- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Multiple genetic loci confer susceptibility to breast and ovarian cancers. We have previously developed a model (BOADICEA) under which susceptibility to breast cancer is explained
by mutations in _BRCA1_ and _BRCA2_, as well as by the joint multiplicative effects of many genes (polygenic component). We have now updated BOADICEA using additional family data from two UK
population-based studies of breast cancer and family data from _BRCA1_ and _BRCA2_ carriers identified by 22 population-based studies of breast or ovarian cancer. The combined data set
includes 2785 families (301 _BRCA1_ positive and 236 _BRCA2_ positive). Incidences were smoothed using locally weighted regression techniques to avoid large variations between adjacent
intervals. A birth cohort effect on the cancer risks was implemented, whereby each individual was assumed to develop cancer according to calendar period-specific incidences. The fitted model
predicts that the average breast cancer risks in carriers increase in more recent birth cohorts. For example, the average cumulative breast cancer risk to age 70 years among _BRCA1_
carriers is 50% for women born in 1920–1929 and 58% among women born after 1950. The model was further extended to take into account the risks of male breast, prostate and pancreatic cancer,
and to allow for the risk of multiple cancers. BOADICEA can be used to predict carrier probabilities and cancer risks to individuals with any family history, and has been implemented in a
user-friendly Web-based program (http://www.srl.cam.ac.uk/genepi/boadicea/boadicea_home.html). SIMILAR CONTENT BEING VIEWED BY OTHERS VALIDATION OF THE BOADICEA MODEL AND A 313-VARIANT
POLYGENIC RISK SCORE FOR BREAST CANCER RISK PREDICTION IN A DUTCH PROSPECTIVE COHORT Article Open access 06 July 2020 POTENTIAL OF POLYGENIC RISK SCORES FOR IMPROVING POPULATION ESTIMATES OF
WOMEN’S BREAST CANCER GENETIC RISKS Article Open access 06 July 2021 THE ROLE OF POLYGENIC RISK AND SUSCEPTIBILITY GENES IN BREAST CANCER OVER THE COURSE OF LIFE Article Open access 14
December 2020 MAIN The risk of breast cancer in first-degree relatives of women with breast cancer is approximately two times higher than in women from the general population (Collaborative
Group in Hormonal Factors in Breast Cancer, 2001). Mutations in the high-risk breast cancer-susceptibility genes _BRCA1_ and _BRCA2_ account for approximately 15% of this excess familial
risk (Easton, 1999; Peto et al, 1999; Anglian Breast Cancer Study Group, 2000; Dite et al, 2003). We had previously derived a breast cancer susceptibility model, the Breast and Ovarian
Analysis of Disease Incidence and Carrier Estimation Algorithm (BOADICEA), based on segregation analysis of breast and ovarian cancer occurrence in a combined data set, including a
population-based series of 1484 breast cancer cases and 156 multiple case families from the United Kingdom (Antoniou et al, 2002, 2004). According to this model, genetic susceptibility to
breast cancer is explained by the effects of _BRCA1_ and _BRCA2_ mutations, and the residual familial clustering is explained by the joint multiplicative effect of a large number of genes
each of small effect (i.e., by a polygenic component). Direct evidence for the polygenic basis of the residual familial clustering not due to _BRCA1_ and _BRCA2_ mutations has more recently
been provided by the identification of further loci that confer moderate risks, including mutations in _CHEK2_, _ATM_, _PALB2_, _BRIP1_ (The CHEK2 Breast Cancer Case–Control Consortium,
2004; Renwick et al, 2006; Seal et al, 2006; Rahman et al, 2007) and the low-risk variants identified through genome-wide or candidate gene association studies (Cox et al, 2007; Easton et
al, 2007; Hunter et al, 2007; Stacey et al, 2007). The BOADICEA model can be used to estimate the likelihood of carrying a _BRCA1_ or a _BRCA2_ mutation, and the risks of developing breast
or ovarian cancer. However, there are a number of limitations associated with the first version of BOADICEA. The model assumed that a fixed set of calendar period incidences applied to all
cohorts, when breast cancer incidences have been increasing over time (Office for National Statistics, 2001; Tryggvadottir et al, 2006). Moreover, the incidences were assumed to change in
5-year intervals when in reality they change smoothly with age. As part of the model-fitting process, we also estimated the _BRCA1_ and _BRCA2_ breast and ovarian cancer risks, but these
were based on a relatively small number of mutation-carrying families and were therefore imprecise (Antoniou et al, 2002, 2004). Finally, the model took into account only the occurrence of a
first breast or a first ovarian cancer and the risks of second or subsequent cancers were ignored. _BRCA1_ and _BRCA2_ mutations are associated with increased risk of cancer at several
sites other than female breast and ovary. The strongest evidence is for prostate and pancreatic cancer in _BRCA2_ carriers, which has been consistently found in multiple studies (The Breast
Cancer Linkage Consortium, 1999; Tulinius et al, 2002; Edwards et al, 2003; Kirchhoff et al, 2004; van Asperen et al, 2005; Risch et al, 2006; Tryggvadottir et al, 2007). There is also more
limited evidence for an increased risk of cancer of the gall bladder, bile duct, stomach and malignant melanoma (The Breast Cancer Linkage Consortium, 1999; Edwards et al, 2003; Kirchhoff et
al, 2004; van Asperen et al, 2005; Risch et al, 2006). _BRCA1_ carriers have been found to have elevated risks of colorectal, pancreatic, prostate, testicular and uterine cancer (Ford et
al, 1994; Brose et al, 2002; Thompson and Easton, 2002; Risch et al, 2006). The colorectal cancer risk, however, has not been consistently replicated, while the prostate and pancreatic
cancer risks are lower than in _BRCA2_ carriers. In addition, male _BRCA1_ and (to a greater extent) _BRCA2_ carriers are at an increased risk of developing breast cancer (Thorlacius et al,
1997; Thompson and Easton, 2001; Risch et al, 2006; Tai et al, 2007, DF Easton, unpublished data). Incorporation of these additional cancer phenotypes into the model should provide greater
discrimination between _BRCA1_ and _BRCA2_ mutation carriers and noncarriers. In this paper, we have attempted to improve BOADICEA by analysing additional population-based data and by
extending the model to account for the risk of cancer after the first diagnosis and the risks of cancer at sites other than breast and ovary. MATERIALS AND METHODS We combined the data from
the two studies used in the initial model development with data from three additional published population-based studies. Specifically, the present analysis includes the following data sets:
_(a) The Anglian Breast Cancer Study (ABC, now SEARCH)._ The families were identified through 1484 women with breast cancer diagnosed before the age of 55 years and registered in the East
Anglian Cancer Registry between 1991 and 1996. These index cases were invited to provide blood samples and complete an epidemiological questionnaire, including family history of cancer in
all first-degree relatives. The blood samples were tested for germline mutations in _BRCA1_ and _BRCA2_ using conformation-sensitive gel electrophoresis (CSGE). Mutations were confirmed by
sequencing. These data were used in the initial model development and the study is described in more detail elsewhere (Anglian Breast Cancer Study Group, 2000; Antoniou et al, 2001). _(b) UK
National Case–Control Study (UK)._ Women with breast cancer were identified through two UK population-based case–control studies. The first study involved 755 patients diagnosed under the
age of 36 years and registered between 1982 and 1985. The second study included 644 patients diagnosed from age 36 to 45 years and registered between 1988 and 1989. These index cases
provided family history information of breast and ovarian cancer and were later contacted to provide blood samples. DNA was screened for germline mutations in _BRCA1_ and _BRCA2_ by
heteroduplex analysis. Again, mutations were confirmed by sequencing. In all, 617 samples were tested for _BRCA1_ and _BRCA2_ mutations and were included in our analysis. This data set is
described in detail elsewhere (Peto et al, 1999). _(c) The Manchester Study._ Women diagnosed with breast cancer at or before the age of 30 years were recruited via the North West Regional
Cancer Registry (UK) between 1980 and 1997. A total of 99 index cases provided blood samples, which were screened for mutations in _BRCA1_ and _BRCA2_ using a combination of the Protein
truncation test, single-strand conformation polymorphism/heteroduplex analysis and fluorescent chemical cleavage of mismatch analysis. Three-generational pedigrees were constructed through
interviews and were augmented with data from hospital notes. The study is described in Lalloo et al (2003). _(d) Multiple case families: ‘British’ (B) families._ In all, 156 families were
ascertained in response to national publicity in the United Kingdom and by referral by oncologists or general practitioners. Eligibility was restricted to families with at least two breast
cancer cases, one or more diagnosed before the age of 50 years. Occurrence of cancer and follow-up was recorded on all family members. One or more individuals from each family provided blood
