- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
To test the hypothesis that PI3K/Akt/eNOS signaling has a protective role in a murine model of ventilation associated lung injury (VALI) through down-regulation of p38 MAPK signaling.
Male C57BL/J6 (wild-type, WT) or eNOS knockout mice (eNOS−/−) were exposed to mechanical ventilation (MV) with low (LVT, 7 mL/kg) and high tidal volume (HVT, 20 mL/kg) for 0−4 h. A subset of
WT mice was administered the specific inhibitors of PI3K (100 nmol/L Wortmannin [Wort], ip) or of p38 MAPK (SB203580, 2 mg/kg, ip) 1 h before MV. Cultured type II alveolar epithelial cells
C10 were exposed to 18% cyclic stretch for 2 h with or without 20 nmol/L Wort pretreatment. At the end of the experiment, the capillary leakage in vivo was assessed by extravasation of Evans
blue dye (EBD), wet/dry weight ratio and lung lavage protein concentration. The lung tissue and cell lysate were also collected for protein and histological review.
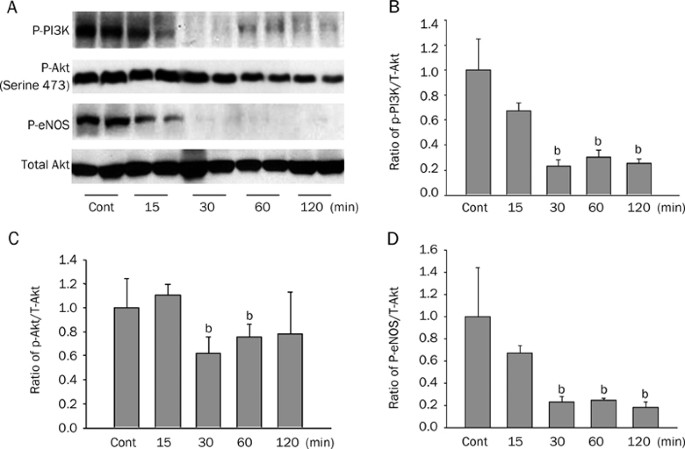
MV decreased PI3K/Akt phosphorylation and eNOS expression but increased phospho-p38 MAPK expression along with a lung leakage of EBD. Inhibitions of phospho-Akt by Wort worsen the lung
edema, whereas inhibition of p38 MAPK kinase restored activation of Akt together with alleviated capillary leakage. eNOS−/− mice showed an exacerbated lung edema and injury. The stretched
C10 cells demonstrated that Wort diminished the activation of Akt, but potentiated phosphorylation of MAPK p38.
Our results indicate that PI-3K/Akt/eNOS pathway has significant protective effects in VALI by preventing capillary leakage, and that there is a cross-talk between PI3K/Akt and p38 MAPK
pathways in vascular barrier dysfunction resulting from VALI.
Acute lung injury (ALI) is a complex syndrome marked by increased vascular permeability resulting in tissue edema and profound hypoxia1, 2. Mechanical ventilation (MV) is often a necessary
treatment for respiratory failure and a supportive measure in critically ill patients. However, it is well known that ventilation with high tidal volume (HVT MV) or pressure may be harmful,
causing damage to previously healthy lungs or worsening already injured ones. This is especially the case for permeability whereby exacting mechanical stress on various components of the
respiratory system can lead to ventilation associated lung injury (VALI). We performed the study described herein in an effort to elucidate the underlying mechanisms of VALI and investigate
potential therapies against VALI.
Akt (protein kinase B), a multifunctional serine-threonine protein kinase, is a major downstream signaling molecule of phosphoinositide 3-kinase (PI3K). It is also an upstream enzyme of
eNOS. Its activation has been implicated in various endothelial functions, including cell survival, migration, and activation of nitric oxide synthase3, 4, 5. Studies in vitro have shown
that Akt can directly phosphorylate endothelial nitric oxide (NO) synthase (eNOS) and activate the enzyme, leading to NO production6. Thus the PI3K/Akt/eNOS signaling pathway is critical for
maintenance of endothelial vascular tone, and integrity7, 8. We have previously demonstrated that NOS signaling and p38 MAPK are significantly implicated in the development of acute lung
injury9, 10. Though the PI3K/Akt and MAPKs, which include the ERK1/2, JNK, and p38 MAPK subgroups, are probably the best-characterized survival-related or death-associated signaling
pathways11, 12, 13, 14, 15 respectively, their functional roles in the VALI, especially the interplay between these two pathways, have not yet been elucidated. The emerging evidence has
triggered interest on the crosstalk link between PI3K/Akt and MAPKs signaling16, 17, 18, 19.
