- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Epigenetic alterations in the 11p15.5 imprinted gene cluster are frequent in human cancers and are associated with disordered imprinting of insulin-like growth factor (_IGF_)_2_ and
_H19_. Recently, an imprinted gene cluster at 14q32 has been defined and includes two closely linked but reciprocally imprinted genes, _DLK1_ and _GTL2_, that have similarities to _IGF2_
and _H19_, respectively. Both _GTL2_ and _H19_ are maternally expressed RNAs with no protein product and display paternal allele promoter region methylation, and _DLK1_ and _IGF2_ are both
paternally expressed. To determine whether methylation alterations within the 14q32 imprinted domain occur in human tumorigenesis, we investigated the status of the _GTL2_ promoter
differentially methylated region (DMR) in 20 neuroblastoma tumours, 20 phaeochromocytomas and, 40 Wilms' tumours. Hypermethylation of the _GTL2_ promoter DMR was detected in 25% of
neuroblastomas, 10% of phaeochromocytoma and 2.5% of Wilms' tumours. Tumours with _GTL2_ promoter DMR hypermethylation also demonstrated hypermethylation at an upstream intergenic DMR
thought to represent a germline imprinting control element. Analysis of neuroblastoma cell lines revealed that _GTL2_ DMR hypermethylation was associated with transcriptional repression of
_GTL2_. These epigenetic findings are similar to those reported in Wilms' tumours in which _H19_ repression and DMR hypermethylation is associated with loss of imprinting (LOI,
biallelic expression) of _IGF2_. However, a neuroblastoma cell line with hypermethylation of the _GTL2_ promoter and intergenic DMR did not show LOI of _DLK1_ and although treatment with a
demethylating agent restored _GTL2_ expression and reduced _DLK1_ expression. As described for _IGF2_/_H19_, epigenetic changes at _DLK1_/_GTL2_ occur in human cancers. However, these
changes are not associated with _DLK1_ LOI highlighting differences in the imprinting control mechanisms operating in the _IGF2-H19_ and _DLK1-GTL2_ domains. GTL2 promoter and intergenic DMR
hypermethylation is associated with the loss of _GTL2_ expression and this may contribute to tumorigenesis in a subset of human cancers. SIMILAR CONTENT BEING VIEWED BY OTHERS ATLAS OF
IMPRINTED AND ALLELE-SPECIFIC DNA METHYLATION IN THE HUMAN BODY Article Open access 11 March 2025 GENOMIC IMPRINTING IN MOUSE BLASTOCYSTS IS PREDOMINANTLY ASSOCIATED WITH H3K27ME3 Article
Open access 21 June 2021 GENETIC AND EPIGENETIC FEATURES OF BILATERAL WILMS TUMOR PREDISPOSITION IN PATIENTS FROM THE CHILDREN’S ONCOLOGY GROUP AREN18B5-Q Article Open access 18 December
2023 MAIN Genomic imprinting is a process whereby imprinted genes demonstrate parent-of-origin differences in allelic expression. Although only a minority of genes are imprinted, many of
those identified to date have a role in the regulation of cell growth and differentiation and aberrant imprinting frequently leads to abnormal pre- and/or postnatal growth (Reik and Walter,
2001). Furthermore, disordered imprinting has been implicated in the pathogenesis of paediatric and adult cancers. Imprinted genes frequently occur in clusters. In particular, the imprinted
gene cluster at 11p15.5, which contains the paternally expressed insulin-like growth factor (_IGF_)_2_ and the maternally expressed _CDKN1C_ and _H19_ genes has been implicated in disorders
of growth and in neoplasia (Maher and Reik, 2000; Feinberg et al, 2002). For example, germline-inactivating mutations in _CDKN1C_ and epigenetic alterations leading to the loss of imprinting
(LOI) (biallelic expression) of _IGF2_ and silencing of _CDKN1C_ or _H19_ cause Beckwith–Wiedemann syndrome, a congenital overgrowth disorder associated with susceptibility to embryonal
tumours (Maher and Reik, 2000; Weksberg et al, 2003). In addition, epigenetic alterations at _IGF2_ and _H19_ have been implicated in the pathogenesis of sporadic childhood (e.g. Wilms'
tumour) and adult (e.g. colorectal) cancers (Cui et al, 2001; Nakagawa et al, 2001). _IGF2_ and _H19_ are closely linked and reciprocally imprinted, and epigenetic alterations in human
cancers at an intergenic differentially methylated region (DMR) are associated with LOI of _IGF2_ and silencing at _H19_. In the past decade, the imprinted gene clusters at 11p15.5 and 15q13