samples, which were analysed for _BRCA1_ and _BRCA2_ mutations using CSGE (Antoniou et al, 2002). This set of families was also used in the initial development of BOADICEA. _(e)
Meta-analysis families (BRCA families)._ This data set included pedigree data from _BRCA1_ and _BRCA2_ mutation carriers identified in 22 population-based studies of breast or ovarian cancer
patients reported by Antoniou et al (2003). A study was eligible for the meta-analysis if it was based on mutation testing of a series of index cases diagnosed with either breast (male or
female) or epithelial ovarian cancer and who were unselected for family history of cancer. In each study, the index cases had to be tested for _BRCA1_ and/or _BRCA2_ mutations by systematic
screening, and family history information had to be available on all first-degree relatives of identified mutation carriers. To avoid replication, the families of mutation carriers
identified through the ABC, UK and Manchester studies were not considered to be part of the BRCA families for the present analysis. A total of 429 families of _BRCA1_ and _BRCA2_ mutation
carriers were included in the present analysis. _BRCA1_ and _BRCA2_ mutations were considered to be disease causing if they were classified pathogenic according to the generally accepted
criteria (http://research.nhgri.nih.gov/projects/bic/). For consistency across the population-based studies (ABC, United Kingdom, Manchester and BRCA), family history information was
restricted to the first-degree relatives of the index cases. SEGREGATION ANALYSIS Model fitting was performed using complex segregation analysis of breast and ovarian cancer occurrences in
the combined set of families described above. Individuals were followed from birth and were censored at the age of cancer occurrence, age at death or at the age of 70 years whichever
occurred first. Female patients with no age information or no year of birth were censored at age 0 (692 female patients in the combined data set). In the initial development of BOADICEA, a
number of different genetic models were investigated for the genetic susceptibility to breast cancer (Antoniou et al, 2002). It was found that the most parsimonious model was one that
incorporated the simultaneous effects of _BRCA1_, _BRCA2_ and a polygenic component representing the combined multiplicative effect of multiple loci of small effect. This model is consistent
with the recent discovery of multiple low-risk susceptibility genes (and the failure to identify any further ‘high-risk’ loci by linkage) (Smith et al, 2006; Easton et al, 2007; Hunter et
al, 2007; Stacey et al, 2007). Although it is likely that the genetic causes of breast cancer are more complicated, for the present analysis we focused only on polygenic models for the
residual familial clustering of breast cancer other than that due to mutations in _BRCA1_ and _BRCA2_. The breast cancer incidence for individual _i_ at age _t_ was assumed to be birth
cohort specific, and to depend on the underlying _BRCA1_ or _BRCA2_ genotype and polygenotype through a model of the form _λ__i_(_t_)=_λ_0(_t_)exp(_G__i_(_t_)+_P__i_(_t_)), where _λ_0(_t_)
is the baseline incidence for the cohort, _G__i_(_t_) represents the major gene effect at age _t_ (_BRCA1_ carrier, _BRCA2_ carrier or noncarrier) and _P__i_(_t_) is the polygenic effect
assumed to be normally distributed with mean zero and variance _σ_2(_t_). The polygenic component was approximated by the hypergeometric polygenic model (Antoniou et al, 2001; Lange, 2002).
More details about the implementation of this approximation in MENDEL can be found in Antoniou et al (2001). Under the above model, the polygenotype is assumed to modify the disease risk in
_BRCA1_ and _BRCA2_ carriers. In this context, that is, when _P__i_(_t_) acts on _BRCA1_ and _BRCA2_ background, the polygenic component is referred to as the ‘modifying’ component. In this
analysis, we generalised the model to allow for different polygenic and modifying variances in mutation carriers and noncarriers. We also fitted models in which the polygenic and modifying
variance was age dependent. CALENDAR PERIOD- AND COHORT-SPECIFIC INCIDENCES The breast and ovarian cancer incidences were assumed to be calendar period and cohort specific, based on the
incidences for England and Wales (Cancer in five continents volumes I–VIII, Doll et al, 1966, 1970; Waterhouse et al, 1976, 1982; Muir et al, 1987; Parkin et al, 1992, 2002). Five birth
cohorts were assumed for this purpose (<1920, 1920–1929, 1930–1939, 1940–1949, and 1950 or after), and the incidences were derived by assuming that the female patient was born at the
midpoint of the relevant birth cohort (1915 for the first cohort and 1955 for the last cohort). The overall incidences were constrained to agree with the population incidences for each
cohort separately (Antoniou et al, 2001). The rate ratios associated with the major gene and polygenic effects (_G__i_(_t_), _P__i_(_t_)) were assumed not to vary by birth cohort. Owing to
this constraint, the estimated incidences for _BRCA1_ and _BRCA2_ carriers and noncarriers were themselves cohort specific. INCIDENCE SMOOTHING Published incidences are reported in 5-year
intervals, which can result in large variations in the incidences between adjacent age intervals. This is particularly an issue for _BRCA1_ and _BRCA2_ carriers for whom incidence increases
rapidly with age. Since it is more plausible to assume that incidences vary continuously with age, we smoothed the population incidences using locally weighted regression techniques
(Royston, 1991). This method was chosen because it follows the ‘locality’ of the data as opposed to polynomial smoothing, which is a global graduation technique, is influenced by the extreme
points (young and old ages) and is not as flexible. Smoothing was carried out using the statistical software Stata (Stata Corporation, College Station, Texas, USA). The method involves
running a regression at each age _t__i_, using the data for age _t__i_ and a small amount of data near _t__i_. The proportion of data (bandwidth) used in the regression specifies the degree
of smoothness. Various degrees of smoothness were investigated and the resulting set of incidences was compared with the original set of incidences for adherence. As a smoothness criterion,
we used the sum of the absolute values of the third-order finite differences of the smoothed incidences: where Smaller values of the sum (*) correspond to smoother incidence curves.
Consistency of the model with the data was assessed using a _χ_2 test statistic, treating the smoothed incidences as expected values. However, formal tests of significance were not performed
because of the difficulty in determining the correct number of degrees of freedom for the test. _BRCA1_ AND _BRCA2_ RELATIVE RISKS Two types of models were assumed for the _BRCA1_ and
_BRCA2_ log-relative risks (_G__i_(_t_)) for both breast and ovarian cancer. Our primary analysis involved fitting models in which the relative risks are assumed to be constant within each
decade of age (20–29, 30–39, 40–49, 50–59 and 60–69 years). Once the most parsimonious model for the form of the polygenic and modifying variance was chosen, we fitted additional models in
which the log-relative hazards were piecewise linear functions of age (see Appendix), so that the resulting _BRCA1_ and _BRCA2_ incidences were continuous functions of age. ADJUSTMENT FOR
ASCERTAINMENT For the ABC, UK and Manchester families, we adjusted for ascertainment by maximising the conditional likelihood of observing the phenotypes and genotypes in the families, given
the disease status and age at diagnosis of the index case. For the _BRCA1_- and _BRCA2_-positive families, we maximised the conditional likelihood of observing the disease phenotypes and
genotypes in the family, given the disease status, age at diagnosis and mutation status of the index case. Since the ‘B’ families were identified through multiple-affected individuals, we
maximised the likelihood of all phenotypes and genotypes in the family conditional on all the phenotypic information for the family. SENSITIVITY OF THE MUTATION TESTING To allow for the fact
that not all mutations could be detected by the screening methods used, we allowed in our analysis for a sensitivity of mutation testing parameter, giving the probability of detecting a
mutation if one exists. We assumed that 70 and 80%, respectively, of the disease-causing mutations in _BRCA1_ and _BRCA2_ could be detected by the methods used. The sensitivity parameter
only applied for the first screened individual from each family (the index cases). Variants of uncertain significance (VUSs) were assumed to be equivalent to a _BRCA1_- and _BRCA2_-negative
test, since this is how they are treated in clinical genetics and in analyses testing the goodness of fit of the model. Although some such variants may be pathogenic, this effect is allowed
for in the model by the mutation sensitivity parameter (i.e., the incomplete sensitivity is partly due to pathogenic mutations classified as VUSs). For the relatives of index patients who
were screened for family-specific mutations, we assumed that the test was 100% sensitive. MODEL COMPARISONS Nested models were compared against each other using the likelihood ratio test.
