- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT AIM: To ascertain the effects of erlotinib on CYP3A, to investigate the amplitude and kinetics of erlotinib-mediated inhibition of seven major CYP isoforms in human liver microsomes
(HLMs) for evaluating the magnitude of erlotinib in drug-drug interaction _in vivo_. METHODS: The activities of 7 major CYP isoforms (CYP1A2, CYP2A6, CYP3A, CYP2C9, CYP2D6, CYP2C8, and
CYP2E1) were assessed in HLMs using HPLC or UFLC analysis. A two-step incubation method was used to examine the time-dependent inhibition of erlotinib on CYP3A. RESULTS: The activity of
CYP2C8 was inhibited with an IC50 value of 6.17±2.0 μmol/L. Erlotinib stimulated the midazolam 1′-hydroxy reaction, but inhibited the formation of 6β-hydroxytestosterone and oxidized
nifedipine. Inhibition of CYP3A by erlotinib was substrate-dependent: the IC50 values for inhibiting testosterone 6β-hydroxylation and nifedipine metabolism were 31.3±8.0 and 20.5±5.3
μmol/L, respectively. Erlotinib also exhibited the time-dependent inhibition on CYP3A, regardless of the probe substrate used: the value of _K_I and _k_inact were 6.3 μmol/L and 0.035 min−1
for midazolam; 9.0 μmol/L and 0.045 min−1 for testosterone; and 10.1 μmol/L and 0.058 min−1 for nifedipine. CONCLUSION: The inhibition of CYP3A by erlotinib was substrate-dependent, while
its time-dependent inhibition on CYP3A was substrate-independent. The time-dependent inhibition of CYP3A may be a possible cause of drug-drug interaction, suggesting that attention should be
paid to the evaluation of erlotinib's safety, especially in the context of combination therapy. SIMILAR CONTENT BEING VIEWED BY OTHERS A PRELIMINARY EXPLORATION OF LIVER MICROSOMES AND
PBPK TO UNCOVER THE IMPACT OF CYP3A4/5 AND CYP2C19 ON TACROLIMUS AND VORICONAZOLE DRUG-DRUG INTERACTIONS Article Open access 21 February 2025 SPECIES DIFFERENCES IN THE CYP3A-CATALYZED
METABOLISM OF TPN729, A NOVEL PDE5 INHIBITOR Article 24 June 2020 MODULATION OF CYP2C9 ACTIVITY AND HYDROGEN PEROXIDE PRODUCTION BY CYTOCHROME _B_5 Article Open access 23 September 2020
INTRODUCTION Erlotinib (Tarceva; Genentech Inc, San Francisco, CA, USA) is an orally available, reversible human epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor1, 2. It
received approval from the US Food and Drug Administration in November 2004 for the second-line treatment of locally advanced or metastatic non-small cell lung cancer after the failure of at
least 1 previous chemotherapeutic regimen3, 4. Erlotinib was also approved by the United States for the treatment of locally advanced, unresectable or metastatic pancreatic cancer in
combination with gemcitabine5. In addition, clinical trials in a number of other solid tumors are also underway6, 7, 8. Erlotinib is considered better tolerated and less toxic than cytotoxic
drugs, with the most common adverse reactions in patients being rash and diarrhea9. However, erlotinib is frequently involved in clinical drug-drug interactions (DDIs). With 45 interactions
reported before 2007, the DDI frequency of erlotinib was just second to that of ifosfamide and paclitaxel among all antineoplastic drugs10. Co-administration of erlotinib has been reported
to enhance carboplatin exposure11 and increase the serum concentration of phenytoin12. A case of rhabdomyolysis was reported due to the interaction of erlotinib with simvastatin13.
International Normalized Ratio (INR) elevations and bleeding events associated with erotinib-warfarin co-administration have been reported4. Because the drugs involved usually had narrow
therapeutic indices, DDIs might impair the clinical safety of erlotinib. One of the major reasons for clinical DDIs has been recognized to be inhibition or induction of drug metabolism
enzymes. Erlotinib is extensively metabolized, predominantly by CYP3A4/5 and to a lesser extent by CYP1A2 and the extrahepatic isoform CYP1A114. As for the influence of erlotinib on the
catalytic activity of CYP3A, conflicting data have been published concerning its clinical consequences. Li _et al_ found that erlotinib stimulated CYP3A-mediated midazolam metabolism in
liver and intestinal microsomes15. Nevertheless, in a cell-based CYP3A activity assay, erlotinib was shown to decrease the formation of 1′-hydroxymidazolam, showing the potency to inhibit
CYP3A activity16. As for the phase II enzymes, erlotinib was shown to exhibit inhibition activity on human UDP-glucuronosyltransferase (UGT) 1A117. The effects of erlotinib on other phase I
CYP isoforms are still unknown. Thus, the current data were insufficient to explain the widespread DDI cases. Ascertaining the effect of erlotinib on major CYP isoforms will benefit the
clinical safety evaluation of erlotinib in combination with other drugs. The aim of this study was to ascertain the effect of erlotinib on CYP3A activity and to investigate the amplitude and
