- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT ATRX is a SWI/SNF-like chromatin remodeling protein mutated in several X-linked mental retardation syndromes. Gene inactivation studies in mice demonstrate that ATRX is an essential
protein and suggest that patient mutations likely retain partial activity. ATRX associates with the nuclear matrix, pericentromeric heterochromatin, and promyelocytic leukemia nuclear
bodies (PML-NBs) in a speckled nuclear staining pattern. Here, we used GFP–ATRX fusion proteins to identify the specific domains of ATRX necessary for subnuclear targeting and the effect of
patient mutations on this localization. We identified two functional nuclear localization signals (NLSs) and two domains that target ATRX to nuclear speckles. One of the latter domains is
responsible for targeting ATRX to PML-NBs. Surprisingly, this domain encompassed motifs IV–VI of the SNF2 domain suggesting that in addition to chromatin remodeling, it may also have a role
in subnuclear targeting. More importantly, four different patient mutations within this domain resulted in an ∼80% reduction in the number of transfected cells with ATRX nuclear speckles and
PML colocalization. These results demonstrate that patient mutations have a dramatic effect on subnuclear targeting to PML-NBs. Moreover, these findings support the hypothesis that ATRX
patient mutations represent functional hypomorphs and suggest that loss of proper targeting to PML-NBs is an important contributor to the pathogenesis of the ATR-X syndrome. SIMILAR CONTENT
BEING VIEWED BY OTHERS THE CHROMATIN REMODELLER ATRX FACILITATES DIVERSE NUCLEAR PROCESSES, IN A STOCHASTIC MANNER, IN BOTH HETEROCHROMATIN AND EUCHROMATIN Article Open access 17 June 2022
STRUCTURAL BASIS FOR NUCLEAR IMPORT SELECTIVITY OF PIONEER TRANSCRIPTION FACTOR SOX2 Article Open access 04 January 2021 PHF6 COOPERATES WITH SWI/SNF COMPLEXES TO FACILITATE TRANSCRIPTIONAL
PROGRESSION Article Open access 24 August 2024 INTRODUCTION The ATR-X syndrome (OMIM no. 301040) is a severe X-linked mental retardation (XLMR) syndrome associated with a characteristic
facial appearance, urogenital abnormalities and _α_-thalassemia.1, 2 Mutations in the ATRX gene have been identified as the cause of the ATR-X syndrome,3 other XLMR disorders with similar
features but lacking thalassemia,4 and the acquired form of _α_-thalassemia myelodysplasia syndrome (ATMDS).5, 6, 7, 8 The ATRX gene encodes a chromatin remodeling protein with an
ATPase/helicase domain of the SNF2 family and a plant homeodomain (PHD) zinc finger.9 ATRX mutations are predominantly missense mutations with most identified in the PHD (∼60%) or SNF2
domains (∼25%).10 While it is presumed that ATRX mutations are associated with reduced function, few studies have addressed structure/function relationships. Subcellular localization and
initial biochemical studies with ATRX indicate that it likely has a repressive role on nuclear processes. It is localized to pericentromeric heterochromatin and can physically interact with
proteins that reside within heterochromatin, including HP1 and EZH2.11, 12, 13, 14 The ATRX protein also localizes to PML-NBs where it interacts with Daxx, a transcriptional and apoptotic
regulator.12, 15, 16 ATRX is also involved in the regulation of methylation at ribosomal DNA (rDNA) repeats.17 Given that ATRX acts at multiple locations within the nucleus, some mutations
are likely to alter subnuclear localization thereby reducing function. In this regard, immunoblots of protein isolated from a panel of ATR-X patients with unique missense mutations have
