- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Nitric oxide (NO) is a short-lived free radical produced endogenously in biological tissues by nitric oxide synthases (NOSs)1, 2. Three NOS isoforms, namely NOS1 or neuronal NOS (nNOS), NOS2
or inducible NOS (iNOS), and NOS3 or endothelial NOS (eNOS) are present in most cell types, including cardiac myocytes and vascular endothelial cells. Vascular relaxation to mediators such
as acetylcholine or increased blood flow depends on NO produced by the eNOS. The discovery of NO as the endothelium-derived relaxing factor (EDRF) and its crucial function as a signaling
molecule in cardiovascular system was awarded the Nobel Prize in Physiology or Medicine in 19981. Under normal circumstances the main function of eNOS is to produce NO. It catalyzes the
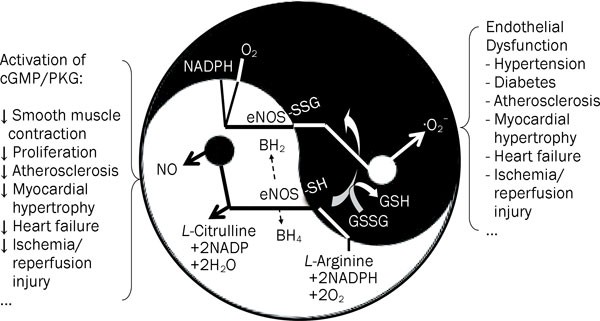
conversion of _L_-arginine (_L_-Arg) to _L_-citrulline and NO via electron transfer from the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) through a flavin containing
reductase domain to oxygen bound at the heme of an oxygenase domain containing tetrahydrobiopterin (BH4) and _L_-Arg binding sites (Figure 1). The eNOS-derived NO activates the guanylate
cyclase/cGMP/protein kinase G (PKG) pathway and modulates protein properties and function through nitrosylation of tyrosine and thiol-groups of cysteine in proteins, which are usually
effective protection mechanisms against oxidative stress. Under pathophysiological conditions such as hypertension, diabetes, septic shock and atherosclerosis, oxidative stress alters many
functions of the endothelium and leads to endothelial dysfunction when the endothelium fails to serve its normal physiologic and protective mechanisms. A common feature of endothelial
dysfunction is the reduced bioavailability of NO and increased production of superoxide (·O2−) and other reactive oxygen species (ROS) in the vasculature3. Multiple mechanisms may underlie
the impaired NO availability4. These include a reduction in the expression level of eNOS mRNA or protein, changes in subcellular compartmentalization of eNOS activity, and compromised
availability of the substrates and/or enzymatic cofactors for eNOS5, 6. Depletion of the substrate _L_-arginine, accumulation of methylarginines, and oxidation of the cofactor BH4 of eNOS
can uncouple the electron transfer reactions and revert eNOS to function as an NADPH oxidase, thus producing ·O2− instead of NO (Figure 1). The rapid reaction of NO with ·O2− can form the
most potent oxidant peroxynitrite anion (OONO−) and causes cellular injury associated with many pathophysiologic conditions, such as hypertension, atherosclerosis, diabetes, myocardial
hypertrophy, heart failure, and ischemia/reperfusion injury7. The precise molecular mechanisms underlying the “switch” of the eNOS function from NO synthesis to ·O2− production under
oxidative stress conditions, however, are still not fully understood. Recently, Chen _et al_ reported that _S_-glutathionylation of eNOS may be a unique mechanism for the redox regulation of
eNOS8. It has been demonstrated previously that cysteine residues are critical for the maintenance of normal eNOS function9. Protein _S_-glutathionylation has been known as a specific
post-translational modification of protein cysteine residues by adding the tripeptide glutathione through reversible thiol-disulfide exchange with oxidized glutathione (GSSG) or reaction of
oxidant-induced protein thiyl radicals with reduced glutathione (GSH). Under oxidative stress, therefore, protein _S_-glutathionylation can serve to prevent irreversible oxidation of protein
thiols10. _S_-glutathionylation has now emerged as a potential mechanism for dynamic, post-translational regulation of a variety of regulatory, structural, and metabolic proteins.