(Prader-Willi and Angelman syndrome region) have been the subject of intense investigation. Recently, an imprinted gene cluster at 14q32 (distal mouse chromosome 12) was identified. The
first imprinted 14q32 gene identified was _GTL2_/_Gtl2_, which, like _H19_, is a maternally expressed RNA with no apparent open reading frame (Miyoshi et al, 2000). Subsequently, a
paternally expressed gene, _DLK1_/_Dlk1_, which encodes an EGF-like membrane protein similar to the _Drosophila Delta_ gene was identified upstream of _GTL_/_Gtl2_ (Kobayashi et al, 2000;
Schmidt et al, 2000; Takada et al, 2000; Wylie et al, 2000). There are a number of similarities between the _IGF2_/_H19_ and the _DLK1_/_GTL2_ domains. In both cases, two closely linked
genes are similarly organised, reciprocally imprinted and developmentally regulated; _H19_ and _GTL2_ do not encode known proteins, and paternally methylated DMRs associated with both genes
have been implicated in the regulation of their imprinting (Paulsen et al, 2001; Takada et al, 2002). In sheep, a single-nucleotide (nt) substitution is associated with altered postnatal
expression of _GTL2_ and _DLK1_, although their imprinting is not perturbed. This mutation results in the Callipyge muscle hypertrophy phenotype (Charlier et al, 2001; Freking et al, 2002;
Georges et al, 2003). In human mutations or epigenetic alterations of _DLK1_ and _GTL2_ have not been described, although distinct phenotypes associated with maternal and paternal disomy for
chromosome 14 are reported (Georgiades et al, 2000; Eggermann et al, 2001; Kurosawa et al, 2002). SAGE analysis demonstrated high expression of _DLK1_ in a subset of neuroblastoma (van
Limpt et al, 2000), but a follow-up study did not find any genetic alterations (e.g. mutation, duplication or rearrangement) or LOI in neuroblastomas with _DLK1_ overexpression (van Limpt et
al, 2003). In addition, downregulation of _GTL2_ has been reported in pituitary adenomas with ectopic expression of _GTL2_ inhibiting tumour growth _in vitro_, suggesting a tumour
suppressor function for that gene (Zhang et al, 2003). We have investigated neuroblastoma and other tumours to determine whether epigenetic alterations within the _DLK1-GTL2_ imprinted
domain are a feature of human neoplasia. We investigated neuroblastoma and phaeochromocytoma as _DLK1_ is highly expressed in the adrenal medulla. Neuroblastoma may also demonstrate 14q
allele loss and Wilms' tumour was of interest as epigenetic alterations at the _IGF2/H19_ locus are common in this tumour. MATERIALS AND METHODS MULTIPLEX METHYLATION POLYMERASE CHAIN
REACTION (MPCR) ASSAY Methylation-specific PCR with oligonucleotide primers specific for the methylated and unmethylated copies of the _GTL2_ promoter on chromosome 14q32 were performed
using previously published primers (Murphy et al, 2003): MSM-UF (5′-GAG GAT GGT TAG TTA TTG GGG T-3′; nt position 65912 (GenBank accession no. AL117190)); MSM-UR (5′-CCA CCA TAA CCA ACA CCC
TAT AAT CAC A-3′; nt 65931–66004); MSM-MF (5′-GTT AGT AAT CGG GTT TGT CGG C-3′; nt 64450–64471) and MSM-MR (5′-AAT CAT AAC TCC GAA CAC CCG CG-3′; nt 64587–64609). The multiplex mPCR
conditions used were initial denaturation at 95°C for 3 min; five cycles of 95°C for 30 s, 70°C for 30 s, 72°C for 30 s; five cycles of 95°C for 30 s, 65°C for 30 s, 72°C for 30 s; followed
by 30 cycles of 95°C for 30 s, 60°C for 30 s, 72°C for 30 s and a final extension of 72°C for 5 min. PCR amplification were performed using 0.05 U _μ_l−1 _Taq_ polymerase in (NH4)2SO4 buffer
with 3.0 mM MgCl2 (MBI Fermentas, St Leon-Rot, Germany). PCR products (160 bp for methylated allele and 120 bp for unmethylated) were separated on a 3% agarose gel, stained with ethidium
bromide and visualised under UV illumination. ANALYSIS OF DNA METHYLATION USING BISULPHITE SEQUENCING Genomic DNA was treated with sodium bisulphite using previously published method (Herman
et al, 1996). DNA (0.5–1 _μ_g) was denatured at 37°C for 10 min in 0.3 M NaOH followed by sulphonation of unmethylated cytosines by incubation in 3.12 M sodium bisulphite containing 1 M
hydroquinone (pH 5) at 95°C for 30 s and 50°C for 15 min for 20 cycles. The resulting sulphonated DNA was purified using the Wizard DNA clean-up system (Promega, Southampton, UK) according
to the manufacturer's instructions and DNA was eluted with 50 _μ_l of distilled water. Following elution, DNA was desulphonated in 0.3 M NaOH for 5 min at room temperature, ethanol