The Akaike Information Criterion (AIC) was used to compare non-nested models (Akaike, 1974). PARAMETER ESTIMATION The models were parameterised in terms of the polygenic and modifying
variances, the _BRCA1_ and _BRCA2_ allele frequencies in the population and the natural logarithm of the ratios of the breast and ovarian cancer incidences in _BRCA1_ and _BRCA2_ mutation
carriers to the population incidences (relative hazards). Parameters were estimated by maximum likelihood, and their variances were obtained from the observed information matrix. To obtain
confidence intervals for parameters with restricted ranges (e.g., allele frequencies) we used transformations to obtain parameters that are likely to be more normally distributed (Antoniou
et al, 2001). RESULTS Table 1 summarises the total number of families by the mutation status of the index case. In all, 2785 families were included in the analysis, of which 301 were _BRCA1_
positive and 236 were _BRCA2_ positive. Table 2 shows the number of breast and ovarian cancer patients by age at diagnosis for the relatives of the index cases. The smoothed incidences used
in the segregation analyses were based on a bandwidth of 0.2. This value provided a compromise between smoothness and adherence to the original published incidences (data not shown). The
graduated calendar- and cohort-specific incidences included the age-specific features for all cohorts observed in the general population (data not shown) (Office for National Statistics,
2001). On the basis of the graduated incidences, the risk of breast cancer in the general population by the age of 80 years is 8.9, 9.8, 10.4, 10.9 and 11.0% for female patients born prior
to 1920, between 1920–1929, 1930–1939, 1940–1949, and 1950 and after, respectively. We first fitted three models, with different assumptions about the polygenic and modifying variances
(Table 3). For these models, the _BRCA1_ and _BRCA2_ relative hazards were assumed to be constant within each 10-year interval: 20–29, 30–39, 40–49, 50–59 and 60–69 years. In the first
model, the polygenic and modifying variances were constrained to be equal. In the second model, the polygenic variance was allowed to be different from the modifying variance, but the latter
was constrained to be the same for _BRCA1_ and _BRCA2_ carriers. In the third model, the polygenic variance and the _BRCA1_ and _BRCA2_ modifying variances were all allowed to vary. In each
of these models, the polygenic and modifying variance were assumed to be constant with age. We found no evidence that the _BRCA1_- and _BRCA2_-modifying variances were different from each
other (_P_=0.76). The modifying variance was estimated to be somewhat lower than the polygenic variance (1.55 _vs_ 2.02), but the difference was not significant (_P_=0.63). When a single
polygenic/modifying variance was assumed, it was estimated to be 1.99 (95% CI: 1.54–2.57). This model had the lowest AIC value of the three models (7948.284). The _BRCA1_ and _BRCA2_
parameter estimates for this model are given in Table 4. We then fitted three further models, for which the polygenic variance was allowed to vary by age group (20–29, 30–39, 40–49, 50–59
and 60–69 years; Table 3). When the modifying variance was restricted to be equal to the polygenic variance, this model did not fit significantly better than the model with the same constant
polygenic and modifying variance (_P_=0.30). The other two models assumed separate constant modifying variances, which were either constrained to be equal or different among _BRCA1_ and
_BRCA2_ mutation carriers. These models also did not improve the fit significantly, compared with the model with a constant polygenic/modifying variance (_P_=0.53 and 0.65). The latter two
models assumed constant modifying variances because models with varying modifying variances resulted in unbounded estimates. Despite the lack of a significant improvement in fit, the
parameter estimates suggest that the polygenic variance may decrease with age. We explicitly allowed for this hypothesis by fitting a model in which the polygenic and modifying variances
were the same linear function of age, that is, _σ__p_2(_t_)=_σ__m_2(_t_)=_α_+_βt_, where _t_ represents the age in years, and estimated the parameters _α_ (=4.86, 95% CI: 1.8–7.9) and _β_
(=−0.06, 95% CI: −0.12 to −0.0002). Compared with the model with a constant variance, there was some marginal evidence that this model fitted better (_P_=0.049). To investigate further the
properties of these models, we computed the age-specific familial relative risks (FRRs) for an individual with an affected mother predicted by these two models as described elsewhere
(Antoniou et al, 2004). We then contrasted these against the observed FRR estimated by epidemiological studies (Collaborative Group in Hormonal Factors in Breast Cancer, 2001). The predicted
FRRs were closer to the observed values for the model with a polygenic/modifying variance that decreased with age than the model with a constant variance (Table 5). When a constant
polygenic variance was assumed, the predicted FRRs decreased markedly at young ages in line with the observed values, but were still high at ages 55 years and older. The model with a
linearly decreasing polygenic variance predicted an FRR, which was close to the observed values at all ages. _BRCA1_ AND _BRCA2_ INCIDENCE SMOOTHING The model with a linearly decreasing
polygenic/modifying variance was extended to allow for the _BRCA1_ and _BRCA2_ log-relative hazards to be piecewise linear functions of age (see Materials and Methods, Appendix and Table 6).
The allele frequencies of _BRCA1_ and _BRCA2_ mutations in the general population were estimated to be 0.06% (95% CI: 0.04–0.10%) and 0.10% (95% CI: 0.07–0.16%), respectively. These
correspond to population carrier frequencies of 0.12% for _BRCA1_ and 0.20% for _BRCA2_. The average cumulative risks of breast and ovarian cancer in _BRCA1_ and _BRCA2_ mutation carriers
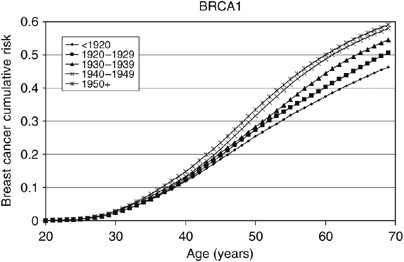
based on this model are shown in Figures 1, 2, 3 and 4. The average cumulative risk of breast cancer in _BRCA1_ carriers over all possible modifiers was estimated to be 46% by the age of 70
years for women born before 1920, rising to 59% for women born after 1950. On the basis of the 5th and 95th percentiles of the distribution of the polygenic/modifying component, the
estimated cumulative breast cancer risks were 7.2 and 98%, respectively, for carriers born before 1920, rising to 12.0 and 99.9% for carriers born after 1950. The average cumulative risks of
breast cancer in _BRCA2_ mutation carriers by the age of 70 years were estimated to be lower, 39% for women born before 1920 (6.3 and 93% at the 5th and 95th percentiles, respectively)
rising to 51% for those born after 1950 (9.8 and 98.7% at the 5th and 95th percentiles, respectively). The estimated ovarian cancer risks were highest for women born between 1930 and 1939,
but the variation across the birth cohorts was smaller than for breast cancer. For _BRCA1_ mutation carriers, the ovarian cancer risk by the age of 70 years was estimated to be 33% for women
born prior to 1920, rising to 36% for those born between 1920 and 1939, and then dropping to 34%. For _BRCA2_ mutation carriers, the corresponding risks were estimated to be 11, 12 and 11%,
respectively. The predicted age-specific FRRs under this model are shown in Table 5. These are generally similar to those generated using piecewise constant hazard ratios, but slightly
closer to the observed values. RISKS OF CANCERS AT OTHER SITES AND RISKS OF SECOND CANCERS Since reliable data on these additional cancer types were not available in the main data set used
to derive BOADICEA, we used instead estimated risks derived from the largest published studies (see Table 7). We incorporated only prostate and pancreatic cancer, and male breast cancer, for
which the evidence for association and estimates of risk were most reliable. We assumed that conditional on the genotype, the age-specific probability of developing a particular cancer was
independent of the probability of developing any other type of cancer. The incidences of prostate and pancreatic cancer for _BRCA1_ and _BRCA2_ mutation carriers were obtained by multiplying
the cohort and calendar period age-specific incidence rates from the general population. Noncarriers were assumed to develop these cancers according to the population incidences. The risk
of breast cancer in female first-degree relatives of male breast cancer patients has been estimated to be 2.4 times greater than the risk in the general population (Basham et al, 2002),
consistent with a common genetic susceptibility to male and female breast cancer. However, _BRCA1_ and _BRCA2_ alone cannot explain all the observed FRRs (Basham et al, 2002). To allow for
this residual effect, we assumed that the polygenic component in the model also applied to male patients. The male breast cancer polygenic variance was chosen such that the predicted FRR to
daughters of male breast cancer patients was equal to 2.4. In choosing this, the overall male breast cancer incidences over the _BRCA1_, _BRCA2_ (Table 7) and polygenic effects were
constrained to agree with the population incidences. On the basis of this approach, the male breast cancer polygenic variance was chosen to be 1.96, close to that estimated for female
patients at young ages. We have also extended the model to allow for the risks of other cancers after the first cancer diagnosis, including the risk of contralateral breast cancer. We
assumed that the increased risk of contralateral breast cancer, or of any other cancer after the first diagnosis (relative to the population rates), was entirely due to the susceptibility as
defined by the model (i.e., no additional variation in risk). On the basis of this assumption, the contralateral breast cancer incidence after the first breast cancer, given the genotype,
is half the breast incidence assumed in the standard model (since only one breast is at risk). Similarly, the incidence of a cancer at another site, after a first cancer diagnosis, was
assumed to be the same as if the preceding cancer had not occurred, consistent with the assumption that the site-specific cancer risks are independent conditional on the genotype. The
transition model for female patients in this extended BOADICEA model is depicted in Figure 5. A similar model applies to male patients (including prostate cancer but excluding ovarian
cancer). DISCUSSION In this report, we have updated and extended our previously published model BOADICEA using additional data. There are several additional features in the updated model.