kinetics of erlotinib-mediated inhibition of seven major CYP isoforms in HLMs. An _in vivo_ magnitude of interaction will be extrapolated from the _in vitro_ inhibition kinetic data to help
explain the clinical DDIs associated with erlotinib. MATERIALS AND METHODS CHEMICALS AND REAGENTS Erlotinib (OSI-774, >99%) was purchased from Nanjing Ange Pharmaceutical Co, Ltd
(Nanjing, China). _D_-glucose-6-phosphate, glucose-6-phosphate dehydrogenase, NADP+, corticosterone, phenacetin, acetaminophen, 7-hydroxycoumarin, 4′-hydroxydiclofenac, sulfaphenazole,
8-methoxypsoralen, clomethiazole, montelukast, nifedipine, oxidized nifedipine, midazolam, 1′-OH-midazolam, troleandomycin (TAO), 6-hydroxychlorzoxazone, 7-hydroxycoumarin, paclitaxel,
6β-hydroxytestosterone and furafylline were purchased from Sigma-Aldrich (St Louis, MO, USA). Testosterone was from Acros Organics (Morris Plains, NJ, USA). Coumarin, diclofenac,
dextromethorphan and ketoconazole were from ICN Biomedicals (Aurora, OH, USA). Human liver microsomes (HLMs) were prepared according to the method described by Guengerich (1989) and other
previous reports18, 19. Protein concentrations were determined using bovine serum albumin as a standard20. Millipore water (Millipore, Bedford, MA, USA) and HPLC grade methanol and
acetonitrile (Tedia, Fairfield, OH, USA) were used throughout; other reagents were of the highest grade commercially available. PROBE SUBSTRATE ASSAYS FOR MAJOR CYP ISOFORMS Probe reactions
for CYP3A, CYP1A2, CYP2C8, CYP2A6, CYP2C9, CYP2D6, and CYP2E1 were testosterone 6β-hydroxylation, phenacetin _O_-demethylation, paclitaxel 6α-hydroxylation, coumarin 7-hydroxylation,
diclofenac 4′-hydroxylation, dextromethorphan _O_-demethylation and chlorzoxazone 6-hydroxylation activities separately. Midazolam 1′-hydroxylation and nifedipine oxidation reactions were
also performed to examine the effect of erlotinib on CYP3A activity. The basic incubation system contained 100 mmol/L potassium phosphate buffer (pH 7.4), a NADPH-generating system (1 mmol/L
NADP+, 10 mmol/L glucose-6-phosphate, 1 unit/mL of glucose-6-phosphate dehydrogenase and 4 mmol/L MgCl2) and the appropriate concentrations of HLMs, the appropriate probe substrate and
erlotinib (or a positive control inhibitor) in a final volume of 200 μL. After a 3-min preincubation at 37 °C, the reaction was initiated by adding the NADPH-generating system and terminated
by adding 100 μL acetonitrile (10% trichloroacetic acid for CYP2A6) with internal standard. The mixture was centrifuged at 20 000×_g_ for 10 min, and an aliquot of supernatant was then
transferred to a 0.3-mL auto-injector vial for HPLC or UFLC analysis. The incubation conditions, including substrate and protein concentrations and incubation times, have been reported21,
22. The HPLC system (SHIMADZU, Kyoto, Japan) consisted of a SCL-10A system controller, two LC-10AT pumps, a SIL-10A autoinjector, and a SPD-10AVP UV detector or a RF-10AXL fluorescence
detector. HPLC separation was achieved using a C18 column (150 mm×4.6 mm ID, 5 μm, Shimadzu) at a flow rate of 1 mL/min. A Shimadzu Prominence UFLCTM system with a Shim-pack XR-ODS (75.0
mm×2.0 mm ID, 2.2 μm, Shimadzu) analytical column was used. The eluent flow rate was 0.3 mL/min and the column temperature was maintained at 40 °C. Analysis conditions for the P450 isoforms
are shown in Table 1. All analytical methods were shown to be precise and accurate. The intra- and inter-day precisions were less than 15%, with accuracy in the range of 86.7%–112.5%19, 21,
22, 23. ENZYME INHIBITION EXPERIMENTS Marker assays for each CYP isoform were performed in the presence of 100 μmol/L erlotinib to evaluate its inhibitory effect toward the seven major human
CYP isoforms. The concentrations of positive control inhibitors used are as follows21, 24, 25: 1 μmol/L ketoconazole for CYP3A, 10 μmol/L furafylline for CYP1A2, 10 μmol/L sulfaphenazole
for CYP2C9, 5 μmol/L montelukast for CYP2C8, 2.5 μmol/L 8-methoxypsoralen for CYP2A6, 10 μmol/L quinidine for CYP2D6 and 50 μmol/L clomethiazole for CYP2E1. For CYP isoforms that were
strongly inhibited, the concentrations at which the enzymes were 50% inhibited (IC50 values) were determined using various concentrations of erlotinib for CYP3A and for CYP2C8. Inhibition
constant (_K_i) values were determined by incubating various probe substrates (5–50 μmol/L paclitaxel, 30–100 μmol/L testosterone or 5–50 μmol/L nifedipine) in the presence or absence of
erlotinib. _K_i values were calculated by nonlinear regression using the equations for competitive inhibition (eq 1), noncompetitive inhibition (eq 2), or mixed inhibition (eq 3)17,
ACTIVATION OF MIDAZOLAM METABOLISM BY ERLOTINIB Midazolam was incubated in pooled HLMs (0.1 mg/mL) for 10 min in the presence or absence of erlotinib. To investigate the concentration
dependence of midazolam metabolism activation phenomena, various concentrations of erlotinib (1–20 μmol/L) were incubated with midazolam at different concentrations (2–20 μmol/L).