shown that many samples have reduced ATRX protein levels.13, 18 Nonetheless, other experiments have demonstrated a more direct role of mutations on protein function. Baculovirus expressed
ATRX protein containing engineered patient mutations within the SNF2 domain were shown to have attenuated ATPase activity compared to the wild-type (WT) protein.15 Similarly, expression of
the PHD domain in bacteria demonstrated that patient mutations impaired DNA-binding activity.18 Collectively, these studies suggest that some mutations may interfere directly with function
while others may have an indirect effect through altered protein stability or aberrant subnuclear targeting of the protein. Here, we utilized GFP–fusion proteins to identify the domains
responsible for nuclear localization, heterochromatin binding and subnuclear targeting to the PML-NBs. Moreover, we demonstrate that patient mutations within one domain reduces the
efficiency of targeting to PML-NBs. MATERIALS AND METHODS CONSTRUCTION OF GFP-TAGGED PROTEIN SUBDOMAINS The full-length ATRX-EGFP construct was generated by cutting the HA-ATRX-pCAGGS
construct19 with _Xho_I and _Sst_II and ligating to EGFP-C2. _Eco_RI, _Sac_I-_Pst_I and _Pst_I-_Sst_II fragments were cut from HA-ATRX-pCAGGs and ligated to the EGFP-C2 linearized vector to
create the L16, 85/30 and 8R/4 constructs, respectively. The 8R4-EGFPC2 construct was put back in frame by cutting with _Xho_I and filling the ends with Klenow enzyme. The L3, L5, L6 and L8
constructs were created by cutting 85/30-C2 with _Sac_I-_Hin_dIII, _Eco_RI-_Pst_I, _Sac_I-_Afl_II and _Hin_dII-_Eco_RI, respectively. Construct L9 was generated by deleting the _Bam_HI
fragment from the 85/30+8R4 vector. Constructs L18 and L19 were obtained by isolating the _Bam_HI and _Eco_RI-_Bam_HI fragments from 8R4-C2, respectively and religating downstream of the
nuclear localization signal in the linearized L8 construct. We obtained L20 by cutting a _Sac_I fragment from L19 and religating to the EGFP-C3 vector. L24 was generated by removing an
_Afl_II-_Sal_I fragment from the L16 construct. Finally, L25 was obtained by cutting the L16 construct with _Afl_II and _Xho_I, treating with Klenow followed by religation. SITE-DIRECTED
MUTAGENESIS Mutant constructs were generated by a PCR-based site-directed mutagenesis method (Stratagene QuickChange kit) using the L16, L2 and L19 GFP-tagged constructs as templates.
Template DNA (20 ng) was amplified using the appropriate primers (100 ng each forward and reverse, see Table 1) using the PCR in a 50-_μ_l reaction with the following conditions: 95°C for 30
s, then 95°C for 30 s, 55°C for 1 min and 68°C for 14 min for 12 cycles. The samples were then incubated for 1 h at 37°C with _Dpn_I to digest parental non-mutated double-stranded DNA. The
product (1 _μ_l) was transformed in XL-2 Blue competent cells. Plasmid DNA was obtained from the resulting colonies and screened using the new or abolished restriction enzyme sites
introduced by silent mutations (see Table 1 below). TRANSFECTIONS AND IMMUNOFLUORESCENCE HeLa cells were grown in EMEM supplemented with 10% fetal bovine serum in 5% CO2 at 37°C. Plasmid DNA
for each GFP deletion construct was isolated using the Midi-Preparation kit from Qiagen and verified by sequence analysis. Transient transfection was performed using the Lipofectamine 2000
reagent (Life Technologies Inc.) according to the manufacturer's protocol. Depending on the level of cell death observed with some of the constructs, cells were processed after 16–24 h
by fixing in cold ethanol/methanol (3:1) and counterstaining with DAPI. Immunofluorescence images were captured using a Zeiss Axioplan 2 microscope outfitted with an AxioCam camera and