The breast and ovarian cancer incidences in _BRCA1_ and _BRCA2_ mutation carriers are now based on a much larger number of mutation-carrying families and are, therefore, more reliable; the
variance of the polygenic component is now age dependent as opposed to constant; and the incidences vary gradually with age and are cohort and calendar period specific. In addition, the
model has been extended to allow for the risks of male breast, prostate and pancreatic cancer, and the risks of other cancers after a first diagnosis. We have shown that the updated version,
with a polygenic variance that decreases linearly with age, predicts accurately the FRR of breast cancer observed in epidemiological studies (Collaborative Group in Hormonal Factors in
Breast Cancer, 2001). Such a model is consistent with the hypothesis that a large number of variants increase the rate at which key mutational events occur, and the increased relative risk
conferred by at least some variants is higher at younger ages. Direct evidence for a polygenic component is provided by the recent identification of at least seven common variants through
genome-wide association and candidate gene studies (Cox et al, 2007; Easton et al, 2007; Hunter et al, 2007; Stacey et al, 2007). For the two strongest associations identified by Easton et
al (2007) (_FGFR2_ and _TNRC9_), the per allele odds ratio was higher below the age of 40 years, although not significantly so. Other studies have found that the relative risks associated
with _CHEK2_ 1100delC and _ATM_ mutations are somewhat higher at young ages (The CHEK2 Breast Cancer Case–Control Consortium, 2004; Thompson et al, 2005). We investigated models that allowed
for different polygenic (modifying) variances for _BRCA1_ and _BRCA2_ mutation carriers. The point estimate of the _BRCA1_ variance was lower than of the polygenic variance for noncarriers,
while that of the _BRCA2_ variance was more similar to the latter. This would be consistent with recent findings that the established breast cancer susceptibility variants at the _FGFR2_
and _MAP3K1_ loci are associated with the risk of breast cancer in _BRCA2_ but not _BRCA1_ carriers (Antoniou et al, in press b). The latter observations may reflect differences in
susceptibility by tumour characteristics, since some loci confer susceptibility only to ER-positive disease (Garcia-Closas, in press). However, the power to distinguish between these models,
even with the current large data set, was limited, and we could not reject a model with equal polygenic and modifying variances. This would be consistent with the hypothesis that most
‘polygenes’ confer similar relative risks in _BRCA1_ carriers, _BRCA2_ carriers and noncarriers. As part of the model fitting, we have also re-estimated the breast and ovarian cancer risks
in _BRCA1_ and _BRCA2_ carriers. The majority of the current data came from families of unselected series of cases that had been previously used to estimate the _BRCA1_ and _BRCA2_
penetrance (Antoniou et al, 2003). The present analyses differed in the following three important respects: they were cohort and calendar period specific; they were based on incidences,
which vary continuously with age; and, most importantly, they were estimated while allowing for other residual familial effects (i.e., polygenic-modifying component). Although a direct
comparison with the breast cancer estimates in the original report is not strictly valid, the present breast cancer estimates are generally lower. There is a straightforward explanation for
this difference. The present breast cancer risks (Figures 1, 2, 3 and 4) represent the risks averaged over all possible polygenic and modifying effects and would therefore be applicable to a
randomly chosen _BRCA1_ or _BRCA2_ mutation carrier (i.e., for a carrier without any knowledge of her family history). In contrast, the estimates in Antoniou et al (2003) represent the
breast cancer risks among _BRCA1_ and _BRCA2_ mutation carriers who have an affected first-degree relative (affected with breast cancer in most cases). Under the BOADICEA model, these women
would be expected to have higher than average breast cancer risks, as demonstrated elsewhere (Antoniou et al, 2004) and as other researchers have pointed out (Begg, 2002). An important
consequence of these arguments is that there is no single set of penetrance estimates that applies to all carriers. The range of breast cancer cumulative risks given by the distribution of
the polygenic/modifying component implies that the cancer risks depend strongly on the genotypes at modifying loci – not just the presence of a mutation – and these can be much higher or
much lower than the average estimates. To utilise the full range of these risk estimates would require the modifying genes to be identified, but, in the meantime, the results indicate that
family history in addition to mutation status should be taken into account in genetic counselling. The BOADICEA model allows this level of sophistication. This feature is also consistent
with the recent results of Begg et al (2008), who also demonstrated that breast cancer risks vary between families where a _BRCA1_ or _BRCA2_ mutation has been identified. The updated
version of BOADICEA was one of the models used in a recent validation study of _BRCA1_ and _BRCA2_ carrier prediction algorithms (Antoniou et al, in press a) using a large series of families
seen in the UK genetics clinics. The BOADICEA model was compared against other models including the genetic risk models BRCAPRO (Parmigiani et al, 1998) and IBIS (Tyrer et al, 2004). It was
found to be the most accurate model in terms of predicting the observed number of mutations in total, and across the whole range of probabilities of being a mutation carrier, and had the
highest power to discriminate between mutation carriers and noncarriers. This, taken together with the observation that BOADICEA also predicts well the FRRs of breast cancer, provides
confidence that BOADICEA is a well-validated and well-calibrated model that can be a useful tool for genetic counselling individuals with family history of breast cancer. However, further
validation studies will be important to evaluate the ability of this (and other) models to predict the prospective risk of developing breast or ovarian cancer. The current version of
BOADICEA has now been implemented as a user-friendly Web-based program (http://www.srl.cam.ac.uk/genepi/boadicea/boadicea_home.html). Users can either create a pedigree online, or can upload
a pedigree file. The program allows for families of any size or structure; pedigrees built online are restricted to first- and second-degree relatives but uploaded files can be of arbitrary
complexity. It has been shown that relatives more distant than second degree can provide important information for risk models (Antoniou et al, 2005; Barcenas et al, 2006). The BOADICEA
returns both predicted probabilities of carrying a _BRCA1_ or a _BRCA2_ mutation, and risks (by the age of up to 80 years) of developing breast or ovarian cancer for unaffected individuals,
or the risk of contralateral breast cancer or ovarian cancer for those who have already developed a first breast cancer. The code has also been modified to allow for the possibility that the
individual is of Ashkenazi Jewish origin. This case requires separate consideration owing to the high prevalence of three founder mutations in this population (Satagopan et al, 2001), so
that estimates based on allele frequencies for the United Kingdom would be misleading. For this purpose, we used the _BRCA1_ and _BRCA2_ mutation prevalence for young controls (_BRCA1_: 1.6%
and _BRCA2_: 1.2%) reported in Satagopan et al (2001). The cancer risks among _BRCA1_ and _BRCA2_ mutation carriers, and the polygenic variance, were assumed to be the same in the Ashkenazi