1′-hydroxymidazolam was measured using a validated method based on UFLC as described in Table 1. To further explore the potential mechanism of activation phenomena, kinetic data were fit to
a two-site model26, where _S_ is the substrate, _B_ is the effector, _V_max and _K_m are the kinetic constants for substrate metabolism, _K_B is the binding constant for the effector, α is
the change in _K_m resulting from the effector binding, and β is the change in _V_max from the effector binding. For activation, α<1 and/or β>1. SINGLE POINT INACTIVATION EXPERIMENTS
Single point inactivation experiments were used as previously reported to determine NADPH-dependent and preincubation-dependent inhibition by erlotinib27. Briefly, erlotinib was incubated
with pooled HLMs (1 mg/mL) in the absence and presence of the NADPH-generating system for 30 min at 37 °C. For CYP3A and CYP2C8, the concentration of erlotinib utilized was ten times the
concentration that gave 25% inhibition under conditions of reversible inhibition. For other CYP isoforms, 50 μmol/L of erlotinib was used. Moreover, midazolam was also used as a probe
substrate to perform single point inactivation experiments for CYP3A, and 50 μmol/L of erlotinib was used. After incubation, an aliquot (20 μL) was transferred to another incubation tube
(final volume 200 μL) containing an NADPH-generating system and probe substrates whose concentrations were proximal to _K_m values. Further incubations were performed to measure residual
activity. INACTIVATION CONSTANT (_K_I AND _K_INACT) ASSAYS To determine the _K_I and _k_inact values for the inactivation of CYP3A, five concentrations of erlotinib (0, 5, 10, 20 and 50
μmol/L) were incubated for 0 to 30 min with pooled HLMs (1 mg/mL) at 37 °C. After preincubation, an aliquot (20 μL) was transferred to another incubation tube (final volume 200 μL)
containing an NADPH-generating system and different probe substrates for CYP3A to measure residual activity. Substrate concentrations of four times the _K_m were selected to minimize the
reversible inhibition caused by erlotinib. The concentrations used for different probe substrates were as follows: 400 μmol/L testosterone, 20 μmol/L midazolam and 60 μmol/L nifedipine. To
determine the _k_obs (observed inactivation rate) values, the decrease in natural logarithm of the activity over time was plotted for each erlotinib concentration, and the _k_obs values were
described as the negative slopes of the lines. Inactivation kinetic parameters were calculated using nonlinear regression of the data according to equation (5): where [I] is the initial
inhibitor concentration, _k_inact is the maximal inactivation rate constant and _K_I is the inhibitor concentration required for half the maximal rate of inactivation. The unbound _K_I
(_K_I, u) was calculated according to equation (6), where _f_u, m is the free fraction of erlotinib in the microsomes. _f_u, m is predicted according to equation (7) as previously
reported28. The terms are defined as follows: _C_mic is the microsomal protein concentration used in the preincubation, and logP is the log of the octanol-water (pH 7.4) partition (P)
coefficient of the erlotinib. A concentration of 1 mg/mL was used for _C_mic in this experiment, and LogP is approximately 2.7, according to the literature4. Thus, _f_u, m is calculated to
be 44.2%. QUANTITATIVE PREDICTION OF THE DDI POTENTIAL OF ERLOTINIB (AUCI/AUC) The equations (8) and (9) were utilized to predict the interaction potential of erlotinib caused by reversible
inhibition and TDI of CYP3A. Equation (10) was used to predict the interaction potential by reversible inhibition of CYP2C8. All equations were adapted as reported27, 29. The terms are
defined as follows: AUCi/AUC is the predicted ratio of _in vivo_ exposure of the interacting drug with co-administration of erlotinib _versus_ that in the control situation,
_f_m(CYP3A)/_f_m(CYP2C8) is the portion of total clearance of the interacting drug to which CYP3A/CYP2C8 contributes, _k_deg(CYP3A) is the first-order rate constant of _in vivo_ degradation
of CYP3A, _k_inact is the maximum inactivation rate constant, _K_I, u is the unbound _K_I, _K_i is the reversible inhibition constant, and [I]_in vivo_ is the _in vivo_ concentration of
erlotinib at the enzyme active site. The general assumption is that only unbound drug is available for interaction with the enzyme active site. However, at present, there is no consensus on
the _in vivo_ precipitant concentration that should be used. According to a recent publication, the reversible inhibition portion performed the best when the unbound portal vein
concentration was used for [I]_in vivo_, while for irreversible inactivation and induction the unbound systemic concentration was the best. Thus, in this research, the unbound portal vein
concentration (0.16, 0.18, and 0.31 μmol/L) was used for the reversible inhibition portion (for 50, 100, and 150 mg/d doses, respectively), while the unbound systemic concentration (0.07,
0.19, and 0.13 μmol/L) was adopted to avoid over-prediction of irreversible inactivation30. The values for [I]_in vivo_ were derived from references17, 31. A _k_deg(CYP3A) of 0.000321 min-1
was adopted, in accordance with Obach _et al_27. The values of _f_m(CYP3A) and _f_m(CYP2C8) were arbitrarily set to be 0.1–1 to predict the DDI risk for all possible coadministered drugs17.