precipitated and resuspended in 50 _μ_l distilled water. METHYLATION STATUS OF _GTL2_ AND _DLK1_ DMRS Following multiplex PCR methylation screening (see above), the methylation status of
three areas of the CpG-rich region upstream of _GTL2_ were analysed in detail (G1, position 65897–66197; G2, position 66541–66920; G3, position 67541–67910; GeneBank accession no. AL117190;
relative to _GTL2_ transcription start site, G1=−363 bp → −161 bp upstream; G2=+293 bp → +673 bp downstream; G3=+1213 bp → +1583 bp downstream _GTL2_ transcription start site; GeneBank
accession no. AY314975) and a region upstream _DLK1_ promoter (D1, position 140551–140810; GeneBank accession no. AL132711) and two downstream regions (D2, position 141271–141420; D3,
position 141571–141894; GeneBank accession no. AL132711). Except for D1 and D2 regions, bisulphite-modified DNA was amplified using two rounds of nested PCR (95°C for 15 min followed by 35
cycles of 95°C for 30 s, 55–58°C for 30 s, 72°C for 30 s and a final extension of 72°C for 5 min) using HotStar _Taq_ DNA polymerase (Qiagen, Crawley, UK). Primer sets were designed to
amplify DNA fragments containing both methylated and unmethylated CpG dinucleotides. The primer sequences are: G1F (5′-TTA GGT GTG GGA TTT GYG TTT YGA TAG TT-3′); G1R (5′-CAA AAA AAA TAA TCT
CTA ACR TCA ACR CAT TCT ACT A-3′); G1FN (5′-GGT TAT TGG TYG TTT GAG GAY GGT TAG TT-3′); G2F (5′-TTA GGG TTT TTT TTT GGA GGG TTT AGT-3′); G2R (5′-AAA ACT AAT CCA TAA AAA CTA CTA ACA AAT-3′);
G2RN (5′-ACC TAA AAT CCA CAC TAC ACT AAA CCT ATA-3′); G3F (5′-AGA GGG AAT AGT TTT GAG ATT TTT YGG ATT TAT-3′); G3R (5′-ATC CTC CAA ACA CCR CTA TCA CRC ATA TAA-3′); G3RN (5′-ATA ATC TCR AAA
CRA AAA ACA AAA CCT ATA-3′); D1F (5′-TAT ATA GTG GGT ATT TTA ATT GTT TTT TAT-3′); D1R (5′-TAA AAA AAC AAA CCC ATA AAC ATC CCC AAA-3′); D2F (5′-TTG GTA ATT AGT ATT TTT TAT TTT TA-3′); D2R
(5′-ACT TTT ATC ACA AAT AAC ATA CAT AAA C-3′); D3F (5′-TTG TTT ATG TAT GTT ATT GTG GAT-3′); D3R (5′-TAA AAT CCC RAA CAC ACR TAC AAT AAT-3′); D3FN (5′-GTT AAG GTT TTG ATT GAG ATG TTG TGT
G-3′); and D3RN (5′-TAT ACC CCT AAC CAT AAA AAC RCA AA-3′). Amplification products were purified from agarose gel using QiaQuick gel extraction kit (Qiagen, Crawley, UK), cloned into pGem
T-Easy vector system (Promega, Southampton, UK). In all, eight to 10 individual clones for each DMR regions were sequenced using BigDye terminator cycle sequencing kit V 1.1/3.1
(Perkin-Elmer/Applied Biosystems, Warrington, UK) and run on an ABI377/3730. METHYLATION STATUS OF THE INTERGENIC GERMLINE-DERIVED DIFFERENTIALLY METHYLATED REGION (IG-DMR) A seminested PCR
was performed to amplify the IG-DMR (nt position 51021–51180; GeneBank accession no. AL117190). The primer sequences are: IG-F (5′-TTT TGA GGA GAT TGA TAT TTT TAG TTT TAT T-3′); IG-R (5′-ATA
AAC TAC ACT ACT AAA AAC TAC ATT TAA A-3′); and IG-Fnes (5′-TTA GGT TGG AAT TGT TAA GAG TTT GTG GAT T-3′). PCR was performed as previously described using HotStar _Taq_ DNA polymerase
(Qiagen) with an annealing temperature of 53°C and 1.5 mM MgCl2 for both first-round (primer set IG-F/IG-R) and second-round (primer set IG-Fnes/IG-R) PCRs. PCR products were then cloned and
sequenced as described above. EXPRESSION ANALYSIS: REVERSE TRANSCRIPTASE (RT) AND QUANTITATIVE REAL-TIME PCR RNA (1 _μ_g) was reverse transcribed using Reverse Transcription Systems and
oligo-dT primers (Promega) according to the manufacturer's protocols. cDNA (1 _μ_l) obtained was then used as template for RT–PCR amplification. A single-nucleotide polymorphism (SNP)
previously identified in exon 5 of _GTL2_ and an SNP in exon 5 of _DLK1_ (Wylie et al, 2000) were used to analyse _GTL2_ and _DLK1_ gene expression. _GTL2_ and _DLK1_ cDNA were amplified
using oligonucleotide primers described by Wylie et al (2000), except that for _GTL2_ reverse primer, GTL2RK (5′-TTC CAC GGA GTA GAG CGA GTC A-3′), was used. Amplification products were
purified and sequenced as described above using primers: GTL2FS (5′-ATC CCT TTG GGA AAT TCT CAG G-3′) and DLK1FS (5′-AGG CAC CTG CGT GGA TGA T-3′). For quantitative real-time PCR, total RNA
from cell lines was extracted with the RNAzol B (Biogenesis, Poole, UK) according to the manufacturer's instruction and treated with DNAse 1 (Invitrogen, Paisley, UK). Total RNA (1
_μ_g) was reverse transcribed as described previously. The cDNA was used in triplicate in a real-time PCR analysis using a real-time Thermal Cycler Model 7900 (Applied Biosystems). The