and non-Ashkenazi versions. Similar modification will be required for other populations where the frequencies of _BRCA1_ and _BRCA2_ mutations are different (e.g., the Icelandic). These
will be implemented at a later stage. There are several directions in which our model can be extended, and these may lead to improvements in discriminatory power and more accurate
predictions of cancer risk. Now that some of the loci that may comprise the ‘polygenic’ component have been identified, it will be possible to incorporate their specific effects into the
model, allowing these additional loci to be used in counselling. Other extensions that remain challenges include the incorporation of variation in risk by mutation type; extensions to other
populations with different risks and/or mutation frequencies; inclusion of data on pathological subtypes (for example, ‘basal’ breast cancer); tumour histological characteristics; and the
inclusion of hormonal, reproductive and lifestyle risk factors. CHANGE HISTORY * _ 16 NOVEMBER 2011 This paper was modified 12 months after initial publication to switch to Creative Commons
licence terms, as noted at publication _ REFERENCES * Akaike H (1974) A new look at the statistical model identification. _IEEE Trans Automat Contr_ 19: 716–726 Article Google Scholar *
Anglian Breast Cancer Study Group (2000) Prevalence and penetrance of _BRCA1_ and _BRCA2_ mutations in a population-based series of breast cancer cases. _Br J Cancer_ 83: 1301–1308 Article
PubMed Central Google Scholar * Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, Pasini B, Radice P, Manoukian S, Eccles DM, Tang
N, Olah E, Anton-Culver H, Warner E, Lubinski J, Gronwald J, Gorski B, Tulinius H, Thorlacius S, Eerola H, Nevanlinna H, Syrjakoski K, Kallioniemi OP, Thompson D, Evans C, Peto J, Lalloo F,
Evans DG, Easton DF (2003) Average risks of breast and ovarian cancer associated with _BRCA1_ or _BRCA2_ mutations detected in case series unselected for family history: a combined analysis
of 22 studies. _Am J Hum Genet_ 72: 1117–1130 Article CAS PubMed PubMed Central Google Scholar * Antoniou AC, Durocher F, Smith P, Simard J, Easton DF (2005) _BRCA1_ and _BRCA2_
mutation predictions using the BOADICEA and BRCAPRO models and penetrance estimation in high-risk French-Canadian families. _Breast Cancer Res_ 8: R3 Article PubMed PubMed Central Google
Scholar * Antoniou AC, Hardy R, Walker L, Evans DG, Shenton A, Eeles R, Shanley S, Pichert G, Izatt L, Rose S, Douglas F, Eccles D, Morrison PJ, Scott J, Zimmern RL, Easton DF, Pharoah PDP
. Predicting the likelihood of carrying a _BRCA1_ or _BRCA2_ mutation: validation study of BOADICEA, BRCAPRO, IBIS, Myriad and the Manchester scoring system using data from UK genetics
clinics. _J Med Genet_ (in press a) * Antoniou AC, Pharoah PD, McMullan G, Day NE, Ponder BA, Easton D (2001) Evidence for further breast cancer susceptibility genes in addition to _BRCA1_
and _BRCA2_ in a population-based study. _Genet Epidemiol_ 21: 1–18 Article CAS PubMed Google Scholar * Antoniou AC, Pharoah PD, McMullan G, Day NE, Stratton MR, Peto J, Ponder BJ,
Easton DF (2002) A comprehensive model for familial breast cancer incorporating _BRCA1_, _BRCA2_ and other genes. _Br J Cancer_ 86: 76–83 Article CAS PubMed PubMed Central Google Scholar
* Antoniou AC, Pharoah PP, Smith P, Easton DF (2004) The BOADICEA model of genetic susceptibility to breast and ovarian cancer. _Br J Cancer_ 91: 1580–1590 Article CAS PubMed PubMed
Central Google Scholar * Antoniou AC, Spurdle AB, Sinilnikova OM, Healey S, Pooley KA, Schmutzler RK, Versmold B, Engel C, Meindl A, Arnold N, Hofmann V, Sutter C, Niederacher D, Deissler
H, Caldes T, Kämpjärvi K, Nevanlinna H, Simard J, Beesley J, Chen X, the Kathleen Cuningham Consortium for Research into Familial Breast Cancer, Neuhausen SL, Rebbeck TR, Wagner T, Lynch HT,
Isaacs C, Weitzel J, Ganz PA, Daly MB, Tomlinson G, Olopade OI, Blum JL, Couch FJ, Peterlongo P, Manoukian S, Barile M, Radice P, Szabo CI, Mateus Pereira LH, Greene MH, Rennert G,
Lejbkowicz F, Barnett-Griness O, Andrulis IL, Ozcelik H, OCGN, Gerdes AM, Caligo MA, Laitman Y, Kaufman B, Milgrom R, Friedman E, The Swedish _BRCA1_ and _BRCA2_ Study Collaborators, Domchek
SM, Nathanson KL, Osorio A, Llort G, Milne RL, Benítez J, Hamann U, Hogervorst FBL, Manders P, Ligtenberg MJL, van den Ouweland AMW, The DNA-HEBON Collaborators, Peock S, Cook M, Platte R,
Evans DG, Eeles R, Pichert G, Chu C, Eccles D, Davidson D, Douglas F, EMBRACE, Godwin AK, Barjhoux L, Mazoyer S, Sobol H, Bourdon V, Eisinger F, Chompret A, Capoulade C, Bressac-de
Paillerets B, Lenoir GM, Gauthier-Villars M, Houdayer C, Stoppa-Lyonnet D, GEMO, Chenevix-Trench G, Easton DF on behalf of CIMBA. Common breast cancer predisposition alleles are associated
with breast cancer risk in _BRCA1_ and _BRCA2_ mutation carriers. _Am J Hum Genet_ (in press b) * Barcenas CH, Hosain GM, Arun B, Zong J, Zhou X, Chen J, Cortada JM, Mills GB, Tomlinson GE,
Miller AR, Strong LC, Amos CI (2006) Assessing BRCA carrier probabilities in extended families. _J Clin Oncol_ 24: 354–360 Article PubMed Google Scholar * Basham VM, Lipscombe JM, Ward
JM, Gayther SA, Ponder BA, Easton DF, Pharoah PD (2002) _BRCA1_ and _BRCA2_ mutations in a population-based study of male breast cancer. _Breast Cancer Res_ 4: R2 Article CAS PubMed
Google Scholar * Begg CB (2002) On the use of familial aggregation in population-based case probands for calculating penetrance. _J Natl Cancer Inst_ 94: 1221–1226 Article PubMed Google
Scholar * Begg CB, Haile RW, Borg A, Malone KE, Concannon P, Thomas DC, Langholz B, Bernstein L, Olsen JH, Lynch CF, Anton-Culver H, Capanu M, Liang X, Hummer AJ, Sima C, Bernstein JL
(2008) Variation of breast cancer risk among _BRCA1/2_ carriers. _JAMA_ 299: 194–201 Article CAS PubMed PubMed Central Google Scholar * Brose MS, Rebbeck TR, Calzone KA, Stopfer JE,
Nathanson KL, Weber BL (2002) Cancer risk estimates for _BRCA1_ mutation carriers identified in a risk evaluation program. _J Natl Cancer Inst_ 94: 1365–1372 Article CAS PubMed Google
Scholar * Collaborative Group in Hormonal Factors in Breast Cancer (2001) Familial breast cancer: collaborative reanalysis of individual data from 52 epidemiological studies including 58
209 women with breast cancer and 101 986 women without the disease. _Lancet_ 358: 1389–1399 Article Google Scholar * Cox A, Dunning AM, Garcia-Closas M, Balasubramanian S, Reed MW, Pooley
KA, Scollen S, Baynes C, Ponder BA, Chanock S, Lissowska J, Brinton L, Peplonska B, Southey MC, Hopper JL, McCredie MR, Giles GG, Fletcher O, Johnson N, dos Santos Silva I, Gibson L, Bojesen
SE, Nordestgaard BG, Axelsson CK, Torres D, Hamann U, Justenhoven C, Brauch H, Chang-Claude J, Kropp S, Risch A, Wang-Gohrke S, Schurmann P, Bogdanova N, Dork T, Fagerholm R, Aaltonen K,
Blomqvist C, Nevanlinna H, Seal S, Renwick A, Stratton MR, Rahman N, Sangrajrang S, Hughes D, Odefrey F, Brennan P, Spurdle AB, Chenevix-Trench G, Beesley J, Mannermaa A, Hartikainen J,
Kataja V, Kosma VM, Couch FJ, Olson JE, Goode EL, Broeks A, Schmidt MK, Hogervorst FB, van’t Veer LJ, Kang D, Yoo KY, Noh DY, Ahn SH, Wedren S, Hall P, Low YL, Liu J, Milne RL, Ribas G,
Gonzalez-Neira A, Benitez J, Sigurdson AJ, Stredrick DL, Alexander BH, Struewing JP, Pharoah PD, Easton DF (2007) A common coding variant in CASP8 is associated with breast cancer risk. _Nat
Genet_ 39: 352–358 Article CAS PubMed Google Scholar * Dite GS, Jenkins MA, Southey MC, Hocking JS, Giles GG, McCredie MR, Venter DJ, Hopper JL (2003) Familial risks, early-onset breast
cancer, and _BRCA1_ and _BRCA2_ germline mutations. _J Natl Cancer Inst_ 95: 448–457 Article CAS PubMed Google Scholar * Doll R, Muir CS, Waterhouse JAH (eds) (1970) In _Cancer
Incidence in Five Continents_. Vol. II. UICC: Geneva, Switzerland Book Google Scholar * Doll R, Payne P, Waterhouse J (eds) (1966) In _Cancer Incidence in Five Continents_. Vol. I.