RESULTS INHIBITION OF MAJOR CYP ISOFORMS BY ERLOTINIB Erlotinib with the concentration of 100 μmol/L inhibited the activities of CYP1A2, CYP2C9, CYP2A6, CYP2D6, CYP2C8, and CYP2E1 by 30.0%,
49.0%, 9%, 37%, 76%, and -7%, respectively. For CYP3A, 100 μmol/L erlotinib inhibited 69.3% and 71.6%, respectively, of the enzyme's testosterone 6β-hydroxylation and nifedipine
oxidation activities. However, erlotinib stimulated the midazolam 1′-hydroxy activity by 171%. All positive control inhibitors strongly inhibited the corresponding probe reactions, with less
than 20% of control activity remaining upon inhibition. Further kinetic analysis was conducted for CYP3A (testosterone 6β-hydroxylation, nifedipine oxidation) and CYP2C8 (paclitaxel
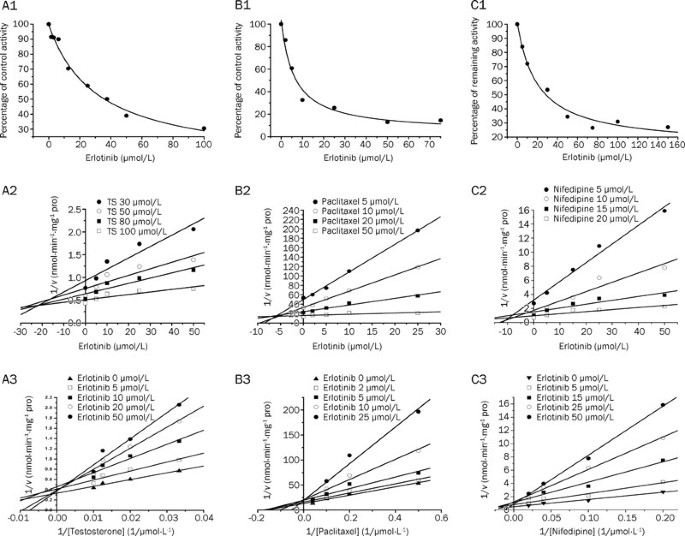
6α-hydroxylation), whose activities were inhibited by more than 50%. As shown in Figure 1, erlotinib inhibited testosterone 6β-hydroxylation in a concentration-dependent manner with an IC50
of 31.3±8.0 μmol/L. Lineweaver-Burk and Dixon plots showed that the inhibition of CYP3A by erlotinib was well fitted to a competitive model of inhibition. The _K_i value was calculated to be
14.1±4.3 μmol/L using a nonlinear regression equation (eq 1). Erlotinib also inhibited the metabolism of nifedipine in a competitive manner with an IC50 of 20.5±5.3 μmol/L. A _K_i value of
4.3±0.9 μmol/L was obtained by nonlinear fitting. The results demonstrated that erlotinib inhibited paclitaxel 6α-hydroxylation in a concentration-dependent manner, with an IC50 of 6.17±2.0
μmol/L. Lineweaver-Burk and Dixon plots suggested that erlotinib also competitively inhibited CYP2C8. The _K_i value was calculated to be 5.8±1.9 μmol/L using a nonlinear regression equation
(eq 1). ACTIVATION OF MIDAZOLAM METABOLISM As shown in Figure 2, using different concentrations of midazolam (2–20 μmol/L), erlotinib stimulated the formation of 1′-OH-midazolam. At a
constant concentration of midazolam, the formation of 1′-OH-midazolam increased with increasing amounts of erlotinib (1–20 μmol/L). The two-site model fitting results are listed in Table 2.
The data fit this model (Table 2) well with an α=0.50 and a β=1.80, indicating a decrease in _K_m and an increase in _V_max, respectively32. These results showed the existence of activation.
TIME- AND NADPH-DEPENDENT INHIBITIONS When erlotinib was pre-incubated with HLMs for 30 min in the presence of NADPH, the percentage of inhibition on CYP3A by erlotinib increased
significantly compared with that without NADPH (using testosterone and nifedipine as probe substrates). When midazolam was used as a probe substrate, interesting results were obtained
(Figure 3). When NADPH was not present during the preincubation process, erlotinib stimulated the metabolism of midazolam, but when NADPH was added to the preincubation, erlotinib showed an
inhibitory effect on the activity of CYP3A. Inactivation kinetic parameters were obtained using different probe substrates. As calculated from the observed inactivation plots (Figure 4),
inactivation parameters (_K_I and _k_inact, respectively) for CYP3A were calculated to be 6.3 μmol/L and 0.035 min-1, 9.0 μmol/L and 0.045 min-1, 10.1 μmol/L and 0.058 min-1 for the probe
substrates midazolam, testosterone and nifedipine, respectively. The inhibition of other isoforms by erlotinib was not time and NADPH dependent (data not shown). _IN VITRO_-_IN VIVO_
EXTRAPOLATION OF DDI MAGNITUDES For reversible inhibition of CYP2C8, adopting a _K_i of 5.8 μmol/L and unbound portal vein concentrations of 0.16, 0.18, and 0.31 μmol/L for 50, 100, and 150
mg/d doses, respectively, the AUCi/AUCs were predicted to be 1.0027–1.0276, 1.0030–1.0310 and 1.0051–1.0534 for an _f_m value between 0.1 and 1. For reversible inhibition of CYP3A (with the
probe substrate testosterone), using a _K_i of 14.1 μmol/L and the same unbound portal vein concentrations as for CYP2C8, the AUCi/AUCs were predicted to be 1.0011–1.0113, 1.0013–1.0128, and
1.0022–1.0220 for an _f_m value between 0.1 and 1, for 50, 100, and 150 mg/d doses, respectively. For the probe substrate nifedipine, using a _K_i of 4.3 μmol/L and the same unbound portal
vein concentration, the corresponding results of AUCi/AUCs were 1.0036–1.0372, 1.0040–1.0419, and 1.0068–1.0721 for an _f_m value between 0.1 and 1, for 50, 100, and 150 mg/d doses,
respectively. For irreversible inactivation of CYP3A (using different probe substrates), unbound systemic concentrations of 0.07, 0.13, and 0.19 μmol/L were adopted to avoid over-prediction
for three oral doses17. With a _K_I, u and a _k_inact of erlotinib for probe substrates midazolam, testosterone and nifedipine being 2.8 μmol/L and 0.035 min-1, 4.0 μmol/L and 0.045 min-1
and 4.5 μmol/L and 0.058 min-1, respectively, the AUC was calculated to increase to 107.9%–372.6%, 109.1%–606.2%, and 109.7%–839.9% for midazolam; 107.7%−345.3%, 108.9%–555.6%, and
109.5%–765.9% for testosterone; 108.0%–381.1%, 109.2%–622.0%, and 109.7%–862.9% for nifedipine. DISCUSSION Human CYP3A is one of the most important CYP isoforms involved in drug clearance
and metabolized more than 50% of the drugs on the market33. Inhibition or stimulation of the catalytic activity of this enzyme could play a key role in clinical DDIs. In previous studies,
conflicting data were obtained about the effects of erlotinib on metabolism of the substrate midazolam mediated by CYP3A34. Thus, rigorously ascertaining erlotinib's effect on CYP3A
will provide important information that may aid in the prevention of clinical DDIs. In this study, our experimental results showed that DDI patterns via modulation of CYP3A by erlotinib are
substrate dependent. Erlotinib stimulated the formation of 1′-hydroxymidazolam in HLMs. However, it inhibited the reactions of testosterone 6β-hydroxylation and nifedipine oxidation.
