Dupasquier S, Garin E, Jallas AC, Combaret V _et al_. Oncogenic cooperation between
H-Twist and N-Myc overrides failsafe programs in cancer cells. _Cancer Cell_ 2004; 6: 625–630. Article CAS PubMed Google Scholar * Maestro R, Dei Tos AP, Hamamori Y, Krasnokutsky S,
Sartorelli V, Kedes L _et al_. _twist_ is a potential oncogene that inhibits apoptosis. _Genes Dev_ 1999; 13: 2207–2217. Article CAS PubMed PubMed Central Google Scholar * Hamamori Y,
Sartorelli V, Ogryzko V, Puri PL, Wu HY, Wang JY _et al_. Regulation of histone acetyltransferases p300 and PCAF by the bHLH protein twist and adenoviral oncoprotein E1A. _Cell_ 1999; 96:
405–413. Article CAS PubMed Google Scholar * Stasinopoulos IA, Mironchik Y, Raman A, Wildes F, Winnard Jr P, Raman V . HOXA5-twist interaction alters p53 homeostasis in breast cancer
cells. _J Biol Chem_ 2005; 280: 2294–2299. Article CAS PubMed Google Scholar * Shiota M, Izumi H, Onitsuka T, Miyamoto N, Kashiwagi E, Kidani A _et al_. Twist and p53 reciprocally
regulate target genes via direct interaction. _Oncogene_ 2008; 27: 5543–5553. Article CAS PubMed Google Scholar * Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C _et al_.
Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. _Cancer Cell_ 2008; 14: 79–89. Article CAS PubMed Google Scholar *
Nowak K, Kerl K, Fehr D, Kramps C, Gessner C, Killmer K _et al_. _BMI1_ is a target gene of E2F-1 and is strongly expressed in primary neuroblastomas. _Nucleic Acids Res_ 2006; 34:
1745–1754. Article CAS PubMed PubMed Central Google Scholar * Cui H, Hu B, Li T, Ma J, Alam G, Gunning WT _et al_. Bmi-1 is essential for the tumorigenicity of neuroblastoma cells. _Am
J Pathol_ 2007; 170: 1370–1378. Article CAS PubMed PubMed Central Google Scholar * Sparmann A, van Lohuizen M . Polycomb silencers control cell fate, development and cancer. _Nat Rev
Cancer_ 2006; 6: 846–856. Article CAS PubMed Google Scholar * Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M . The oncogene and Polycomb-group gene _bmi-1_ regulates cell
proliferation and senescence through the _ink4a_ locus. _Nature_ 1999; 397: 164–168. Article CAS PubMed Google Scholar * Lessard J, Sauvageau G . _Bmi-1_ determines the proliferative
capacity of normal and leukaemic stem cells. _Nature_ 2003; 423: 255–260. Article CAS PubMed Google Scholar * Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL _et al_. Bmi-1 is
required for maintenance of adult self-renewing haematopoietic stem cells. _Nature_ 2003; 423: 302–305. Article CAS PubMed Google Scholar * Molofsky AV, Pardal R, Iwashita T, Park IK,
Clarke MF, Morrison SJ . _Bmi-1_ dependence distinguishes neural stem cell self-renewal from progenitor proliferation. _Nature_ 2003; 425: 962–967. Article CAS PubMed PubMed Central
Google Scholar * Molofsky AV, He S, Bydon M, Morrison SJ, Pardal R . Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the
p16Ink4a and p19Arf senescence pathways. _Genes Dev_ 2005; 19: 1432–1437. Article CAS PubMed PubMed Central Google Scholar * van Lohuizen M, Verbeek S, Scheijen B, Wientjens E, van der
Gulden H, Berns A . Identification of cooperating oncogenes in Eμ-_myc_ transgenic mice by provirus tagging. _Cell_ 1991; 65: 737–752. Article CAS PubMed Google Scholar * Haupt Y,
Alexander WS, Barri G, Klinken SP, Adams JM . Novel zinc finger gene implicated as _myc_ collaborator by retrovirally accelerated lymphomagenesis in μ-_myc_ transgenic mice. _Cell_ 1991; 65:
753–763. Article CAS PubMed Google Scholar * Haupt Y, Bath ML, Harris AW, Adams JM . _bmi-1_ transgene induces lymphomas and collaborates with _myc_ in tumorigenesis. _Oncogene_ 1993;
8: 3161–3164. CAS PubMed Google Scholar * Alkema MJ, Jacobs H, van Lohuizen M, Berns A . Pertubation of B and T cell development and predisposition to lymphomagenesis in E_μBmi_1
transgenic mice require the Bmi1 RING finger. _Oncogene_ 1997; 15: 899–910. Article CAS PubMed Google Scholar * Jacobs JJ, Scheijen B, Voncken JW, Kieboom K, Berns A, van Lohuizen M .
Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. _Genes Dev_ 1999; 13: 2678–2690. Article CAS PubMed PubMed Central Google Scholar *
Datta S, Hoenerhoff MJ, Bommi P, Sainger R, Guo WJ, Dimri M _et al_. Bmi-1 cooperates with H-Ras to transform human mammary epithelial cells via dysregulation of multiple growth-regulatory
pathways. _Cancer Res_ 2007; 67: 10286–10295. Article CAS PubMed PubMed Central Google Scholar * Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M _et al_. Rb
inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. _Nature_ 2004; 430: 797–802. Article CAS PubMed Google Scholar * Strieder V, Lutz W .
E2F proteins regulate _MYCN_ expression in neuroblastomas. _J Biol Chem_ 2003; 278: 2983–2989. Article CAS PubMed Google Scholar * Cui H, Ma J, Ding J, Li T, Alam G, Ding HF . Bmi-1
regulates the differentiation and clonogenic self-renewal of I-type neuroblastoma cells in a concentration-dependent manner. _J Biol Chem_ 2006; 281: 34696–34704. Article CAS PubMed
Google Scholar * White PS, Thompson PM, Gotoh T, Okawa ER, Igarashi J, Kok M _et al_. Definition and characterization of a region of 1p36.3 consistently deleted in neuroblastoma. _Oncogene_
2005; 24: 2684–2694. Article CAS PubMed Google Scholar * Okawa ER, Gotoh T, Manne J, Igarashi J, Fujita T, Silverman KA _et al_. Expression and sequence analysis of candidates for the
1p36.31 tumor suppressor gene deleted in neuroblastomas. _Oncogene_ 2008; 27: 803–810. Article CAS PubMed Google Scholar * Fujita T, Igarashi J, Okawa ER, Gotoh T, Manne J, Kolla V _et
al_. _CHD5_, a tumor suppressor gene deleted from 1p36.31 in neuroblastomas. _J Natl Cancer Inst_ 2008; 100: 940–949. Article CAS PubMed PubMed Central Google Scholar * Thompson PM,
Gotoh T, Kok M, White PS, Brodeur GM . _CHD5_, a new member of the chromodomain gene family, is preferentially expressed in the nervous system. _Oncogene_ 2003; 22: 1002–1011. Article CAS
PubMed Google Scholar * Bagchi A, Papazoglu C, Wu Y, Capurso D, Brodt M, Francis D _et al_. _CHD5_ is a tumor suppressor at human _1p36_. _Cell_ 2007; 128: 459–475. Article CAS PubMed
Google Scholar * Bown N, Cotterill S, Lastowska M, O’Neill S, Pearson AD, Plantaz D _et al_. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. _N Engl J Med_
1999; 340: 1954–1961. Article CAS PubMed Google Scholar * Vandesompele J, Baudis M, De Preter K, Van Roy N, Ambros P, Bown N _et al_. Unequivocal delineation of clinicogenetic subgroups
and development of a new model for improved outcome prediction in neuroblastoma. _J Clin Oncol_ 2005; 23: 2280–2299. Article CAS PubMed Google Scholar * Saito-Ohara F, Imoto I, Inoue J,
Hosoi H, Nakagawara A, Sugimoto T _et al_. PPM1D is a potential target for 17q gain in neuroblastoma. _Cancer Res_ 2003; 63: 1876–1883. CAS PubMed Google Scholar * Li J, Yang Y, Peng Y,
Austin RJ, van Eyndhoven WG, Nguyen KC _et al_. Oncogenic properties of _PPM1D_ located within a breast cancer amplification epicenter at 17q23. _Nat Genet_ 2002; 31: 133–134. Article CAS
PubMed Google Scholar * Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA _et al_. Amplification of _PPM1D_ in human tumors abrogates p53 tumor-suppressor activity.