primer sets are: _DLK1_ (5′-GCG AGG ATG ACA ATG TTT GC-3′ (forward) and 5′-AGC AGG CCC GAA CAT CTC TA-3′ (reverse)) and _GTL2_ (5′-ATC AGC CAA GCT TCT TGG AA-3′ (forward) and 5′-AGC TTC CAT
CCG CAG TTC T-3′ (reverse)). Beta-actin was used as a control for normalisation. PCR was performed in a 25 _μ_l volume that included 12.5 _μ_l of 2 × Cyber green (Applied Biosystems), 25 ng
of cDNA template and 0.0125–0.025 pmol of each primer. Relative RNA quantification was performed using the comparative _C_T method. The fold differences in RNA expression are the mean value
from three independent observations. TREATMENT OF CELL LINES WITH 5-AZA-2′-DEOXYCYTIDINE (5-AZA-DC) 5-Aza-dC (Sigma, Gillingham, Dorset, UK) was freshly prepared in ddH2O at 2 mg ml−1 and
filter sterilised. A total of 1 × 106 cells were plated in 75 cm2 flask in RPMI 1640 medium supplemented with 10% FCS and left to settle for 24 h (day 0). Cells were treated with 2 _μ_ M of
5-aza-dC at day 1 and 4 and harvested at day 5. The culture medium was changed before each treatment and 24 h after treatment. RESULTS EPIGENETIC ANALYSIS OF _DLK1/GTL2_ DOMAIN IN
NEUROBLASTOMA TUMOURS AND CELL LINES The methylation status of the _GTL2_ promoter DMR was assessed in four normal control blood DNA samples and all four cases demonstrated both methylated
and unmethylated allele products. We then analysed tumour DNA from 20 primary neuroblastomas. We found that five of 20 (25%) neuroblastoma tumours demonstrated only products from a
methylated allele at the _GTL2_ promoter DMR. The other 15 tumours showed a normal result with methylated and unmethylated allele products. In four of five tumours with hypermethylation of
the _GTL2_ promoter DMR, heterozygosity at a _DLK1_ SNP (c.564T>C) (_n_=−2) or at closely linked microsatellite markers (D14S598, D14S1426, D14S749 and D14S1006) excluded 14q32 maternal
allele loss as a cause of the _GTL2_ promoter DMR hypermethylation. In order to investigate whether _GTL2_ promoter DMR hypermethylation in tumours was associated with alterations in _DLK_
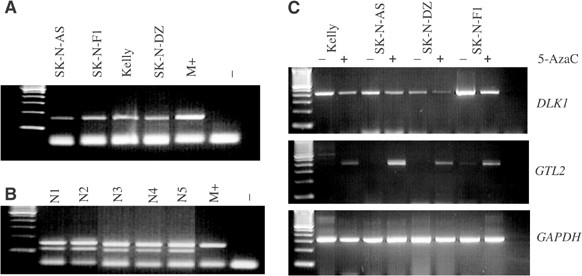
and _GTL2_ expression and imprinting, we examined the _GTL2_ promoter DMR methylation status in four neuroblastoma cell lines (SK-N-F1, SK-N-AS, SK-N-DZ, Kelly). All four neuroblastoma cell
lines demonstrated _GTL2_ promoter DMR hypermethylation (Figure 1A and B). One cell line, SK-N-AS, was heterozygous for a transcribed _DLK1_ SNP, thus enabling us to (a) exclude maternal
allele loss as the cause of 5′ _GTL2_ DMR hypermethylation and (b) perform allele-specific expression studies to investigate _DLK1_ imprinting status. We also analysed _DLK1_ and _GTL2_
expression before and after treatment with the demethylating agent 5-azacytidine (5-AzaC) in the four neuroblastoma cell lines with _GTL2_ promoter DMR hypermethylation. In all cases, there
was silencing of _GTL2_ expression in the untreated cell line and reactivation of _GTL2_ expression after treatment with 5-AzaC (Figure 1C and Table 1). In addition, 5-AzaC treatment
resulted in a reduction in _DLK1_ mRNA expression. Analysis of _DLK1_ allelic expression in the informative SK-N-AS neuroblastoma line demonstrated monoallelic (allele C) _DLK1_ expression
pre- and post-treatment with 5-AzaC (Figure 2). Thus, hypermethylation of the _GTL2_ promoter DMR in the SK-N-AS was associated with the upregulation of _DLK1_ expression and silencing of
_GTL2_ expression, but not LOI of _DLK1_. Hypermethylation of 5′ _GTL2_ promoter was observed in five of 20 (25%) of neuroblastoma tumours. RNA was available from seven of the 20 tumours
(see Figure 3). A low level of _GTL2_ expression was detected in the tumour with hypermethylation of the 5′ _GTL2_ promoter DMR (possibly caused by normal tissue contamination). To establish
the precise epigenetic status of individual CpGs within the _GTL2_ promoter DMR region, bisulphite sequencing was undertaken in four normal control blood DNA samples and in the SK-N-AS
hypermethylated neuroblastoma cell line, which is heterozygous for _DLK1_ SNP. A total of 54 CpGs were analysed in three subregions of the CpG-rich region upstream of _GTL2_ (G1 _n_=12 CpGs,
G2 _n_=19 and G3 _n_=21). Between eight and 10 clones were sequenced from each of the normal blood samples and the methylation status of each of the 54 CpGs determined (see Figure 4C).