Erlotinib's time-dependent inhibition of CYP3A was not substrate dependent. The patterns of interaction between drug compounds and CYP3A were previously shown to be substrate
dependent35. For example, the flavonoid α-naphthoflavone, although well known to activate CYP3A436, may also inhibit the enzyme37, depending on the CYP3A4 substrate. In a recent report,
substrate-dependent phenomena were found for ginsenosides' effects on CYP3A38. Until now, the mechanism of substrate-dependent modulation of CYP3A activity remained unclear. The
relatively large active site cavity and the conformational flexibility of CYP3A were considered the major causes of these phenomena39, 40. CYP3A can bind multiple ligands simultaneously,
resulting in changes in the affinity of the substrate-binding site for different substrates26. This multiple ligand-binding property may contribute to the complex substrate-dependent
effects, but whether conformational changes occurred simultaneously was unknown. In the case of erlotinib, further investigation was needed to explore the molecular and structural basis of
these substrate-dependent effects. The substrate-dependent effects of erlotinib on CYP3A point to the need for greater attention to the safety of combined medications. In the case of
heteroactivation, the clearance of the interacting drug would increase. Alternatively, if erlotinib inhibited the metabolism of the interacting drug, the AUC of the latter drug would
increase. In either case, a different DDI might occur and possibly cause harm to the patient. When substrate-dependent effects may be present, it is prudent to employ a testing strategy
using several probe substrates to evaluate the DDI potential41, 42. Moreover, CYP3A4 and CYP3A5 are the most abundant members of the CYP3A subfamily. A recent study has shown significant
differences between the heteroactivation potential of CYP3A4 and CYP3A5 for CYP3A-mediated carbamazepine 10,11-epoxidation43. Inhibitors of CYP3A usually have different potencies for
inhibition of CYP3A4 and CYP3A5 in terms of both reversible and irreversible inhibition44, 45. Thus, the different expression levels of CYP3A4 and CYP3A5 may contribute to interindividual
variability in erlotinib interactions. The time-dependent inhibition of CYP3A was found to be substrate independent. After preincubation, erlotinib showed enhanced inhibition activity for
the midazolam 1′-hydroxylation, testosterone 6β-hydroxylation and nifedipine oxidation reactions (Figure 3). The TDI parameters (_K_I and _k_inact) were 6.3 μmol/L and 0.035 min-1, 9.0
μmol/L and 0.045 min-1 and 10.1 μmol/L and 0.058 min-1, respectively, for the midazolam 1′-hydroxylation, testosterone 6β-hydroxylation and nifedipine oxidation reactions. When midazolam was
used as the probe substrate, the following similar inactivation kinetic parameters were reported by Li _et al_46: _k_inact=0.09 min-1 and _K_I=22 μmol/L. The discrepancy in parameters may
be due to the differences between labs. It should be noted that tyrosine kinase inhibitors such as dasatinib have been reported to inhibit CYPs via generation of reactive intermediates47,
48. Recently, the bioactivation of erlotinib was also reported; in that study, reactive epoxide and quinone-imine electrophiles were detected, providing a possible mechanism for the time
dependent inhibition of CYP3A46. Based on the results shown above, the conflicting data about the effect of erlotinib on CYP3A can be explained as follows. First, without preincubation the
action of erlotinib on CYP3A was substrate dependent (Figure 3). Erlotinib stimulated the formation of 1′-hydroxymidazolam15. Second, the time-dependent inhibition of CYP3A was actually
substrate independent (Figure 3). In the cell-based CYP3A activity assessment method by Harmsen _et al_, the cells were cultured in medium containing erlotinib for two consecutive days
before measurement of the formation of 1′-hydroxymidazolam16. During the 2-d culture period, time dependent inhibition of CYP3A could occur, thus decreasing the formation of 1′-hydroxy
midazolam. Using the kinetic information we obtained regarding the reversible and time-dependent inhibition of CYP enzymes, the _in vivo_ DDI magnitude of erlotinib was extrapolated. For the
reversible inhibition of CYP3A and 2C8, even using a dose of 150 mg/d and an _f_m of 1, the increase in the AUC was predicted to be no more than 10%. On the contrary, the AUC was predicted
to increase significantly even with the lower oral dose and the smaller _f_m when adopting the TDI prediction equation. The DDI potential of erlotinib on phase II
UDP-Glucuronosyltransferases has been evaluated previously17. The maximum increase in AUC was estimated to be less than 50% for drugs predominantly cleared by UGT1A1, even at a dose of 150
mg/d. Therefore, time-dependent inhibition of CYP3A might be one of the most important factors leading to clinical DDIs. CONCLUSION In conclusion, our results demonstrate that the action of
erlotinib on CYP3A was substrate dependent. It stimulated the metabolism of midazolam and inhibited the formation of 6β-hydroxy testosterone and oxidized nifedipine. In contrast, the
time-dependent inhibition of erlotinib on CYP3A was substrate-independent. Moreover, the time-dependent inhibition of CYP3A was a possible reason for clinical DDIs related to erlotinib.