AxioVision software. For analysis of mutated constructs, at least 300 cells were counted and assessed for localization pattern. RESULTS NUCLEAR LOCALIZATION OF ATRX DELETION CONSTRUCTS To
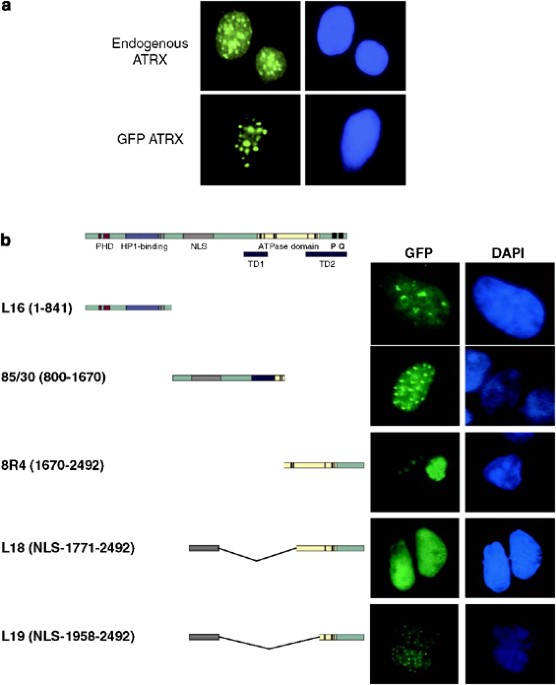
identify the domains responsible for nuclear and subnuclear localization we generated a panel of GFP–ATRX fusion constructs. A full-length GFP–ATRX fusion protein recapitulated the staining
pattern of the endogenous ATRX protein upon transfection into HeLa cells and demonstrating that the tag does not interfere with localization (Figure 1a). The PredictNLS program
(http://cubic.bioc.columbia.edu/predictNLS/) identified putative nuclear localization signals (NLSs) in the ATRX protein sequence encompassing amino acids 685–697, three overlapping sites
within 1126–1196, and individual sites at 1420–1426 and 1928–1938. As such, we divided the _ATRX_ gene into three fragments – L16, 85/30 and 8R/4 for a preliminary analysis of nuclear
localization. We observed that both L16 and 85/30 independently localized to the nucleus with distinct patterns. The L16-staining pattern was diffuse throughout the nucleus with 6–10 intense
staining ‘bundles’ (Figure 1b) consistent with previous reports that this region targets the protein to heterochromatin.13, 20 The 85/30 fusion protein was localized to 20–30 distinct and
smaller nuclear speckles, reminiscent of PML nuclear body staining and consistent with previous reports that ATRX localizes to these structures.12, 15, 16 In contrast, the C-terminal
fragment 8R/4 was restricted to the cytoplasm in a pattern containing one or two large regions of intense perinuclear staining that most likely represents protein aggregates (Figure 1b). It
also suggested that the predicted NLS (1928–1938) located within this fragment does not support nuclear localization on its own. As such, we fused an NLS-containing region of ATRX (924–1214)
in front of the C-terminal fragment (construct L18), resulting in a homogeneous nuclear staining pattern. Further reduction of L18 to generate the C-terminal fragment (L19) demonstrated a
speckled nuclear staining pattern upon transfection (Figure 1b) indicating that the larger fusion protein may have masked the domain responsible for speckle formation. The NLS sequences
between 685–697 and 1126–1196 can function independently to target ATRX to the nucleus. The NLS sequences between residues 1420–1426 (within construct L5; Figure 2) and 1928–1938 were unable
to support nuclear localization of the fusion protein. Moreover, we defined two regions within ATRX that generate nuclear speckles and could potentially target ATRX to PML nuclear bodies.
REFINED MAPPING OF THE NUCLEAR SPECKLE DOMAINS To refine further the domain within 85/30 that results in speckle formation, we generated additional deletion constructs (Figure 2a).
Constructs L3 and L5 did not enter the nucleus (Figure 2b and data not shown), thereby refining the NLS to amino acids 924–1214. The L9 and L2 constructs each retained the nuclear
speckle-staining pattern in HeLa cells and in 293 cells (Figure 2b and data not shown). We occasionally observed significant staining in the cytoplasm of HeLa cells with the L2 construct
that was not observed in 293 cells (data not shown). However, upon longer incubation before fixation, the cytoplasmic staining in the HeLa cells dissipated suggesting that high expression
levels of L2 may overwhelm the transport machinery in this cell type. The L8 and L6 fusion proteins always showed a nuclear-staining pattern with only a few large dots of intense staining.