Comparison of the CpG methylation status of the _GTL2_ promoter DMR subregions in controls and the neuroblastoma cell line revealed that within subregions G1 and G2, there was extensive
methylation of CpGs in SK-N-AS compared to normal control DNAs. However, in subregion G3, there was little difference between the CpG methylation status of normal control DNAs and the
SK-N-AS cell line DNA. METHYLATION ANALYSIS OF THE _GTL2_ PROMOTER DMR IN PHAEOCHROMOCYTOMA AND WILMS' TUMOUR Methylation analysis of the _GTL2_ DMR was then undertaken in 20
phaeochromocytomas and 40 Wilms' tumours. _GTL2_ promoter DMR hypermethylation was detected in two phaeochromocytomas (10%) and one Wilms' tumour (2.5%). Genotyping with 14q32
microsatellite markers (D14S598, D14S1426, D14S1006, D14S749) excluded 14q32 allele loss in all three tumours. METHYLATION ANALYSIS OF IG-DMR Recently, an IG-DMR was demonstrated to function
as an imprinting control element for all imprinted genes on the maternal chromosome only (Lin et al, 2003). To determine whether _GTL2_ promoter DMR hypermethylation was associated with
epigenetic alterations at the upstream IG-DMR, we analysed the methylation status of the eight CpGs within the IG-DMR in four normal control DNAs and in four neuroblastoma cell lines with 5′
_GTL2_ DMR methylation. Eight to 12 individual clones for each were analysed for IG-DMR CpGs methylation status. In the four normal control samples, 32% of IG-DMR CpGs sequenced were
methylated, while in the SK-N-AS neuroblastoma cell line with _GTL2_ promoter DMR hypermethylation, all eight CpGs (_n_=12 clones) were methylated. In addition, the three neuroblastoma cell
lines with 5′ _GTL2_ DMR hypermethylation (but uninformative for _DLK1_ imprinting status) showed almost complete methylation at all IG-DMR CpGs analysed (see Figure 4B). Similarly, analysis
of the IG-DMR CpG methylation status in four primary tumours (two neuroblastoma, one phaeochromocytoma and one Wilms' tumour) with _GTL2_ promoter DMR hypermethylation demonstrated
heavy CpG methylation (although less complete than in the neuroblastoma cell lines, possibly because of normal tissue contamination). In contrast, cell lines and tumours with _GTL2_ promoter
DMR and IG-DMR hypermethylation did not show any differences from normal controls at the CpG-rich region upstream of the _DLK1_ promoter nor at the two CpG-rich regions downstream of the
_DLK1_ promoter. DISCUSSION We detected epigenetic changes within the _GTL2_ promoter DMR (and the IG-DMR) in 25% of neuroblastoma tumours, 10% of phaeochromocytoma and 2.5% of Wilms'
tumours analysed. In most cases, allele loss was excluded as a cause of the apparent hypermethylation. All cases with _GTL2_ promoter DMR methylation also demonstrated hypermethylation at
the upstream IG-DMR region, but epigenetic alterations were not detected in the three CpG-rich regions close to _DLK1_. Analysis of _DLK1_ and _GTL2_ expression in four neuroblastoma cell
lines with _GTL2_ DMR hypermethylation demonstrated repression of _GTL2_ transcription and re-expression of _GTL2_ after treatment with 5-AzaC. In most cell lines, re-expression of _GTL2_
after treatment with 5-AzaC was associated with downregulation of _DLK1_ expression. Furthermore, in an informative neuroblastoma cell line (SK-N-AS), there was no evidence of LOI of _DLK1_.
The absence of LOI of DLK1 is consistent with other reports of monoallelic _DLK1_ expression in neuroblastoma, brain tumours and lymphoma (van Limpt et al, 2003; Yin et al, 2004). The
_DLK1/GTL2_ imprinted domain has similarities to the _IGF2/H19_ gene at 11p15.5. In both cases, two closely linked genes are reciprocally imprinted, and both _H19_ and _GTL2_ do not appear
to encode a protein. Like _H19_ and _Igf2_, _Gtl2_ and _Dlk_ transcripts can be found in the same tissues during development (Takada et al, 2000). In Beckwith–Wiedemann syndrome and sporadic
Wilms' tumour, hypermethylation of the _H19_ promoter DMR (and the upstream ‘CTCF box’ DMR) is associated with silencing of _H19_ expression (Cui et al, 2001; Nakagawa et al, 2001). We
identified hypermethylation of the _GTL2_ DMRs in tumours and cell lines, so there are apparent similarities between epigenetics at the _H19_ and _GTL2_ loci in human neoplasia, although
the frequency of _GTL2_ hypermethylation in Wilms' tumour is much less than that at _H19_. However, whereas _de novo_ hypermethylation of the _H19_ promoter and CTCF box DMRs is
associated with _H19_ silencing and LOI of IGF2, LOI of _DLK1_ was not associated with _GTL2_ promoter DMR and IG-DMR hypermethylation. In mice, deletion of the unmethylated copy of the
IG-DMR leads to bidirectional LOI of all genes in the imprinted cluster after maternal inheritance, but imprinting is unaltered after paternal transmission of the deleted methylated copy
