- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Few data are available on population genetic structure in nematode species, and little of the available data allows direct comparison of the genetic structures of species having different
life cycles. Here we use mtDNA sequence data to describe the genetic structure of a heterorhabditid nematode, and compare results to published data on other nematode species. Heterorhabditis
marelatus is a parasite of soil-dwelling insects. Its life cycle and local ecology should result in small effective population sizes and restricted gene flow. As predicted, H. marelatus
shows much lower mtDNA diversity within populations and over the species as a whole, and has a much more strongly subdivided population structure, than parasites of mobile vertebrate hosts.
From data such as these we can begin to generalize about the effects of life cycle variation on genetic structure in different nematode species.
We still know little about the population genetic structure of most parasite species, the exceptions being mostly species of medical or agricultural importance (e.g. Lymbery et al., 1990;
Tibayrenc et al., 1991; Day et al., 1992; Anderson et al., 1995; Blouin et al., 1995; Dybdahl & Lively, 1996; Babiker & Walliker, 1997; Blair et al., 1997). This oversight is surprising
because data on genetic structure are necessary for understanding important evolutionary processes such as adaptation to host defences, host-race formation, speciation, and the evolution of
resistance to drugs or vaccines. Nematodes in particular are a grossly understudied taxon. Even though nematodes are one of the most species-rich, ecologically diverse and economically
important taxa, we have information on genetic structure for only a handful of nematode species, and almost all of these are human or agricultural parasites (recently reviewed in Anderson et
al., 1998). Virtually nothing is known of the genetic structure of any free-living nematode species (including Caenorhabditis elegans). Thus, more comparative studies on genetic structure
in nematode species are clearly needed. Indeed, Hughes et al. (1997) specifically called for more data on nematodes in their recent review of patterns of population differentiation in
different taxa.
Parasitic nematodes display a wide variety of life cycles and life histories. For example, they parasitize almost all groups of plants and animals, and occur in virtually every marine,
terrestrial and freshwater habitat. Their breeding system can be obligately or facultatively amphimictic (two distinct sexes), parthenogenetic or hermaphroditic. They range from highly
host-specific to undiscriminating, and vary in the presence or absence of free-living stages and intermediate hosts. How this diversity of life cycles influences genetic structure in
different nematode species is unknown. We currently have too few comparative data from which to make any but the simplest predictions.
What is needed are comparative studies of the genetic structure of nematode species that differ in key features of the life cycle, using similar sampling designs and the same molecular
markers. For example, using mtDNA sequence data Blouin et al. (1995) showed that host mobility has a large effect on genetic structure in trichostrongylid species that parasitize different
species of ruminants. We see the effect of differences in population size in comparisons between trichostrongylids and Ascaris species (Anderson et al., 1998). Both have similar life cycles
(simple, one-host, obligately outcrossing, with a mobile vertebrate host), but differ by orders of magnitude in population sizes, and correspondingly in levels of both mtDNA and nuclear
intron diversity. In contrast, plant parasitic nematodes having a predominantly parthenogenetic mode of reproduction show much lower overall mtDNA diversity than either Ascaris or the
trichostrongylids (Meloidogyne spp.; Hugall et al., 1994). Here we used mtDNA sequence data to describe the genetic structure of a species that parasitizes soil-dwelling insects.
Heterorhabditis marelatus is in the family Heterorhabditidae, one of two main families of entomopathogenic nematodes (Gaugler & Kaya, 1990). Entomopathogenic nematodes are obligate parasites
of soil-dwelling insects. Infective juveniles (IJs) of these species actively seek insect hosts in soil. After penetrating a host, IJs release a symbiotic gut bacterium (Photorhabdus spp.)
that rapidly kills the host, usually within 24–48 h. Nematodes reproduce within the cadaver, and large numbers of IJs escape into the soil to seek additional hosts.
Heterorhabditis marelatus occurs along the Pacific coast from the San Francisco Bay area (D. Strong, pers. comm.) to at least southern Washington (personal observation). Populations occur in
sandy soils under vegetation, usually behind the dunes of sandy beaches, and up to a few hundred metres inland. On the Pacific coast their habitat is subdivided into what is essentially a
linear series of habitat islands separated by stretches of rocky shoreline. Here we refer to the nematodes inhabiting a continuous stretch of suitable habitat (usually a discrete beach) as a
population.