_Nat Genet_ 2002; 31: 210–215. Article CAS PubMed Google Scholar * Rauta J, Alarmo EL, Kauraniemi P, Karhu R, Kuukasjarvi T, Kallioniemi A . The serine-threonine protein phosphatase
_PPM1D_ is frequently activated through amplification in aggressive primary breast tumours. _Breast Cancer Res Treat_ 2006; 95: 257–263. Article CAS PubMed Google Scholar * Hirasawa A,
Saito-Ohara F, Inoue J, Aoki D, Susumu N, Yokoyama T _et al_. Association of 17q21-q24 gain in ovarian clear cell adenocarcinomas with poor prognosis and identification of _PPM1D_ and
_APPBP2_ as likely amplification targets. _Clin Cancer Res_ 2003; 9: 1995–2004. CAS PubMed Google Scholar * Mendrzyk F, Radlwimmer B, Joos S, Kokocinski F, Benner A, Stange DE _et al_.
Genomic and protein expression profiling identifies CDK6 as novel independent prognostic marker in medulloblastoma. _J Clin Oncol_ 2005; 23: 8853–8862. Article CAS PubMed Google Scholar
* Castellino RC, De Bortoli M, Lu X, Moon SH, Nguyen TA, Shepard MA _et al_. Medulloblastomas overexpress the p53-inactivating oncogene _WIP1/PPM1D_. _J Neurooncol_ 2008; 86: 245–256.
Article CAS PubMed Google Scholar * Fiscella M, Zhang H, Fan S, Sakaguchi K, Shen S, Mercer WE _et al_. Wip1, a novel human protein phosphatase that is induced in response to ionizing
radiation in a p53-dependent manner. _Proc Natl Acad Sci USA_ 1997; 94: 6048–6053. Article CAS PubMed PubMed Central Google Scholar * Takekawa M, Adachi M, Nakahata A, Nakayama I, Itoh
F, Tsukuda H _et al_. p53-inducible Wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. _EMBO J_ 2000; 19: 6517–6526. Article CAS
PubMed PubMed Central Google Scholar * Lu X, Nannenga B, Donehower LA . PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. _Genes Dev_ 2005; 19: 1162–1174.
Article CAS PubMed PubMed Central Google Scholar * Fujimoto H, Onishi N, Kato N, Takekawa M, Xu XZ, Kosugi A _et al_. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1
phosphatase. _Cell Death Differ_ 2006; 13: 1170–1180. Article CAS PubMed Google Scholar * Yoda A, Xu XZ, Onishi N, Toyoshima K, Fujimoto H, Kato N _et al_. Intrinsic kinase activity and
SQ/TQ domain of Chk2 kinase as well as N-terminal domain of Wip1 phosphatase are required for regulation of Chk2 by Wip1. _J Biol Chem_ 2006; 281: 24847–24862. Article CAS PubMed Google
Scholar * Oliva-Trastoy M, Berthonaud V, Chevalier A, Ducrot C, Marsolier-Kergoat MC, Mann C _et al_. The Wip1 phosphatase (PPM1D) antagonizes activation of the Chk2 tumour suppressor
kinase. _Oncogene_ 2007; 26: 1449–1458. Article CAS PubMed Google Scholar * Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C _et al_. Wip1 phosphatase modulates ATM-dependent
signaling pathways. _Mol Cell_ 2006; 23: 757–764. Article CAS PubMed Google Scholar * Shreeram S, Hee WK, Demidov ON, Kek C, Yamaguchi H, Fornace Jr AJ _et al_. Regulation of
ATM/p53-dependent suppression of myc-induced lymphomas by Wip1 phosphatase. _J Exp Med_ 2006; 203: 2793–2799. Article CAS PubMed PubMed Central Google Scholar * Yamaguchi H, Durell SR,
Chatterjee DK, Anderson CW, Appella E . The Wip1 phosphatase PPM1D dephosphorylates SQ/TQ motifs in checkpoint substrates phosphorylated by PI3K-like kinases. _Biochemistry_ 2007; 46:
12594–12603. Article CAS PubMed Google Scholar * Lu X, Ma O, Nguyen TA, Jones SN, Oren M, Donehower LA . The Wip1 phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop.