(Lin et al, 2003). This asymmetric regulation of imprinting distinguishes the imprinting control mechanisms in the 11p15.5 and the 14q32 imprinted gene clusters. The Callipyge phenotype
demonstrates complex inheritance patterns. Thus, while paternal inheritance of the Callipyge mutation is associated with muscle hypertrophy, and maternal transmission is not, surprisingly,
homozygotes do not demonstrate the phenotype. This unusual inheritance pattern (known as polar overdominance) has been attributed to the Callipyge mutation affecting a long-range control
element resulting in enhanced expression _in cis_ of all genes in the heterozygote sheep (but with no change in their imprinting) (Charlier et al, 2001; Freking et al, 2002; Georges et al,
2003). Furthermore, the absence of a mutant phenotype in the homozygotes suggests that overexpression of imprinted genes on the maternal chromosome abrogates the functional defect caused by
the paternal chromosome _in trans_. Hence, when the Callipyge mutation is paternally inherited, _DLK1_ transcription is upregulated, while _GTL2_ is upregulated after maternal transmission
(Charlier et al, 2001; Freking et al, 2002; Georges et al, 2003). In homozygotes, it is postulated that _GTL2_-associated transcripts negatively regulate _DLK1_ transcripts; however, the
precise mechanism is not known. Our finding that hypermethylation of the _GTL2_ promoter and upstream DMR is associated with silencing of _GTL2_ and upregulation of _DLK1_ without LOI would
be consistent with a model, whereby _GTL2_-associated transcripts negatively regulate _DLK1_ transcription and further analysis of neuroblastoma cell lines with GTL2 hypermethylation and
silencing may provide important insights into the mechanisms of imprinting control in the 14q32 imprinted domain. _DLK1_ encodes a transmembrane protein with six EGF-like repeats that shows
homology to the _Delta_ gene in _Drosophila melanogaster_, which is involved in the Notch signalling pathway. The precise function of the DLK1 protein is unclear, but _DLK1_ expression is
upregulated in myelodysplastic syndrome, a slowly progressing haematological malignancy, and in uterine leiomyomata (compared to normal myometrium) (Miyazato et al, 2001; Tsibris et al,
2002). In addition, overexpression of the _DLK1_ protein is reported to prevent adipocyte differentiation of 3T3-L1 cells in response to IGF1 or insulin. These latter effects were attributed
to changes in the activation levels and kinetics of extracellular-regulated kinase/mitogen-activated protein kinase (Ruiz-Hidalgo et al, 2002). However, although it might be postulated that
increased DLK1 expression might be pro-oncogenic, there is no direct evidence for this. Indeed, van Limpt et al (2003) suggested that neuroblastomas DLK1 expression increases during
chromaffin cell lineage differentiation and high DLK1 expression in neuroblastoma cell lines may merely indicate that the neuroblastoma has developed from a later stage chromaffin cell
precursor. Hence, upregulation of DLK1 expression associated with _GTL2_ promoter and intergenic DMR hypermethylation may be coincidental to tumorigenesis. _GTL2_ promoter and intergenic DMR
hypermethylation was associated with transcriptional silencing of _GTL2_ and treatment with 5-AzaC resulted in re-expression of _GTL2_. Recently, an isoform of _GTL2_, _MEG3a_, was reported
to be expressed highly in normal pituitary and brain and in other tissues including adrenal (Zhang et al, 2003), but not in many pituitary tumours. Furthermore, transfection of MEG3a into a
HeLa cell line inhibited cell proliferation in the _in vitro_ colony formation and growth rate assays. Hence, although neither GTL2 nor MEG3a have been shown to encode an expressed protein,
_GTL2_ promoter and intergenic DMR hypermethylation might promote tumorigenesis by downregulating _GTL2_ expression. These findings are similar to those reported for _H19_ (Hao et al,
1993), although the precise role of H19 silencing _per se_ in Wilms' tumour and tumour susceptibility in Beckwith–Wiedemann syndrome has not been unequivocally established (Tycko,
2000). Thus, further investigations of the potential roles of _GTL2_, _MEG3a_ and epigenetic changes in the _DLK1_/_GTL2_ domain are indicated. CHANGE HISTORY * _ 16 NOVEMBER 2011 This paper
was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication _ REFERENCES * Charlier C, Segers K, Karim L, Shay T, Gyapay G, Cockett
N, Georges M (2001) The Callipyge mutation enhances the expression of coregulated imprinted genes in _cis_ without affecting their imprinting status. _Nat Genet_ 27: 367–369 Article CAS
PubMed Google Scholar * Cui H, Niemitz EL, Ravenel JD, Onyango P, Brandenburg SA, Lobanenkov VV, Feinberg AP (2001) Loss of imprinting of insulin-like growth factor-II in Wilms' tumor