The purpose of this study is to identify the protective role of PI3K/Akt/eNOS signaling in the context of the lung vascular permeability in response to mechanical stress and to ascertain if
this protective effect occurs via p38 MAPK signaling in murine HVT MV and cyclic stretch models. Our results indicate that the PI3K/Akt/eNOS pathway is involved in the process of HVT MV with
a change in phosphorylated forms in intact lungs. Furthermore, genetic deletion of eNOS exacerbates vascular dysfunction as assessed by Evans Blue Dye (EBD). Finally, the findings that
pharmacologic antagonism of PI3K with Wort potentiates phosphorylation of p38 MAPK in response to cyclic stretch in C10 epithelial cells and that inhibition of p38 MAPK facilitates the
activation of Akt, a survival signaling protein, suggest interplay between the Akt and p38 MAPK pathways in vascular barrier dysfunction20.
CMRL 1066 medium was purchased from Invitrogen (Carlsbad, CA), and fetal bovine serum was obtained from Hyclone (Logan, UT) and Invitrogen (Carlsbad, CA). Wortmannin was obtained from
Calbiochem (San Diego, CA). Phospho-specific antibodies of PI3K, Akt, and eNOS as well as p38 MAP kinase were obtained from Cell Signaling Technology (Beverly, MA). SB203580 and other
reagents used in this study were obtained from Sigma (St Louis, MO).
Male C57BL/6J mice aged 10 weeks (23−25 g, Jackson Laboratory, Bar Harbor, ME) were studied in a pathogen-free facility under a protocol approved by the Johns Hopkins University Department
of Laboratory Animal Medicine. Anesthesia was induced with sodium pentobarbital (50 mg/kg, ip). Animals then underwent tracheotomy and intubation prior to exposure to MV (Harvard Apparatus,
Boston, MA) with room air for 0 (Control, Cont), 2, or 4 h with either 7 mL/kg or 20 mL/kg tidal volume (LVT or HVT). The respiratory rate was set at 160 breaths per minute for all tidal
volumes together with an adjustment of the dead space to maintain arterial pH between 7.35 and 7.45. Airway pressures were continuously measured and remained around 0−2 cm H2O. A 500-μL
bolus of lactated Ringer's solution was given intravenously at the initiation of MV to maintain adequate mean arterial blood pressure (about 80 Torr) during 4 h of ventilation. The adequacy
of MV settings on gas exchange was evaluated according to the mean blood pressure (MBP) and the arterial blood gases obtained via catheterization of a femoral artery. Rectal temperature was
maintained near 37 °C using a heating lamp and pad. At the end of MV, the animals were administered an intraperitoneal lethal dose of the anesthetic agent before the lungs were harvested.
To assess the effect of MV on the signaling of PI3K/Akt/eNOS and p38 MAPK, mice were treated with either a specific inhibitor of p38 MAPK, SB203580 (2 mg/kg, ip), or a specific inhibitor of
PI3K, Wort (100 nmol/L, ip)21, 1 h before exposure to MV. SB203580 and Wort were dissolved in DMSO first and then diluted further by saline. All doses, route of administration, and timing of
delivery of pharmacological agents as well as the solvent were based on known half-life of agents and preliminary experiments that demonstrated efficacy22, 23.
Acute changes in vascular permeability were evaluated by using bronchoalveolar lavage (BAL) fluid albumin concentrations and EBD assay in intact mice. Pulmonary edema formation was also
assessed using lung wet-to-dry weight (W/D) ratios. EBD (20 mg/kg) was injected into the external jugular vein 60 min prior to termination of the experiment to determine vascular leak as
previously described24, 25. To determine wet-to-dry lung weight ratio, as a separate measure of pulmonary edema, briefly the left lung was excised and immediately weighed on pre-tared dishes
for determination of wet lung weight. The samples were then dried in an oven (Fisher Isotemp) of 60 °C for 7 d and weighed several times until the weight became consistent26.
C10, a murine nonmalignant alveolar type II–like epithelial cell line, was cultured in CMRL 1066 medium supplemented with 10% fetal bovine serum (FBS) and antibiotics. Cells were seeded at
standard densities (8×105 cells/well) onto collagen I–coated BioFlex plates, and once confluence was reached, the plates were mounted onto the FX-4000T Flexercell Tension Plus system
(Flexercell International, McKeesport, PA) equipped with a 25-mm BioFlex loading station. This system provides uniform radial and circumferential strain across a membrane surface along all
radii. Cells were subjected to 18% elongation for the desired time at 25 cycles per minute. Cells grown on BioFlex plates and simultaneously placed in a cell culture incubator were
considered as static controls. At the end of the experiment, the cells were harvested for protein analysis.
Aliquots from cell lysates and tissue homogenates were immunoblotted using native and phosphospecific antibodies as previously described9. The blots were then visualized with the ECL Western
Blot Detection Kit (Amersham, Piscataway, NJ, USA).
Data are shown as mean±standard deviation (SD), for each experimental condition. Comparisons between groups were performed using t-tests or one-way ANOVA with Bonferroni correction. Analysis
of variance for comparison of the different groups was used with significance set at P