Cancer patients often receive multiple concurrent medications and should be carefully monitored for possible DDIs. A better understanding of the modulatory effects of erlotinib on the major
CYP isoforms could inform clinical safety evaluations of drug combinations. AUTHOR CONTRIBUTION Ling YANG and Pei-pei DONG designed research; Pei-pei DONG, Yu-xi MAO, Liang-liang ZHU,
Yan-qing QU, and Wei LI performed research; Ling YANG, Chang-xiao LIU, and Li-ming WANG contributed new analytical tools and reagents; Pei-pei DONG, Zhong-ze FANG, Yan-yan ZHANG, and
Guang-bo GE analyzed data; and Pei-pei DONG wrote the paper. REFERENCES * Ciardiello F, Tortora G . A novel approach in the treatment of cancer: Targeting the epidermal growth factor
receptor. _Clin Cancer Res_ 2001; 7: 2958–70. CAS PubMed Google Scholar * Smith J . Erlotinib: Small-molecule targeted therapy in the treatment of non-small-cell lung cancer. _Clin Ther_
2005; 27: 1513–34. Article CAS PubMed Google Scholar * Shepherd FA, Pereira JR, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, _et al_. Erlotinib in previously treated non-small-cell lung
cancer. _N Engl J Med_ 2005; 353: 123–32. Article CAS PubMed Google Scholar * Cohen MH, Johnson JR, Chen YF, Sridhara R, Pazdur R . FDA drug approval summary: Erlotinib (Tarceva (R))
tablets. _Oncologist_ 2005; 10: 461–6. Article CAS PubMed Google Scholar * Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, _et al_. Erlotinib plus gemcitabine compared
with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada clinical trials group. _J Clin Oncol_ 2007; 25: 1960–6.
Article CAS PubMed Google Scholar * Vasey PA, Gore M, Wilson R, Rustin G, Gabra H, Guastalla JP, _et al_. A phase Ib trial of docetaxel, carboplatin and erlotinib in ovarian, fallopian
tube and primary peritoneal cancers. _Br J Cancer_ 2008; 98: 1774–80. Article CAS PubMed PubMed Central Google Scholar * Lin CC, Calvo E, Papadopoulos KP, Patnaik A, Sarantopoulos J,
Mita AC, _et al_. Phase I study of cetuximab, erlotinib, and bevacizumab in patients with advanced solid tumors. _Cancer Chemother Pharmacol_ 2009; 63: 1065–71. Article CAS PubMed Google
Scholar * Thomas F, Rochaix P, White-Koning M, Hennebelle I, Sarini J, Benlyazid A, _et al_. Population pharmacokinetics of erlotinib and its pharmacokinetic/pharmacodynamic relationships
in head and neck squamous cell carcinoma. _Eur J Cancer_ 2009; 45: 2316–23. Article CAS PubMed Google Scholar * Gridelli C, Rossi A, Malone P, Colantuoni G, Del Gaizo F, Ferrara C, _et
al_. Erlotinib in non-small-cell lung cancer. _Expert Opin Pharmacother_ 2007; 8: 2579–92. Article CAS PubMed Google Scholar * Schwiertz V, Bertin C, Henry A, Charpiat B . Number and
nature of drug interactions concerning antineoplastic drugs. _Bull Cancer_ 2007; 94: 477–82. CAS PubMed Google Scholar * Patnaik A, Wood D, Tolcher AW, Hamilton M, Kreisberg JI, Hammond
LA, _et al_. Phase 1, pharmacokinetic, and biological study of erlotinib in combination with paclitaxel and carboplatin in patients with advanced solid tumors. _Clin Cancer Res_ 2006; 12:
7406–13. Article CAS PubMed Google Scholar * Grenader T, Gipps M, Shavit L, Gabizon A . Significant drug interaction: Phenytoin toxicity due to erlotinib. _Lung Cancer_ 2007; 57: 404–6.
Article PubMed Google Scholar * Veeraputhiran M, Sundermeyer M . Rhabdomyolysis resulting from pharmacologic interaction between erlotinib and simvastatin. _Clin Lung Cancer_ 2008; 9:
232–34. Article CAS PubMed Google Scholar * Ling J, Johnson KA, Miao Z, Rakhit A, Pantze MP, Hamilton M, _et al_. Metabolism and excretion of erlotinib, a small molecule inhibitor of
epidermal growth factor receptor tyrosine kinase, in healthy male volunteers. _Drug Metab Dispos_ 2006; 34: 420–6. CAS PubMed Google Scholar * Li J, Zhao M, He P, Hidalgo M, Baker SD .