commonly involves altered methylation but not mutations of CTCF or its binding site. _Cancer Res_ 61: 4947–4950 CAS PubMed Google Scholar * Eggermann T, Mergenthaler S, Eggermann K,
Albers A, Linnemann K, Fusch C, Ranke MB, Wollmann HA (2001) Identification of interstitial maternal uniparental disomy (UPD) (14) and complete maternal UPD(20) in a cohort of growth
retarded patients. _J Med Genet_ 38: 86–89 Article CAS PubMed PubMed Central Google Scholar * Feinberg AP, Cui H, Ohlsson R (2002) DNA methylation and genomic imprinting: insights from
cancer into epigenetic mechanisms. _Semin Cancer Biol_ 12: 389–398 Article CAS PubMed Google Scholar * Freking BA, Murphy SK, Wylie AA, Rhodes SJ, Keele JW, Leymaster KA, Jirtle RL,
Smith TP (2002) Identification of the single base change causing the Callipyge muscle hypertrophy phenotype, the only known example of polar overdominance in mammals. _Genome Res_ 12:
1496–1506 Article CAS PubMed PubMed Central Google Scholar * Georges M, Charlier C, Cockett N (2003) The Callipyge locus: evidence for the trans interaction of reciprocally imprinted
genes. _Trends Genet_ 19: 248–252 Article CAS PubMed Google Scholar * Georgiades P, Watkins M, Surani MA, Ferguson-Smith AC 2000 Parental origin-specific developmental defects in mice
with uniparental disomy for chromosome 12. _Development_ 127: 4719–4728 CAS PubMed Google Scholar * Hao Y, Crenshaw T, Moulton T, Newcomb E, Tycko B (1993) Tumour-suppressor activity of
H19 RNA. _Nature_ 365: 764–767 Article CAS PubMed Google Scholar * Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB (1996) Methylation-specific PCR: a novel PCR assay for
methylation status of CpG islands. _Proc Natl Acad Sci USA_ 93: 9821–9826 Article CAS PubMed PubMed Central Google Scholar * Kobayashi S, Wagatsuma H, Ono R, Ichikawa H, Yamazaki M,
Tashiro H, Aisaka K, Miyoshi N, Kohda T, Ogura A, Ohki M, Kaneko-Ishino T, Ishino F (2000) Mouse Peg9/Dlk1 and human PEG9/DLK1 are paternally expressed imprinted genes closely located to the
maternally expressed imprinted genes: mouse Meg3/Gtl2 and human MEG3. _Genes Cells_ 5: 1029–1037 Article CAS PubMed Google Scholar * Kurosawa K, Sasaki H, Sato Y, Yamanaka M, Shimizu M,
Ito Y, Okuyama T, Matsuo M, Imaizumi K, Kuroki Y _et al_ (2002) Paternal UPD14 is responsible for a distinctive malformation complex. _Am J Med Genet_ 110: 268–272 Article PubMed Google
Scholar * Lin SP, Youngson N, Takada S, Seitz H, Reik W, Paulsen M, Cavaille J, Ferguson-Smith AC (2003) Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the
_Dlk1-Gtl2_ imprinted cluster on mouse chromosome 12. _Nat Genet_ 35: 97–102 Article CAS PubMed Google Scholar * Maher ER, Reik W (2000) Beckwith–Wiedemann syndrome: imprinting in
clusters revisited. _J Clin Invest_ 105: 247–252 Article CAS PubMed PubMed Central Google Scholar * Miyazato A, Ueno S, Ohmine K, Ueda M, Yoshida K, Yamashita Y, Kaneko T, Mori M,
Kirito K, Toshima M, Nakamura Y, Saito K, Kano Y, Furusawa S, Ozawa K, Mano H (2001) Identification of myelodysplastic syndrome-specific genes by DNA microarray analysis with purified
hematopoietic stem cell fraction. _Blood_ 98: 422–427 Article CAS PubMed Google Scholar * Miyoshi N, Wagatsuma H, Wakana S, Shiroishi T, Nomura M, Aisaka K, Kohda T, Surani MA,
Kaneko-Ishino T, Ishino F (2000) Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. _Genes
Cells_ 5: 211–220 Article CAS PubMed Google Scholar * Murphy SK, Wylie AA, Coveler KJ, Cotter PD, Papenhausen PR, Sutton PR, Shaffer LG, Jirtle RL (2003) Epigenetic detection of human
chromosome 14 uniparental disomy. _Hum Mutat_ 22: 92–97 Article CAS PubMed Google Scholar * Nakagawa H, Chadwick RB, Peltomaki P, Plass C, Nakamura Y, de La Chapelle A (2001) Loss of
imprinting of the insulin-like growth factor II gene occurs by biallelic methylation in a core region of H19-associated CTCF-binding sites in colorectal cancer. _Proc Natl Acad Sci USA_ 98:
591–596 Article CAS PubMed Google Scholar * Paulsen M, Takada S, Youngson NA, Benchaib M, Charlier C, Seger K, Georges M, Ferguson-Smith AC (2001) Comparative sequence analysis of the
imprinted _Dlk1-Gtl2_ locus in three mammalian species reveals highly conserved genomic elements and refines comparison with the _Igf2-H19_ region. _Genome Res_ 11: 2085–2094 Article CAS
PubMed PubMed Central Google Scholar * Reik W, Walter J (2001) Genomic imprinting: parental influence on the genome. _Nat Rev Genet_ 2: 21–32 Article CAS PubMed Google Scholar *
Ruiz-Hidalgo MJ, Gubina E, Tull L, Baladron V, Laborda J (2002) DLK modulates mitogen-activated protein kinase signaling to allow or prevent differentiation. _Exp Cell Res_ 274: 178–188
Article CAS PubMed Google Scholar * Schmidt JV, Matteson PG, Jones BK, Guan XJ, Tilghman SM (2000) The Dlk1 and Gtl2 genes are linked and reciprocally imprinted. _Genes Dev_ 14:
1997–2002 CAS PubMed PubMed Central Google Scholar * Takada S, Paulsen M, Tevendale M, Tsai CE, Kelsey G, Cattanach BM, Ferguson-Smith AC (2002) Epigenetic analysis of the _Dlk1-Gtl2_
imprinted domain on mouse chromosome 12: implications for imprinting control from comparison with _Igf2-H19_. _Hum Mol Genet_ 11: 77–86 Article CAS PubMed Google Scholar * Takada S,
Tevendale M, Baker J, Georgiades P, Campbell E, Freeman T, Johnson MH, Paulsen M, Ferguson-Smith AC (2000) Delta-like and gtl2 are reciprocally expressed, differentially methylated linked
imprinted genes on mouse chromosome 12. _Curr Biol_ 10: 1135–1138 Article CAS PubMed Google Scholar * Tsibris JC, Segars J, Coppola D, Mane S, Wilbanks GD, O’Brien WF, Spellacy WN (2002)
Insights from gene arrays on the development and growth regulation of uterine leiomyomata. _Fertil Steril_ 78: 114–121 Article PubMed PubMed Central Google Scholar * Tycko B (2000)
Epigenetic gene silencing in cancer. _J Clin Invest_ 105: 401–407 Article CAS PubMed PubMed Central Google Scholar * van Limpt V, Chan A, Caron H, Sluis PV, Boon K, Hermus MC, Versteeg
R (2000) SAGE analysis of neuroblastoma reveals a high expression of the human homologue of the _Drosophila_ _Delta_ gene. _Med Pediatr Oncol_ 35: 554–558 Article CAS PubMed Google
Scholar * van Limpt VAE, Chan AJ, van Sluis PG, Caron HN, van Noesel CJM, Versteeg R (2003) High delta-like expression in a subset of neuroblastoma cell lines corresponds to a
differentiated chromaffin cell type. _Int J Cancer_ 105: 61–69 Article CAS PubMed Google Scholar * Weksberg R, Smith AC, Squire J, Sadowski P (2003) Beckwith–Wiedemann syndrome
demonstrates a role for epigenetic control of normal development. _Hum Mol Genet_ 12: R61–R68 Article CAS PubMed Google Scholar * Wylie AA, Murphy SK, Orton TC, Jirtle RL (2000) Novel
imprinted DLK1/GTL2 domain on human chromosome 14 contains motifs that mimic those implicated in IGF2/H19 regulation. _Genome Res_ 10: 1711–1718 Article CAS PubMed PubMed Central Google
Scholar * Yin D, Xie D, De Vos S, Liu G, Miller CW, Black KL, Koeffler HP (2004) Imprinting status of DLK1 gene in brain tumors and lymphomas. _Int J Oncol_ 24: 1011–1015 CAS PubMed
Google Scholar * Zhang X, Zhou Y, Mehta KR, Danila DC, Scolavino S, Johnson SR, Klibanski A (2003) A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. _J Clin
Endocrinol Metab_ 88: 5119–51126 Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We thank AICR British Heart Foundation and Cancer Research UK for financial
support. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Paediatrics and Child Health, Section of Medical and Molecular Genetics, University of Birmingham, The Medical School,
Edgbaston, Birmingham, B15 2TT, UK D Astuti, F Latif, K Wagner, D Gentle, W N Cooper & E R Maher * Cancer Research UK Renal Molecular Oncology Research Group, University of Birmingham,
The Medical School, Edgbaston, Birmingham, B15 2TT, UK F Latif, D Gentle & E R Maher * TBA, D Catchpoole * Department of Paediatric Oncology Birmingham Children's Hospital and
Department of Paediatrics and Child Health, University of Birmingham, B15 2TT R Grundy * Department of Anatomy, University of Cambridge, Downing Street, Cambridge, CB2 3DY, UK A C
Ferguson-Smith Authors * D Astuti View author publications You can also search for this author inPubMed Google Scholar * F Latif View author publications You can also search for this author
inPubMed Google Scholar * K Wagner View author publications You can also search for this author inPubMed Google Scholar * D Gentle View author publications You can also search for this
author inPubMed Google Scholar * W N Cooper View author publications You can also search for this author inPubMed Google Scholar * D Catchpoole View author publications You can also search
for this author inPubMed Google Scholar * R Grundy View author publications You can also search for this author inPubMed Google Scholar * A C Ferguson-Smith View author publications You can
also search for this author inPubMed Google Scholar * E R Maher View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to E
R Maher. RIGHTS AND PERMISSIONS From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported
License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Astuti, D., Latif, F., Wagner,
K. _et al._ Epigenetic alteration at the _DLK1-GTL2_ imprinted domain in human neoplasia: analysis of neuroblastoma, phaeochromocytoma and Wilms' tumour. _Br J Cancer_ 92, 1574–1580
(2005). https://doi.org/10.1038/sj.bjc.6602478 Download citation * Received: 17 September 2004 * Revised: 16 December 2004 * Accepted: 01 February 2005 * Published: 29 March 2005 * Issue
Date: 25 April 2005 * DOI: https://doi.org/10.1038/sj.bjc.6602478 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * imprinting * DLK1 * GTL2 *
methylation

:max_bytes(150000):strip_icc():focal(511x0:513x2)/ciara-1-1024-d8ce712a8ca345168c0601a9af511592.jpg)