Taken together, these findings demonstrate that the region between amino acids 1498–1651 contains a domain essential for the formation of nuclear speckles, which we named as targeting domain
1 (TD1). Similarly, we digested the L19 fusion construct with _Sac_I resulting in the removal of the region encoding the last 253 amino acids (L20). The L20 construct demonstrated the
nuclear speckle pattern thereby delimiting the C-terminal speckle domain between residues 1965–2239 (TD2). We utilized these smaller constructs to examine whether they targeted the ATRX
protein to PML nuclear bodies. THE C-TERMINAL END OF ATRX TARGET PML-NBS The nuclear speckles that we observed with the L2 and L19 constructs are similar in size and frequency to that
observed for PML nuclear bodies. Moreover, we and others have shown that ATRX localizes to PML nuclear bodies.12, 15, 16, 21, 22 To determine whether TD1 and TD2 both can target ATRX to PML
nuclear bodies, we transfected HeLa cells with the L2 and L19 constructs and examined their colocalization with endogenous PML and Daxx (Figure 3a), or with ectopically expressed PML tagged
with the fluorescent DsRed protein (Figure 3b). The L2 fragment showed almost complete exclusion from endogenous or overexpressed PML (Figure 3b and c). In contrast, the L19 fragment always
showed a high incidence of colocalization with endogenous or overexpressed PML indicating that the C-terminal end of the ATRX protein confers its ability to associate with PML nuclear bodies
(Figure 3b and c). C-TERMINAL ATRX MUTATIONS DISRUPT THE ASSOCIATION WITH PML-NBS The L19 fragment is large, comprising 534 amino acids and encompassing several functional domains of the
ATRX protein including motifs IV–VI of the ATPase domain, the P domain which has weak homology to the SWI/SNF family, and a stretch of glutamine residues named the Q domain.9 A number of
naturally occurring ATR-X patient mutations have been identified in the L19 fragment containing the PML-interacting C terminus of the ATRX protein (Figure 4a), which we have renamed
targeting domain 2 (TD2). To assess the functional importance of clinical mutations for the interaction of ATRX with PML, we introduced four specific mutations in TD2 in different conserved
domains (Figure 4a). Ped 23 (D2035V) and Ped 3 (Y2084H) missense mutations flank motif IV of the ATPase domain. Ped 17 (R2386X) is a nonsense mutation that is located within the P element
and truncates the last 106 amino acids of the ATRX protein. The mutation identified in Ped 26 was a 2-kb genomic deletion that helped localize the ATRX gene.23 The deletion removed the
acceptor splice site and the first ∼2 kb of the last exon and we recapitulated that deletion by creating a nonsense mutation (L2401X) that deletes the remaining 91 amino acids. Transfection
of each mutated L19 construct was analyzed for speckle formation by fluorescence microscopy (Figure 4b). While all cells transfected with L19 displayed a speckled pattern of GFP
fluorescence, this was not the case for the mutated TD2 constructs which primarily presented with a uniform nuclear-staining pattern (Figure 4b). To quantify this result 300 transfected
cells (from five independent transfections) for each mutant construct were scored for uniform or speckled pattern. Overall, the uniform pattern of localization was observed in approximately
83% of the cells whereas only ∼17% of the cells transfected with mutant constructs had the characteristic speckled pattern (Table 2), indicating that these mutations interfere with
localization to PML nuclear bodies or, alternatively, affect retention of the protein within these domains. Nonetheless, the dramatic effect of ATR-X clinical mutations on PML targeting
suggests that these interactions are crucial for the normal function of ATRX in the nucleus. To determine whether the changes in subnuclear localization were not restricted to the truncated
ATRX proteins we introduced two patient mutations into the full-length GFP-ATRX construct, namely the Ped 23 and a PHD zinc-finger mutation that should not affect localization to the PML
nuclear body. In addition, we generated a construct containing a mutation in the active site of the ATPase domain (K1600R; named Mu) that we have previously shown to represent an ATPase-dead
mutation.15 As shown in Figure 5 and Table 2, we observed a predominant speckled pattern in cells transfected for the WT and PHD mutant constructs, whereas there was an increased frequency
of cells with a uniform staining pattern when the Ped 23 and Mu mutants were introduced. Taken together, these results suggest that the SNF2 domain may have a role, in addition to nucleosome