Springer–Verlag: Berlin, Germany Book Google Scholar * Easton DF (1999) How many more breast cancer predisposition genes are there? _Breast Cancer Res_ 1: 14–17 Article CAS PubMed
PubMed Central Google Scholar * Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R, Wareham N, Ahmed S, Healey CS, Bowman R,
Meyer KB, Haiman CA, Kolonel LK, Henderson BE, Le Marchand L, Brennan P, Sangrajrang S, Gaborieau V, Odefrey F, Shen CY, Wu PE, Wang HC, Eccles D, Evans DG, Peto J, Fletcher O, Johnson N,
Seal S, Stratton MR, Rahman N, Chenevix-Trench G, Bojesen SE, Nordestgaard BG, Axelsson CK, Garcia-Closas M, Brinton L, Chanock S, Lissowska J, Peplonska B, Nevanlinna H, Fagerholm R, Eerola
H, Kang D, Yoo KY, Noh DY, Ahn SH, Hunter DJ, Hankinson SE, Cox DG, Hall P, Wedren S, Liu J, Low YL, Bogdanova N, Schurmann P, Dork T, Tollenaar RA, Jacobi CE, Devilee P, Klijn JG,
Sigurdson AJ, Doody MM, Alexander BH, Zhang J, Cox A, Brock IW, MacPherson G, Reed MW, Couch FJ, Goode EL, Olson JE, Meijers-Heijboer H, van den OA, Uitterlinden A, Rivadeneira F, Milne RL,
Ribas G, Gonzalez-Neira A, Benitez J, Hopper JL, McCredie M, Southey M, Giles GG, Schroen C, Justenhoven C, Brauch H, Hamann U, Ko YD, Spurdle AB, Beesley J, Chen X, Mannermaa A, Kosma VM,
Kataja V, Hartikainen J, Day NE, Cox DR, Ponder BA (2007) Genome-wide association study identifies novel breast cancer susceptibility loci. _Nature_ 447: 1087–1093 Article CAS PubMed
PubMed Central Google Scholar * Edwards SM, Kote-Jarai Z, Meitz J, Hamoudi R, Hope Q, Osin P, Jackson R, Southgate C, Singh R, Falconer A, Dearnaley DP, Ardern-Jones A, Murkin A, Dowe A,
Kelly J, Williams S, Oram R, Stevens M, Teare DM, Ponder BA, Gayther SA, Easton DF, Eeles RA (2003) Two percent of men with early-onset prostate cancer harbor germline mutations in the
_BRCA2_ gene. _Am J Hum Genet_ 72: 1–12 Article CAS PubMed Google Scholar * Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE (1994) Risks of cancer in _BRCA1_-mutation carriers. Breast
Cancer Linkage Consortium. _Lancet_ 343: 692–695 Article CAS PubMed Google Scholar * Garcia-Closas M, Hall P, Nevanlinna H, Pooley K, Morrison J, Richesson D, Bojesen SE, Nordestgaard
BG, Axelsson CK, Arias JI, Milne RL, Ribas G, González-Neira A, Benítez J, Zamora P, Brauch H, Justenhoven C, Hamann U, Ko YD, Bruening T, Haas S, Dörk T, Schürmann P, Hillemanns P,
Bogdanova N, Bremer M, Karstens JH, Fagerholm R, Aaltonen K, Aittomäki K, von Smitten K, Blomqvist C, Mannermaa A, Uusitupa M, Eskelinen M, Tengström M, Kosma VM, Kataja V, Chenevix-Trench
G, Spurdle AB, Beesley J, Chen X, Australian Ovarian Cancer Management Group, The Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer, Devilee P, van Asperen
CI, Jacobi CE, Tollenaar RAEM, Huijts P, Klijn J, Chang-Claude J, Kropp S, Slanger T, Flesch-Janys D, Mutschelknauss E, Salazar R, Wang-Gohrke S, Couch F, Goode EL, Olson JE, Vachon C,
Fredericksen ZS, Giles GG, Baglietto L, Severi G, Hopper JL, English DR, Southey MC, Haiman CA, Henderson BE, Kolonel LN, Marchand LL, Stram DO, Hunter DJ, Hankinson SE, Cox DG, Tamimi R,
Kraft P, Sherman M, Chanock S, Lissowska J, Brinton L, Peplonska B, Klijn JGM, Hooning M, Meijers-Heijboer H, Collee JM, van den Ouweland A, Uitterlinden A, Liu J, Lin LY, Yuqing L,
Humphreys K, Czene K, Cox A, Balasubramanian SP, Cross SS, Reed MWR, Blows F, Driver K, Dunning A, Ponder BAJ, Sangrajrang S, Brennan P, McKay J, Odefrey F, Gabrieau V, Sigurdson A, Doody M,
Struewing J, Alexander B, Easton DF, Pharoah PDP . Heterogeneity of breast cancer associations with five susceptibility loci by clinical and pathological characteristics. _PLoS Genet_ (in
press) * Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, Wang J, Yu K, Chatterjee N, Orr N, Willett WC, Colditz GA, Ziegler RG,
Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Hayes RB, Tucker M, Gerhard DS, Fraumeni Jr JF, Hoover RN, Thomas G, Chanock SJ (2007) A genome-wide association study
identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. _Nat Genet_ 39: 870–874 Article CAS PubMed PubMed Central Google Scholar * Kirchhoff T, Kauff
ND, Mitra N, Nafa K, Huang H, Palmer C, Gulati T, Wadsworth E, Donat S, Robson ME, Ellis NA, Offit K (2004) BRCA mutations and risk of prostate cancer in Ashkenazi Jews. _Clin Cancer Res_
10: 2918–2921 Article CAS PubMed Google Scholar * Lalloo F, Varley J, Ellis D, Moran A, O’Dair L, Pharoah P, Evans DG (2003) Prediction of pathogenic mutations in patients with
early-onset breast cancer by family history. _Lancet_ 361: 1101–1102 Article CAS PubMed Google Scholar * Lange K (2002) _Mathematical and Statistical Methods for Genetic Analysis_.