Like most nematodes, Heterorhabditis have minimal powers of dispersal on their own. Gene flow on a regional scale will depend on the opportunities for nematodes to be transported either in
infected hosts, phoretically (by hitching a ride on nonparasitized hosts), or passively through the movement of wind and water. Infective juveniles are susceptible to UV light and to
desiccation, so they cannot be exposed to the air for long (Downes & Griffin, 1996; Strong et al., 1996). Heterorhabditis are tolerant of salt water, so movement along shore by ocean
currents might occur in coastal species like H. marelatus (Griffin et al., 1994). Transport in infected insects is possible, but heterorhabditids specialize on buried insects (as opposed to
insects walking on the soil surface), and hosts are killed rapidly following infection. Thus, the first prediction is that gene flow is very restricted on a regional scale.
We also predict that H. marelatus populations will have small effective sizes, for two reasons. First, on a local scale (a few to tens of metres) the distribution of nematodes is very
clumped and patchy, and patches go extinct and are recolonized at high rates (Stuart & Gaugler, 1994; Strong et al., 1996). Secondly, it is likely that each patch consists of very closely
related individuals descended from one or a few maternal founders. An infective juvenile that enters a host must reproduce hermaphroditically. Its offspring then mature into separate males
and females who reproduce for one or more generations before producing infective juveniles that leave the host. A single infection can produce hundreds of thousands of IJs, and these tiny
nematodes cannot move far on their own. Thus, patches probably contain the descendants of one or a few maternal founders. This sort of metapopulation patch structure should result in very
small mitochondrial effective sizes within populations (McCauley, 1991; Caballero & Hill, 1992; Harrison & Hastings, 1996).
Therefore, H. marelatus should show lower overall genetic diversity, and a more strongly subdivided genetic structure, than obligately outcrossing parasites of mobile vertebrates, such as
the trichostrongylids. To test this hypothesis, we used mtDNA sequence data to describe the genetic structure of H. marelatus populations along the Pacific coast of California and Oregon,
and compared these data to the data from trichostrongylids (for which the same gene and sampling scheme were used, making the two datasets directly comparable).
To describe population structure in H. marelatus, we sequenced 474 bp of the 3′ end of the mitochondrial ND4 gene. ND4 codes for a membrane spanning polypeptide of the hydrophobic subunit of
NADH dehydrogenase complex I, and has been shown to be an excellent marker for population genetics studies in nematodes (Blouin et al., 1998). We sequenced each of nine or 10 individuals
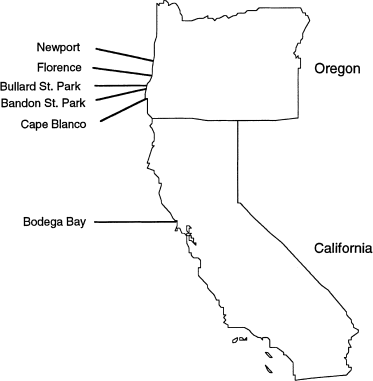
per population, in six populations from coastal Oregon and California (Fig. 1). At each site we collected soil samples from an area spanning several hectares. We baited each soil sample with
waxworms (Galleria mellonella), and isolated a single first generation hermaphroditic nematode from infected hosts. To avoid sampling related individuals from the same patch of soil, we
made sure that no samples were taken any closer than several metres apart, and sequenced only one individual per soil sample. This region of the ND4 gene was used so that the results could
be directly compared to those of Blouin et al. (1995), who used the same gene and sample sizes to study the genetic structures of four species of trichostrongylid nematodes that parasitize
ruminants in North America. Here the geographical scale over which we sampled H. marelatus populations (Oregon and northern California) is about the same as that over which populations of
two of the trichostrongylids were sampled (south-eastern U.S. for Mazamastrongylus odocoilei and Haemonchus placei), and is smaller than the scale over which populations of the other two
were sampled (entire U.S. for Haemonchus contortus and Teladorsagia circumcincta).
Sampling sites. Partial ND4 gene sequences in Heterorhabditis marelatus were obtained from nine or 10 nematodes from each site (Table 1).