Differential metabolism of gefitinib and erlotinib by human cytochrome P450 enzymes. _Clin Cancer Res_ 2007; 13: 3731–7. Article CAS PubMed Google Scholar * Harmsen S, Meijerman I,
Beijnen JH, Schellens JHM . Nuclear receptor mediated induction of cytochrome P450 3A4 by anticancer drugs: a key role for the pregnane X receptor. _Cancer Chemother Pharmacol_ 2009; 64:
35–43. Article CAS PubMed Google Scholar * Liu Y, Ramirez J, House L, Ratain MJ . Comparison of the drug-drug interactions potential of erlotinib and gefitinib via inhibition of
UDP-glucuronosyltransferases. _Drug Metab Dispos_ 2010; 38: 32–9. Article PubMed PubMed Central Google Scholar * Zhang YY, Liu Y, Zhang JW, Ge GB, Liu HX, Wang LM, _et al_. C-7
configuration as one of determinants in taxanes metabolism by human cytochrome P450 enzymes. _Xenobiotica_ 2009; 39: 903–14. Article CAS PubMed Google Scholar * Liang SC, Ge GB, Liu HX,
Zhang YY, Wang LM, Zhang JW, _et al_. Identification and characterization of human udp-glucuronosyltransferases responsible for the _in vitro_ glucuronidation of daphnetin. _Drug Metab
Dispos_ 2010; 38: 973–80. Article CAS PubMed Google Scholar * Lowry OH, Rosebrough NJ, Farr AL, Randall RJ . Protein measurement with the folin phenol reagent. _J Biol Chem_ 1951; 193:
265–75. CAS PubMed Google Scholar * Liu Y, Ma H, Zhang JW, Deng MC, Yang L . Influence of ginsenoside Rh-1 and F-1 on human cytochrome P450 enzymes. _Planta Med_ 2006; 72: 126–31. Article
CAS PubMed Google Scholar * Zhang JW, Liu Y, Li W, Hao DC, Yang L . Inhibitory effect of medroxyprogesterone acetate on human liver cytochrome P450 enzymes. _Eur J Clin Pharmacol_ 2006;
62: 497–502. Article CAS PubMed Google Scholar * Dong PP, Ge GB, Zhang YY, Ai CZ, Li GH, Zhu LL, _et al_. Quantitative structure-retention relationship studies for taxanes including
epimers and isomeric metabolites in ultra fast liquid chromatography. _J Chromatogr A_ 2009; 1216: 7055–62. Article CAS PubMed Google Scholar * Liu YT, Hao K, Liu XQ, Wang GJ .
Metabolism and metabolic inhibition of gambogic acid in rat liver microsomes. _Acta Pharmacol Sin_ 2006; 27: 1253–58. Article CAS PubMed Google Scholar * Fang ZZ, Zhang YY, Ge GB, Huo H,
Liang SC, Yang L . Time-dependent inhibition (TDI) of CYP3A4 and CYP2C9 by noscapine potentially explains clinical noscapine-warfarin interaction. _Br J Clin Pharmacol_ 2010; 69: 193–9.
Article CAS PubMed PubMed Central Google Scholar * Korzekwa KR, Krishnamachary N, Shou M, Ogai A, Parise RA, Rettie AE, _et al_. Evaluation of atypical cytochrome P450 kinetics with
two-substrate models: Evidence that multiple substrates can simultaneously bind to cytochrome P450 active sites. _Biochemistry_ 1998; 37: 4137–47. Article CAS PubMed Google Scholar *
Obach RS, Walsky RL, Venkatakrishnan K . Mechanism-based inactivation of human cytochrome P450 enzymes and the prediction of drug-drug interactions. _Drug Metab Dispos_ 2007; 35: 246–55.
Article CAS PubMed Google Scholar * Austin RP, Barton P, Cockroft SL, Wenlock MC, Riley RJ . The influence of nonspecific microsomal binding on apparent intrinsic clearance, and its
prediction from physicochemical properties. _Drug Metab Dispos_ 2002; 30: 1497–503. Article CAS PubMed Google Scholar * Brown HS, Ito K, Galetin A, Houston JB . Prediction of _in vivo_
drug-drug interactions from _in vitro_ data: impact of incorporating parallel pathways of drug elimination and inhibitor absorption rate constant. _Br J Clin Pharmacol_ 2005; 60: 508–18.
Article CAS PubMed PubMed Central Google Scholar * Fahmi OA, Hurst S, Plowchalk D, Cook J, Guo F, Youdim K, _et al_. Comparison of different algorithms for predicting clinical drug-drug
interactions, based on the use of cyp3a4 _in vitro_ data: predictions of compounds as precipitants of interaction. _Drug Metab Dispos_ 2009; 37: 1658–66. Article CAS PubMed Google
Scholar * Yamamoto N, Horiike A, Fujisaka Y, Murakami H, Shimoyama T, Yamada Y, _et al_. Phase I dose-finding and pharmacokinetic study of the oral epidermal growth factor receptor tyrosine
kinase inhibitor Ro50-8231 (erlotinib) in Japanese patients with solid tumors. _Cancer Chemother Pharmacol_ 2008; 61: 489–96. Article CAS PubMed Google Scholar * Hutzler JM, Kolwankar
D, Hummel MA, Tracy TS . Activation of CYP2C9-mediated metabolism by a series of dapsone analogs: Kinetics and structural requirements. _Drug Metab Dispos_ 2002; 30: 1194–200. Article CAS
PubMed Google Scholar * Rendic S . Summary of information on human CYP enzymes: Human P450 metabolism data. _Drug Metab Rev_ 2002; 34: 83–448. Article CAS PubMed Google Scholar * van
Erp NP, Gelderblom H, Guchelaar HJ . Clinical pharmacokinetics of tyrosine kinase inhibitors. _Cancer Treat Rev_ 2009; 35: 692–706. Article CAS PubMed Google Scholar * Wang RW, Newton
DJ, Liu N, Atkins WM, Lu AYH . Human cytochrome P-450 3A4: _In vitro_ drug-drug interaction patterns are substrate-dependent. _Drug Metab Dispos_ 2000; 28: 360–6. CAS PubMed Google Scholar
* Schwab GE, Raucy JL, Johnson EF . Modulation of rabbit and human hepatic cytochrome-p-450-catalyzed steroid hydroxylations by alpha-naphthoflavone. _Mol Pharmacol_ 1988; 33: 493–9. CAS
PubMed Google Scholar * Yun CH, Wood M, Wood AJJ, Guengerich FP . Identification of the pharmacogenetic determinants of alfentanil metabolism-cytochrome p-450-3a4 — an explanation of the
variable elimination clearance. _Anesthesiology_ 1992; 77: 467–74. Article CAS PubMed Google Scholar * Hao M, Zhao YQ, Chen PZ, Huang H, Liu H, Jiang HL, _et al_. Structure-activity