Springer-Verlang: New York Book Google Scholar * Muir C, Waterhouse J, Mack T, Powell J, Whelan S (eds) (1987) In _Cancer Incidence in Five Continents_. Vol. V. IARC Scientific
Publications: Lyon, France Google Scholar * Office for National Statistics (2001) _Cancer Trends in England and Wales 1950–1999: Studies on Medical and Population Subjects No. 66_. Quinn M,
Babb P, Brock A, Kirby L, Jones J (eds). The Stationary Office: London Google Scholar * Parkin DM, Muir CS, Whelan SL, Gao YT, Ferlay J, Powell J (eds) (1992) In _Cancer Incidence in Five
Continents_. Vol. VI. IARC Scientific Publications: Lyon, France Google Scholar * Parkin DM, Whelan SL, Ferlay J, Teppo L, Thomas DB (eds) (2002) In _Cancer Incidence in Five Continents_.
Vol. VIII. IARC Scientific Publications: Lyon, France Google Scholar * Parmigiani G, Berry D, Aguilar O (1998) Determining carrier probabilities for breast cancer-susceptibility genes
_BRCA1_ and _BRCA2_. _Am J Hum Genet_ 62: 145–158 Article CAS PubMed PubMed Central Google Scholar * Peto J, Collins N, Barfoot R, Seal S, Warren W, Rahman N, Easton DF, Evans C, Deacon
J, Stratton MR (1999) Prevalence of _BRCA1_ and _BRCA2_ gene mutations in patients with early-onset breast cancer. _J Natl Cancer Inst_ 91: 943–949 Article CAS PubMed Google Scholar *
Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, Evans DG, Eccles D, Easton DF, Stratton MR (2007)
PALB2, which encodes a _BRCA2_-interacting protein, is a breast cancer susceptibility gene. _Nat Genet_ 39: 165–167 Article CAS PubMed Google Scholar * Renwick A, Thompson D, Seal S,
Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N (2006) ATM mutations that cause
ataxia-telangiectasia are breast cancer susceptibility alleles. _Nat Genet_ 38: 873–875 Article CAS PubMed Google Scholar * Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Fan I,
Tang J, Li S, Zhang S, Shaw PA, Narod SA (2006) Population _BRCA1_ and _BRCA2_ mutation frequencies and cancer penetrances: a kin–cohort study in Ontario, Canada. _J Natl Cancer Inst_ 98:
1694–1706 Article CAS PubMed Google Scholar * Royston P (1991) Lowess smoothing. _Stata Tech Bull_ 3: 7–9 Google Scholar * Satagopan JM, Offit K, Foulkes W, Robson ME, Wacholder S, Eng
CM, Karp SE, Begg CB (2001) The lifetime risks of breast cancer in Ashkenazi Jewish carriers of _BRCA1_ and _BRCA2_ mutations. _Cancer Epidemiol Biomarkers Prev_ 10: 467–473 CAS PubMed
Google Scholar * Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, North B, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR,
Rahman N (2006) Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. _Nat Genet_ 38: 1239–1241 Article CAS PubMed Google
Scholar * Smith P, McGuffog L, Easton DF, Mann GJ, Pupo GM, Newman B, Chenevix-Trench G, Szabo C, Southey M, Renard H, Odefrey F, Lynch H, Stoppa-Lyonnet D, Couch F, Hopper JL, Giles GG,
McCredie MR, Buys S, Andrulis I, Senie R, Goldgar DE, Oldenburg R, Kroeze-Jansema K, Kraan J, Meijers-Heijboer H, Klijn JG, van Asperen C, van Leeuwen I, Vasen HF, Cornelisse CJ, Devilee P,
Baskcomb L, Seal S, Barfoot R, Mangion J, Hall A, Edkins S, Rapley E, Wooster R, Chang-Claude J, Eccles D, Evans DG, Futreal PA, Nathanson KL, Weber BL, Rahman N, Stratton MR (2006) A genome
wide linkage search for breast cancer susceptibility genes. _Genes Chromosomes Cancer_ 45: 646–655 Article CAS PubMed PubMed Central Google Scholar * Stacey SN, Manolescu A, Sulem P,
Rafnar T, Gudmundsson J, Gudjonsson SA, Masson G, Jakobsdottir M, Thorlacius S, Helgason A, Aben KK, Strobbe LJ, Albers-Akkers MT, Swinkels DW, Henderson BE, Kolonel LN, Le Marchand L,
Millastre E, Andres R, Godino J, Garcia-Prats MD, Polo E, Tres A, Mouy M, Saemundsdottir J, Backman VM, Gudmundsson L, Kristjansson K, Bergthorsson JT, Kostic J, Frigge ML, Geller F,
Gudbjartsson D, Sigurdsson H, Jonsdottir T, Hrafnkelsson J, Johannsson J, Sveinsson T, Myrdal G, Grimsson HN, Jonsson T, von Holst S, Werelius B, Margolin S, Lindblom A, Mayordomo JI, Haiman
CA, Kiemeney LA, Johannsson OT, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K (2007) Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive
breast cancer. _Nat Genet_ 39: 865–869 Article CAS PubMed Google Scholar * Tai YC, Domchek S, Parmigiani G, Chen S (2007) Breast cancer risk among male _BRCA1_ and _BRCA2_ mutation
carriers. _J Natl Cancer Inst_ 99: 1811–1814 Article CAS PubMed Google Scholar * The Breast Cancer Linkage Consortium (1999) Cancer risks in _BRCA2_ mutation carriers. _J Natl Cancer
Inst_ 91: 1310–1316 Article Google Scholar * The CHEK2 Breast Cancer Case–Control Consortium (2004) CHEK2*1100delC and susceptibility to breast cancer: a collaborative analysis involving
10 860 breast cancer cases and 9065 controls from 10 studies. _Am J Hum Genet_ 74: 1175–1182 Article PubMed Central Google Scholar * Thompson D, Duedal S, Kirner J, McGuffog L, Last J,
Reiman A, Byrd P, Taylor M, Easton DF (2005) Cancer risks and mortality in heterozygous ATM mutation carriers. _J Natl Cancer Inst_ 97: 813–822 Article CAS PubMed Google Scholar *
Thompson D, Easton D (2001) Variation in cancer risks, by mutation position, in _BRCA2_ mutation carriers. _Am J Hum Genet_ 68: 410–419 Article CAS PubMed PubMed Central Google Scholar
* Thompson D, Easton DF (2002) Cancer Incidence in _BRCA1_ mutation carriers. _J Natl Cancer Inst_ 94: 1358–1365 Article CAS PubMed Google Scholar * Thorlacius S, Sigurdsson S,
Bjarnadottir H, Olafsdottir G, Jonasson JG, Tryggvadottir L, Tulinius H, Eyfjord JE (1997) Study of a single _BRCA2_ mutation with high carrier frequency in a small population. _Am J Hum
Genet_ 60: 1079–1084 CAS PubMed PubMed Central Google Scholar * Tryggvadottir L, Sigvaldason H, Olafsdottir GH, Jonasson JG, Jonsson T, Tulinius H, Eyfjord JE (2006) Population-based
study of changing breast cancer risk in Icelandic _BRCA2_ mutation carriers, 1920–2000. _J Natl Cancer Inst_ 98: 116–122 Article CAS PubMed Google Scholar * Tryggvadottir L, Vidarsdottir
L, Thorgeirsson T, Jonasson JG, Olafsdottir EJ, Olafsdottir GH, Rafnar T, Thorlacius S, Jonsson E, Eyfjord JE, Tulinius H (2007) Prostate cancer progression and survival in _BRCA2_ mutation
carriers. _J Natl Cancer Inst_ 99: 929–935 Article CAS PubMed Google Scholar * Tulinius H, Olafsdottir GH, Sigvaldason H, Arason A, Barkardottir RB, Egilsson V, Ogmundsdottir HM,
Tryggvadottir L, Gudlaugsdottir S, Eyfjord JE (2002) The effect of a single _BRCA2_ mutation on cancer in Iceland. _J Med Genet_ 39: 457–462 Article CAS PubMed PubMed Central Google
Scholar * Tyrer J, Duffy SW, Cuzick J (2004) A breast cancer prediction model incorporating familial and personal risk factors. _Stat Med_ 23: 1111–1130 Article PubMed Google Scholar *
van Asperen CJ, Brohet RM, Meijers-Heijboer EJ, Hoogerbrugge N, Verhoef S, Vasen HF, Ausems MG, Menko FH, Gomez Garcia EB, Klijn JG, Hogervorst FB, van Houwelingen JC, van’t Veer LJ, Rookus
MA, van Leeuwen FE (2005) Cancer risks in _BRCA2_ families: estimates for sites other than breast and ovary. _J Med Genet_ 42: 711–719 Article CAS PubMed PubMed Central Google Scholar *
Waterhouse JAH, Muir C, Correa P, Powell J (eds) (1976) In _Cancer Incidence in Five Continents_. Vol. III. IARC Scientific Publications: Lyon, France Google Scholar * Waterhouse JAH, Muir
C, Shanmugaratnam K, Powell J (eds) (1982) In _Cancer Incidence in Five Continents_. Vol. IV. IARC Scientific Publications: Lyon, France Google Scholar Download references ACKNOWLEDGEMENTS