Individual nematodes were crushed with a pestle in 20 μL of a 5% chelex solution, and incubated overnight at 55°C. Four μL of the supernatant was used to amplify the ND4 region in a 25-μL
PCR reaction (1.5 mM MgCl, 0.3 μM primers, GIBCO Taq and buffer) using a Perkin Elmer 9600 thermocycler (94°C denature for 3 min, then 35 cycles of 94°C for 45 s, 54°C for 1 min, 72°C for
1.5 min, then a 7-min extension at 72°C). The PCR product was then run on a 1% agarose gel, isolated using a Supelco GenElute spin column, and sequenced on an ABI 377 automated sequencer
using the PCR primers as sequencing primers. Primers used were:
Heterorhabditis marelatus shows strong differentiation among populations and low genetic diversity, both within populations and in the species as a whole (Table 1). We found only four
distinct haplotypes (labelled A, B, B′ and C) out of 58 sequences in the entire sample (Figs 2 and Figs. 3), and at most two haplotypes in any population (Table 1). This diversity was
strongly structured, with 86% of the total sequence diversity (NST; Lynch & Crease, 1990) and 78% of the haplotype diversity (FST) distributed among populations (Table 1). Even the two
closest populations (Bandon, OR, and Bullard, OR, 8 km apart; Fig. 1) had no haplotypes in common, and the only private allele (Slatkin, 1985) in the sample had a frequency of 8/10 in its
population. Note also that this most geographically restricted allele (allele B′) also appears to be the most recently derived of the four haplotypes (Fig. 3a), and that it occurs in a
population with its parent allele (allele B; Table 1). This pattern is exactly what one expects under restricted gene flow, because the geographical range of a haplotype should be strongly
correlated with its age (Templeton et al., 1995). Finally, the distribution of pairwise sequence differences in H. marelatus clearly differs from that expected in a single population under
drift–mutation equilibrium, there being too few haplotypes, given the distances among them (Fig. 3b; Tajima’s D=3.12, P < 0.01; Tajima, 1989). Assuming neutrality, this pattern is again
consistent with historical subdivision of the species into isolated units.
Alignment of the four ND4 haplotypes observed in Heterorhabditis marelatus. All polymorphisms are in silent sites, except for the substitution in position 268 that separates haplotype B′
from haplotype B.
(a) Network tree showing number of substitutions separating each Heterorhabditis marelatus haplotype from the consensus sequence. All substitutions in each branch are unique to that branch.
(b) Percentage of sequence difference between haplotypes.
The above results are all consistent with the small effective population sizes and low rates of gene flow predicted by the life cycle of H. marelatus. That there are too few haplotypes given
the large genetic distances between them is interesting. A selective sweep cannot be ruled out, but rapid drift within populations, combined with occasional long-distance gene flow, could
produce the same pattern. That two common alleles (A and B) are widespread throughout the species’ range, whereas even adjacent populations can be fixed for different alleles, is consistent
with this scenario. Perhaps migration occurs in an isolation-by-distance fashion on land, and occasionally over long distances via ocean currents. More intensive sampling of populations
throughout the range of the species might well reveal more haplotypes, but the overall pattern of strongly restricted gene flow on a local scale, with widespread common alleles, is unlikely
to change.
Heterorhabditis spp. are extensively studied for their biocontrol abilities, and there is great interest in finding new strains that differ in characters such as host-seeking behaviour,
environmental tolerance and ability to control different pests (Bedding et al., 1983; Kung & Gaugler, 1991; Gaugler et al., 1997). Their symbiotic bacteria are equal partners in killing
insects, and trait variation in the bacteria may be as or more important than in the nematodes. For example, the toxins produced by Photorhabdus spp. are some of the most potent insect
killers known, rivalling the well-known Bacillus thuringensis toxin, and different species and strains of Photorhabdus carry different toxins (Bowen et al., 1998). Consequently, there is
also a major impetus to search for and characterize new strains of symbiotic bacteria, particularly those adapted to unusual hosts or habitats. Nevertheless, surprisingly little work has
been carried out on the basic ecology and genetics of Heterorhabditis or their bacteria in nature (Gaugler & Kaya, 1990; Strong et al., 1996; Gaugler et al., 1997). Ours are the first data
on genetic structure in a heterorhabditid, and there has been no population genetic work on Photorhabdus. Because the symbiotic bacteria can presumably only disperse in association with
their nematode, their population genetic structure should mirror that of the nematode. As part of an unrelated study we recently sequenced 616 bp of the bacterial 16S gene of bacterial
isolates from each of five nematodes from Florence, OR, and from five nematodes from Newport, OR (≈80 km apart; Fig. 1) (unpubl. data). Isolates from the two populations were fixed for
different 16S rRNA haplotypes. Although these data are anecdotal, they suggest that the nematode and their symbiotic bacterium may both have population genetic structures that promote
genetic drift and the opportunity for local adaptation over short distances. Thus it may be fruitful to search for useful new strains of nematode and bacteria (i.e. those adapted to unique
hosts or environmental conditions) over very small geographical scales.