relationship and substrate-dependent phenomena in effects of ginsenosides on activities of drug-metabolizing p450 enzymes. _PLoS One_ 2008; 3: e2697. Article PubMed PubMed Central Google
Scholar * Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, _et al_. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. _Science_ 2004; 305:
683–6. Article CAS PubMed Google Scholar * Tracy TS . Atypical enzyme kinetics: Their effect on _in vitro_-_in vivo_ pharmacokinetic predictions and drug interactions. _Curr Drug Metab_
2003; 4: 341–6. Article CAS PubMed Google Scholar * Nomeir AA, Ruegg C, Shoemaker M, Favreau LV, Palamanda JR, Silber P, _et al_. Inhibition of CYP3A4 in a rapid microtiter plate assay
using recombinant enzyme and in human liver microsomes using conventional substrates. _Drug Metab Dispos_ 2001; 29: 748–53. CAS PubMed Google Scholar * Grimm SW, Einolf HJ, Hall SD, He K,
Lim HK, Ling KHJ, _et al_. The conduct of _in vitro_ studies to address time-dependent inhibition of drug-metabolizing enzymes: a perspective of the pharmaceutical research and
manufacturers of america. _Drug Metab Dispos_ 2009; 37: 1355–70. Article CAS PubMed Google Scholar * Henshall J, Galetin A, Harrison A, Houston JB . Comparative analysis of CYP3A
heteroactivation by steroid hormones and flavonoids in different _in vitro_ systems and potential _in vivo_ implications. _Drug Metab Dispos_ 2008; 36: 1332–40. Article CAS PubMed Google
Scholar * Gibbs MA, Thummel KE, Shen DD, Kunze KL . Inhibition of cytochrome P-450 3A (CYP3A) in human intestinal and liver microsomes: Comparison of K–I values and impact of CYP3A5
expression. _Drug Metab Dispos_ 1999; 27: 180–7. CAS PubMed Google Scholar * McConn DJ, Lin YS, Allen K, Kunze KL, Thummel KE . Differences in the inhibition of cytochromes P450 3A4 and
3A5 by metabolite-inhibitor complex-forming drugs. _Drug Metab Dispos_ 2004; 32: 1083–91. CAS PubMed Google Scholar * Li XH, Kamenecka TM, Cameron MD . Cytochrome P450-mediated
bioactivation of the epidermal growth factor receptor inhibitor erlotinib to a reactive electrophile. _Drug Metab Dispos_ 2010; 38: 1238–45. Article CAS PubMed PubMed Central Google
Scholar * Li X, Kamenecka TM, Cameron MD . Bioactivation of the epidermal growth factor receptor inhibitor gefitinib: implications for pulmonary and hepatic toxicities. _Chem Res Toxicol_
2009; 22: 1736–42. Article CAS PubMed Google Scholar * Li XH, He YJ, Ruiz CH, Koenig M, Cameron MD . Characterization of dasatinib and its structural analogs as cyp3a4 mechanism-based
inactivators and the proposed bioactivation pathways. _Drug Metab Dispos_ 2009; 37: 1242–50. Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This
work was supported by the National Natural Science Foundation of China (No 30630075, 30772608, 30973590, and 81072698), the National Key Technology R&D Program in the 11th Five-year Plan
of China (No 2008ZX10002-019) and the National Science & Technology Pillar Program of China (No 2009BADB9B02). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Laboratory of Pharmaceutical
Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China Pei-pei Dong, Zhong-ze Fang, Yan-yan Zhang, Guang-bo Ge, Yu-xi Mao, Liang-liang
Zhu, Wei Li & Ling Yang * Graduate University of Chinese Academy of Sciences, Beijing, 100049, China Pei-pei Dong, Zhong-ze Fang, Yu-xi Mao, Liang-liang Zhu & Wei Li * the Second
Affiliated Hospital of Dalian Medical University, Dalian, 116027, China Yan-qing Qu & Li-ming Wang * Tianjin Key Laboratory of Pharmacokinetics and Clinical Pharmacology, Tianjin
Institute of Pharmaceutical Research, Tianjin, 300193, China Chang-xiao Liu Authors * Pei-pei Dong View author publications You can also search for this author inPubMed Google Scholar *
Zhong-ze Fang View author publications You can also search for this author inPubMed Google Scholar * Yan-yan Zhang View author publications You can also search for this author inPubMed
Google Scholar * Guang-bo Ge View author publications You can also search for this author inPubMed Google Scholar * Yu-xi Mao View author publications You can also search for this author
inPubMed Google Scholar * Liang-liang Zhu View author publications You can also search for this author inPubMed Google Scholar * Yan-qing Qu View author publications You can also search for
this author inPubMed Google Scholar * Wei Li View author publications You can also search for this author inPubMed Google Scholar * Li-ming Wang View author publications You can also search
for this author inPubMed Google Scholar * Chang-xiao Liu View author publications You can also search for this author inPubMed Google Scholar * Ling Yang View author publications You can
also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Ling Yang. RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE
Dong, Pp., Fang, Zz., Zhang, Yy. _et al._ Substrate-dependent modulation of the catalytic activity of CYP3A by erlotinib. _Acta Pharmacol Sin_ 32, 399–407 (2011).
https://doi.org/10.1038/aps.2010.218 Download citation * Received: 04 June 2010 * Accepted: 02 December 2010 * Published: 04 March 2011 * Issue Date: March 2011 * DOI:
https://doi.org/10.1038/aps.2010.218 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * erlotinib * CYP isoform * human liver microsomes *
substrate-dependent inhibition (or modulation) * drug interaction