_Cancer Cell_ 2007; 12: 342–354. Article CAS PubMed Google Scholar * Bulavin DV, Phillips C, Nannenga B, Timofeev O, Donehower LA, Anderson CW _et al_. Inactivation of the Wip1
phosphatase inhibits mammary tumorigenesis through p38 MAPK–mediated activation of the p16Ink4a-p19Arf pathway. _Nat Genet_ 2004; 36: 343–350. Article CAS PubMed Google Scholar *
Nannenga B, Lu X, Dumble M, Van Maanen M, Nguyen TA, Sutton R _et al_. Augmented cancer resistance and DNA damage response phenotypes in PPM1D null mice. _Mol Carcinog_ 2006; 45: 594–604.
Article CAS PubMed Google Scholar * Demidov ON, Kek C, Shreeram S, Timofeev O, Fornace AJ, Appella E _et al_. The role of the MKK6/p38 MAPK pathway in Wip1-dependent regulation of
_ErbB2_-driven mammary gland tumorigenesis. _Oncogene_ 2007; 26: 2502–2506. Article CAS PubMed Google Scholar * Moll UM, LaQuaglia M, Benard J, Riou G . Wild-type p53 protein undergoes
cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. _Proc Natl Acad Sci USA_ 1995; 92: 4407–4411. Article CAS PubMed PubMed Central Google
Scholar * Wolff A, Technau A, Ihling C, Technau-Ihling K, Erber R, Bosch FX _et al_. Evidence that wild-type p53 in neuroblastoma cells is in a conformation refractory to integration into
the transcriptional complex. _Oncogene_ 2001; 20: 1307–1317. Article CAS PubMed Google Scholar * Moll UM, Ostermeyer AG, Haladay R, Winkfield B, Frazier M, Zambetti G . Cytoplasmic
sequestration of wild-type p53 protein impairs the G1 checkpoint after DNA damage. _Mol Cell Biol_ 1996; 16: 1126–1137. Article CAS PubMed PubMed Central Google Scholar * Nikolaev AY,
Li M, Puskas N, Qin J, Gu W . Parc: a cytoplasmic anchor for p53. _Cell_ 2003; 112: 29–40. Article CAS PubMed Google Scholar * Smart P, Lane EB, Lane DP, Midgley C, Vojtesek B, Lain S .
Effects on normal fibroblasts and neuroblastoma cells of the activation of the p53 response by the nuclear export inhibitor leptomycin B. _Oncogene_ 1999; 18: 7378–7386. Article CAS PubMed
Google Scholar * Becker K, Marchenko ND, Maurice M, Moll UM . Hyperubiquitylation of wild-type p53 contributes to cytoplasmic sequestration in neuroblastoma. _Cell Death Differ_ 2007; 14:
1350–1360. Article CAS PubMed Google Scholar * Rodriguez-Lopez AM, Xenaki D, Eden TO, Hickman JA, Chresta CM . MDM2 mediated nuclear exclusion of p53 attenuates etoposide-induced
apoptosis in neuroblastoma cells. _Mol Pharmacol_ 2001; 59: 135–143. Article CAS PubMed Google Scholar * Wang X, Zalcenstein A, Oren M . Nitric oxide promotes p53 nuclear retention and
sensitizes neuroblastoma cells to apoptosis by ionizing radiation. _Cell Death Differ_ 2003; 10: 468–476. Article CAS PubMed Google Scholar * Stommel JM, Marchenko ND, Jimenez GS, Moll
UM, Hope TJ, Wahl GM . A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. _EMBO J_ 1999; 18:
1660–1672. Article CAS PubMed PubMed Central Google Scholar * Lu W, Pochampally R, Chen L, Traidej M, Wang Y, Chen J . Nuclear exclusion of p53 in a subset of tumors requires MDM2
function. _Oncogene_ 2000; 19: 232–240. Article CAS PubMed Google Scholar * Flaegstad T, Andresen PA, Johnsen JI, Asomani SK, Jorgensen GE, Vignarajan S _et al_. A possible contributory
role of BK virus infection in neuroblastoma development. _Cancer Res_ 1999; 59: 1160–1163. CAS PubMed Google Scholar * Jorgensen GE, Johnsen JI, Ponthan F, Kogner P, Flaegstad T, Traavik
T . Human polyomavirus BK (BKV) and neuroblastoma: mechanisms of oncogenic action and possible strategy for novel treatment. _Med Pediatr Oncol_ 2000; 35: 593–596. Article CAS PubMed
Google Scholar * Sengupta S, Vonesch JL, Waltzinger C, Zheng H, Wasylyk B . Negative cross-talk between p53 and the glucocorticoid receptor and its role in neuroblastoma cells. _EMBO J_