ACA, APC, the BOADICEA development and the SEARCH study were funded by Cancer Research UK. PDPP is a Cancer Research UK Senior Clinical research fellow. DFE is a Cancer Research UK
principal research fellow. The ABCFS was supported by grants from the National Health and Medical Research Council (NHMRC), the Victorian Health Promotion Foundation and the New South Wales
Cancer Council. MCS is an NHMRC senior research fellow and JLH is an Australia fellow of the NHMRC and a Victorian Breast Cancer Research Consortium group leader. The Helsinki Breast Cancer
Study (HEBCS) was supported by the Helsinki University Central Hospital Research Fund, Academy of Finland (110663), Finnish Cancer Society and Sigrid Juselius Fund. We thank Dr Carl
Blomqvist for his kind help with the patient collection. HA Risch was supported, in part, by National Cancer Institute Grant R01 CA 63682, and SA Narod was supported, in part, by National
Cancer Institute Grant R01 CA 63678. JEE/Icelandic study was funded by a US DOD grant and the Icelandic Cancer Society and supported in part by Hungarian Research Grant OTKA T-046570. We
acknowledge the contribution of the Italian Foundation for Cancer Research (FIRC) to our work. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Public Health and Primary Care,
Cancer Research UK, Genetic Epidemiology Unit, University of Cambridge, Cambridge, UK A C Antoniou, A P Cunningham & D F Easton * Section of Epidemiology, Institute of Cancer Research,
Sutton, UK J Peto * Section of Epidemiology, London School of Hygiene and Tropical Medicine, London, UK J Peto * Academic Unit of Medical Genetics and Regional Genetics Service, St
Mary's Hospital, Manchester, UK D G Evans & F Lalloo * Centre for Research on Women's Health, University of Toronto, Toronto, Canada S A Narod & E Warner * Department of
Epidemiology and Public Health, Yale University School of Medicine, New Haven, CT, USA H A Risch * Faculty of Medicine, University of Iceland, Reykjavík, Iceland J E Eyfjord & L
Tryggvadottir * Icelandic Cancer Society, Reykjavík, Iceland J E Eyfjord & L Tryggvadottir * Centre for Molecular, Environmental, Genetic and Analytic Epidemiology, The University of
Melbourne, Melbourne, Victoria, Australia J L Hopper * Department of Pathology, Genetic Epidemiology Laboratory, The University of Melbourne, Melbourne, Victoria, Australia M C Southey *
Department of Oncology, Lund University Hospital, Lund, Sweden H Olsson, O Johannsson & A Borg * Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy B Passini, P Radice & S
Manoukian * IFOM Fondazione Istituto FIRC di Oncologia Molecolare, Milan, Italy P Radice * Wessex Clinical Genetics Service, Princess Anne Hospital, Southampton, UK D M Eccles * Department
of Chemical Pathology, The Chinese University of Hong Kong, Hong Kong, People's Republic of China N Tang * National Institute of Oncology, Budapest, Hungary E Olah * Epidemiology
Division, Department of Medicine, University of California – Irvine, Irvine, CA, USA H Anton-Culver * Department of Genetics and Pathology, International Hereditary Cancer Centre, Pomeranian
Academy of Medicine, Szczecin, Poland J Lubinski, J Gronwald & B Gorski * Laboratory of Cancer Genetics, Institute of Medical Technology, Tampere University Hospital, Tampere, Finland K
Syrjakoski & O-P Kallioniemi * Department of Obstetrics and Gynecology, Helsinki University Central Hospital, Helsinki, Finland H Eerola & H Nevanlinna * Department of Oncology,
Cancer Research UK, Human Cancer Genetics Group, University of Cambridge, Cambridge, UK P D P Pharoah Authors * A C Antoniou View author publications You can also search for this author
inPubMed Google Scholar * A P Cunningham View author publications You can also search for this author inPubMed Google Scholar * J Peto View author publications You can also search for this
author inPubMed Google Scholar * D G Evans View author publications You can also search for this author inPubMed Google Scholar * F Lalloo View author publications You can also search for
this author inPubMed Google Scholar * S A Narod View author publications You can also search for this author inPubMed Google Scholar * H A Risch View author publications You can also search
for this author inPubMed Google Scholar * J E Eyfjord View author publications You can also search for this author inPubMed Google Scholar * J L Hopper View author publications You can also
search for this author inPubMed Google Scholar * M C Southey View author publications You can also search for this author inPubMed Google Scholar * H Olsson View author publications You can
also search for this author inPubMed Google Scholar * O Johannsson View author publications You can also search for this author inPubMed Google Scholar * A Borg View author publications You
can also search for this author inPubMed Google Scholar * B Passini View author publications You can also search for this author inPubMed Google Scholar * P Radice View author publications
You can also search for this author inPubMed Google Scholar * S Manoukian View author publications You can also search for this author inPubMed Google Scholar * D M Eccles View author
publications You can also search for this author inPubMed Google Scholar * N Tang View author publications You can also search for this author inPubMed Google Scholar * E Olah View author
publications You can also search for this author inPubMed Google Scholar * H Anton-Culver View author publications You can also search for this author inPubMed Google Scholar * E Warner View
author publications You can also search for this author inPubMed Google Scholar * J Lubinski View author publications You can also search for this author inPubMed Google Scholar * J
Gronwald View author publications You can also search for this author inPubMed Google Scholar * B Gorski View author publications You can also search for this author inPubMed Google Scholar
* L Tryggvadottir View author publications You can also search for this author inPubMed Google Scholar * K Syrjakoski View author publications You can also search for this author inPubMed
Google Scholar * O-P Kallioniemi View author publications You can also search for this author inPubMed Google Scholar * H Eerola View author publications You can also search for this author
inPubMed Google Scholar * H Nevanlinna View author publications You can also search for this author inPubMed Google Scholar * P D P Pharoah View author publications You can also search for
this author inPubMed Google Scholar * D F Easton View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to A C Antoniou.
APPENDIX APPENDIX To obtain _BRCA1_ and _BRCA2_ incidences as continuous functions of age, we fitted models in which the log-relative hazards (_G_(_t_)) were piecewise linear functions of
age. For example, RIGHTS AND PERMISSIONS From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0
Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Antoniou, A.,
Cunningham, A., Peto, J. _et al._ The BOADICEA model of genetic susceptibility to breast and ovarian cancers: updates and extensions. _Br J Cancer_ 98, 1457–1466 (2008).
https://doi.org/10.1038/sj.bjc.6604305 Download citation * Received: 30 November 2007 * Revised: 14 February 2008 * Accepted: 21 February 2008 * Published: 18 March 2008 * Issue Date: 22
April 2008 * DOI: https://doi.org/10.1038/sj.bjc.6604305 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable
link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * _BRCA1_ * _BRCA2_ * cancer risk model *
genetic testing