Table 2 confirms the prediction that H. marelatus has lower overall diversity (both species-wide and within individual populations) and a more strongly subdivided genetic structure than the
trichostrongylids. Only four unique haplotypes were found in 58 sequences from H. marelatus, whereas samples of 40 trichostrongylid sequences yielded 31–39 unique haplotypes. Within
populations the haplotype and nucleotide diversity is almost an order of magnitude greater in the trichostrongylids than in H. marelatus, and a much greater proportion of the total sequence
diversity is distributed among populations in H. marelatus. Because the geographical scale over which H. marelatus populations were sampled is smaller than that over which some of the
trichostrongylids were sampled, the higher FST in H. marelatus is even more striking. Within each of the four trichostrongylid species Tajima’s D is not significantly different from zero,
indicating tree topologies that are not significantly different from that expected under neutrality in a single population (see figs 2,3,4,5 in Blouin et al., 1995, for haplotype trees). So
in these species we do not see the signature of historical subdivision into isolated populations that is apparent in the tree of H. marelatus haplotypes.
In the trichostrongylids vs. H. marelatus we see two extremes in a spectrum of genetic structures. Trichostrongylids show levels of mtDNA variation that are greater than those typically seen
in other taxa, and the species that infect livestock show exceptionally high rates of gene flow over vast geographical areas (Blouin et al., 1995; also M. Blouin, S. Richter and E. Hoberg,
unpubl. data on Teladorsagia circumcincta from Iceland vs. North America; C. Constant, unpubl. data on Ostertagia ostertagi from Australia vs. North America). In contrast, H. marelatus shows
very low variation within populations and in the species as a whole, and very restricted gene flow on a small scale. In these respects the genetic structure of H. marelatus may be more
similar to that of parthenogens such as Meloidogyne spp. than to that of outcrossing parasites of vertebrates. For example, only six unique mtDNA haplotypes were found in 48 Meloidogyne
individuals sampled from throughout the eastern half of Australia (data from RFLP of entire mtDNA; Hugall et al., 1994). Individual Meloidogyne samples were spread over a wide geographical
area in that study, so we cannot directly compare levels of within- and between-population diversity in Meloidogyne spp. to that in H. marelatus or the trichostrongylids. However, a testable
prediction is that the distribution of mtDNA diversity within and among populations in Meloidogyne will be most similar to that in Heterorhabditis.
Here we designed a study to compare the genetic structures of two groups of nematodes, by using the same molecular marker and similar sampling schemes. Obviously more comparative data such
as these are needed before we can generalize about the effects of life cycle on genetic structure in nematodes. In particular, we need data on species in ‘natural’ habitats (i.e. species
that are not human associates). To our knowledge, there are no data on genetic structure in any free-living species, and of the parasitic species, only three are not parasites of humans or
their domesticated plants or livestock (these include the present data on H. marelatus, mtDNA data on Mazamastrongylus odocoilei, which is a parasite of deer [Blouin et al., 1995], and
allozyme data on Anisakid nematodes of fish and cetaceans [e.g. Paggi et al., 1991; Nascetti et al., 1993;]). Clearly this is a wide-open field of study that deserves more attention.
Thanks to D. Strong, J. Johnston, G. Poinar and D. Anderson for help collecting samples, and to A. Rabe, K. Monsen and A. Giese for comments on an earlier draft. This work was supported by
the OSU Agricultural Research Foundation and by U.S. Department of Agriculture CSREES 96–34354–3072 through the Oregon State University Center for Gene Research and Biotechnology.
Department of Zoology, Oregon State University, Corvallis, 97331, OR, USA
Department of Entomology, Oregon State University, Corvallis, 97331, OR, USA
Anyone you share the following link with will be able to read this content:







:max_bytes(150000):strip_icc():focal(749x0:751x2)/ben-affleck-4-4e7dbffb8964423fa077e339f01816a0.jpg)