2000; 19: 6051–6064. Article CAS PubMed PubMed Central Google Scholar * Ohtsubo C, Shiokawa D, Kodama M, Gaiddon C, Nakagama H, Jochemsen AG _et al_. Cytoplasmic tethering is involved
in synergistic inhibition of p53 by Mdmx and Mdm2. _Cancer Sci_ 2009; 100: 1291–1299. Article CAS PubMed Google Scholar * Zaika A, Marchenko N, Moll UM . Cytoplasmically ‘sequestered’
wild type p53 protein is resistant to Mdm2-mediated degradation. _J Biol Chem_ 1999; 274: 27474–27480. Article CAS PubMed Google Scholar * Chen Z, Lin Y, Barbieri E, Burlingame S, Hicks
J, Ludwig A _et al_. Mdm2 deficiency suppresses MYCN-driven neuroblastoma tumorigenesis _in vivo_. _Neoplasia_ 2009; 11: 753–762. Article CAS PubMed PubMed Central Google Scholar *
Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ _et al_. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. _Genes Dev_ 1998;
12: 2424–2433. Article CAS PubMed PubMed Central Google Scholar * Aslanian A, Iaquinta PJ, Verona R, Lees JA . Repression of the _Arf_ tumor suppressor by E2F3 is required for normal
cell cycle kinetics. _Genes Dev_ 2004; 18: 1413–1422. Article CAS PubMed PubMed Central Google Scholar * Komori H, Enomoto M, Nakamura M, Iwanaga R, Ohtani K . Distinct E2F-mediated
transcriptional program regulates _p14__ARF_ gene expression. _EMBO J_ 2005; 24: 3724–3736. Article CAS PubMed PubMed Central Google Scholar * Barbieri E, Mehta P, Chen Z, Zhang L,
Slack A, Berg S _et al_. MDM2 inhibition sensitizes neuroblastoma to chemotherapy-induced apoptotic cell death. _Mol Cancer Ther_ 2006; 5: 2358–2365. Article CAS PubMed Google Scholar *
Van Maerken T, Ferdinande L, Taildeman J, Lambertz I, Yigit N, Vercruysse L _et al_. Antitumor activity of the selective MDM2 antagonist nutlin-3 against chemoresistant neuroblastoma with
wild-type p53. _J Natl Cancer Inst_ 2009 (in press). Download references ACKNOWLEDGEMENTS The p53 work in our laboratory is supported by the Research Foundation – Flanders (FWO; grants
G.0198.08 and G.0225.09), Cycle for Life – Belgium, the Centrum voor Studie en Behandeling van Gezwelziekten – Ghent, and the Ghent Childhood Cancer Fund. TVM is a research assistant from
the FWO (grant 011F4004). AR is supported by a grant from the Emmanuel van der Schueren Foundation. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Center for Medical Genetics, Ghent
University Hospital, Ghent, Belgium T Van Maerken, J Vandesompele, A Rihani, A De Paepe & F Speleman * Department of Clinical Chemistry, Microbiology and Immunology, Ghent University
Hospital, Ghent, Belgium T Van Maerken Authors * T Van Maerken View author publications You can also search for this author inPubMed Google Scholar * J Vandesompele View author publications
You can also search for this author inPubMed Google Scholar * A Rihani View author publications You can also search for this author inPubMed Google Scholar * A De Paepe View author
publications You can also search for this author inPubMed Google Scholar * F Speleman View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING
AUTHOR Correspondence to T Van Maerken. ADDITIONAL INFORMATION Edited by V De Laurenzi RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Van Maerken, T.,
Vandesompele, J., Rihani, A. _et al._ Escape from p53-mediated tumor surveillance in neuroblastoma: switching off the p14ARF-MDM2-p53 axis. _Cell Death Differ_ 16, 1563–1572 (2009).
https://doi.org/10.1038/cdd.2009.138 Download citation * Received: 06 July 2009 * Accepted: 27 July 2009 * Published: 25 September 2009 * Issue Date: December 2009 * DOI:
https://doi.org/10.1038/cdd.2009.138 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * neuroblastoma * p53 * antitumor barrier * MDM2 * p14ARF