Increasing lines of evidence point to the important role of S-glutathionylation in the regulation of signaling and metabolic pathways in intact cellular systems. Indeed, Chen _et al_ found
that GSSG induced dose-dependent _S_-glutathionylation of human eNOS that was reversed by reducing agents β-mercaptoethanol or dithiothreitol8. _S_-glutathionylation of eNOS reversibly
decreases NOS activity with a concomitant increase in ·O2− generation primarily from the reductase domain. Two highly conserved cysteine residuals are identified as sites of
_S_-glutathionylation and found to be critical for redox-regulation of eNOS function. They further demonstrated that _S_-glutathionylation of eNOS in endothelial cells turned off NO
synthesis and turned on ·O2− generation (Figure 1). This conversion of eNOS function by _S_-glutathionylation is closely associated with impaired endothelium-dependent vasodilatation. In
hypertensive vessels, _S_-glutathionylation of eNOS is increased with impaired endothelium-dependent vasodilatation. Thio-specific reducing agents that reverse the _S_-glutathionylation of
eNOS are able to restore the endothelium-dependent vasodilatation of the hypertensive vessels. Thus, _S_-glutathionylation of eNOS may represent a novel and pivotal molecular switch
providing redox regulation of cellular signaling and endothelial function. Control of this molecular switch will perhaps provide new therapeutic targets and specific strategy to correct
endothelial dysfunction under pathophysiological conditions. Importantly, it may also shed new mechanistic light on the action of some antihypertensive herbal extracts that elicit potent
antioxidant properties and relax blood vessels in an endothelium-dependent and NO-mediated manner. It can be speculated that herbs with varying degrees of redox potential may exert
different, or even opposite, vascular effects by flipping the switch to preferentially cause the formation of NO or ·O2 11, thus lending scientific support for the Yin-Yang temperaments
emphasized in the theory of traditional Chinese medicine. REFERENCES * Furchgott RF . Endothelium-derived relaxing factor: discovery, early studies, and identification as nitric oxide.
_Biosci Rep_ 1999; 19: 235–51. Article CAS PubMed Google Scholar * Murad F . Nitric oxide signaling: would you believe that a simple free radical could be a second messenger, autacoid,
paracrine substance, neurotransmitter, and hormone? _Recent Prog Horm Res_ 1998; 53: 43–59. CAS PubMed Google Scholar * Cai H, Harrison DG . Endothelial dysfunction in cardiovascular
diseases: the role of oxidant stress. _Circ Res_ 2000; 87: 840–4. Article CAS PubMed Google Scholar * Wang Y, Marsden PA . Nitric oxide synthases: gene structure and regulation. _Adv
Pharmacol_ 1995; 34: 71–90. Article CAS PubMed Google Scholar * Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL . Nitric oxide synthase generates superoxide and nitric oxide in
arginine-depleted cells leading to peroxynitrite-mediated cellular injury. _Proc Natl Acad Sci USA_ 1996; 93: 6770–4. Article CAS PubMed PubMed Central Google Scholar * Vasquez-Vivar J
. Tetrahydrobiopterin, superoxide, and vascular dysfunction. _Free Radic Biol Med_ 2009; 47: 1108–19. Article CAS PubMed PubMed Central Google Scholar * Pacher P, Beckman JS, Liaudet L
. Nitric oxide and peroxynitrite in health and disease. _Physiol Rev_ 2007; 87: 315–424. Article CAS PubMed Google Scholar * Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann, C Talukder
MA, _et al_. _S_-glutathionylation uncouples eNOS and regulates its cellular and vascular function. _Nature_ 2010; 468: 1115–8. Article CAS PubMed PubMed Central Google Scholar * Chen
PF, Tsai AL, Wu KK . Cysteine 184 of endothelial nitric oxide synthase is involved in heme coordination and catalytic activity. _J Biol Chem_ 1994; 269: 25062–6. CAS PubMed Google Scholar
* Mieyal JJ, Gallogly MM, Qanungo S, Sabens EA, Shelton MD . Molecular mechanisms and clinical implications of reversible protein _S_-glutathionylation. _Antioxid Redox Signal_ 2008; 10:
1941–88. Article CAS PubMed PubMed Central Google Scholar * Achike FI, Kwan CY . Nitric oxide, human diseases and the herbal products that affect the nitric oxide signalling pathway.
_Clin Exp Pharmacol Physiol_ 2003; 30: 605–15. Article CAS PubMed Google Scholar Download references AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * and the Department of Pharmacology, The
Laboratory of Cardiovascular Phenomics, Center of Biomedical Research Excellence, University of Nevada School of Medicine, Reno, 89557, Nevada, USA Dayue Darrel Duan * Vascular Biology
Research Group, Research Institute of Basic Medical Sciences and Center for Faculty Development, China Medical University, Taichung, Taiwan, China Chiu-yin Kwan Authors * Dayue Darrel Duan
View author publications You can also search for this author inPubMed Google Scholar * Chiu-yin Kwan View author publications You can also search for this author inPubMed Google Scholar
CORRESPONDING AUTHOR Correspondence to Dayue Darrel Duan. RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Duan, D., Kwan, Cy. A molecular switch of “Yin
and Yang”: _S_-glutathionylation of eNOS turns off NO synthesis and turns on superoxide generation. _Acta Pharmacol Sin_ 32, 415–416 (2011). https://doi.org/10.1038/aps.2011.21 Download
citation * Published: 28 March 2011 * Issue Date: April 2011 * DOI: https://doi.org/10.1038/aps.2011.21 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this
content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative

:max_bytes(150000):strip_icc():focal(319x0:321x2)/people_social_image-60e0c8af9eb14624a5b55f2c29dbe25b.png)
.jpg?crop=true&anchor=54,7&q=80&color=ffffffff&u=2xkwh0&w=1988&h=1142)