remodeling, in facilitating targeting to the PML-NB. DISCUSSION Defective chromatin remodeling is now recognized as the cause of numerous mental retardation disorders, including the ATR-X
syndrome.24, 25 For a few syndromes, namely Rett syndrome and Rubinstein-Taybi syndrome, specific neuronal target genes have been identified and their dysregulation shown to contribute to
disease pathophysiology.26, 27, 28, 29, 30 Despite these successes, our understanding of the developmental role of XLMR-associated chromatin remodeling proteins is limited. In this regard,
ATRX is a prime example. It localizes to pericentromeric heterochromatin and PML-NBs and it can physically interact with heterochromatin protein 1 or with Daxx in a protein complex.11, 12,
13, 15, 16, 31 However, it is not clear what the functional implications of these interactions are and, more importantly, how mutations affect these interactions or lead to deficits in
neural development. The goal in the present study was to identify the domains responsible for nuclear and subnuclear localization and to determine whether patient mutations interfered with
targeting. The ATRX protein contains four regions that fulfill the criteria for an NLS motif (amino acids 685–697, three motifs between amino acids 1126–1196, amino acids 1420–1426 and
residues 1928–1938). We demonstrated that two of these regions could function in isolation to target GFP fusion proteins to the nucleus. L16 which contained the first putative NLS located at
amino acids 685–697 was correctly targeted to the nucleus. The 85/30 construct contained two putative NLS sequences but only the one within L8, at residues 1126–1196, conferred nuclear
localization to GFP. Moreover, this fragment could localize other C-terminal ATRX fragments to the nucleus. There are no reported mutations in the two functional NLS sequences we describe
here or in the other putative NLS regions. While NLS mutations may be identified as more patients are characterized, it is equally probable that the outcome of such a mutation is a lethal
phenotype since ATRX ablation in mice results in early embryonic lethality.32 Two different regions of the ATRX protein, TD1 and TD2 resulted in a punctate nuclear-staining pattern, yet only
TD2 colocalized with PML nuclear bodies. The TD1 containing construct L2 encompasses motifs I and Ia of the SNF2 domain and contains the Daxx interaction domain.9, 15 While we previously
demonstrated that the Daxx interaction domain could target ATRX to PML-NBs this was not the case with L2 in the present study. The difference between the two studies was that in Tang _et
al_15 we overexpressed both PML and Daxx in addition to the ATRX fragment (F14). When these experiments were repeated using endogenous levels of PML and Daxx we did not observe localization
of F14 to PML-NBs; such localization required PML and Daxx overexpression (data not shown). Importantly, our present results are consistent with findings for ATRXt, an endogenous ATRX
isoform that truncates prior to the SNF2 domain and does not associate with PML-NBs.33 Nonetheless, some controversy remains as a different study has shown that Daxx is necessary to recruit
ATRX to PML-NBs in mouse fibroblasts.12 While the nature of the L2 speckles remains to be determined they could represent genes targeted for transcriptional regulation by a Daxx/ATRX
complex. Alternatively, it could be a cell type specific difference that promotes ATRX targeting to PML-NBs. Indeed, a recent study has shown that MeCP2 is necessary to target ATRX to
heterochromatin in mature but not immature neurons.34 In contrast, the L19 construct did colocalize with PML-NBs suggesting that the TD2 domain is sufficient for localization of the ATRX
protein to this subnuclear structure. These findings indicate that ATRX could bind directly to other components of PML-NBs, distinct from Daxx, through the TD2 region for targeting to these
foci. The TD2 region also overlaps with the MeCP2 interaction domain suggesting that binding of different factors to this domain may determine the subnuclear localization of ATRX.34
Interestingly, patient mutations resulted in fewer cells with PML colocalization suggesting that alteration of the TD2 domain impairs targeting efficiency or retention within this structure.
How do patient mutations interfere with localization to the PML-NBs and how could this lead to mental retardation? The Ped3 and Ped23 mutations were previously shown to attenuate ATPase
activity15 and they showed reduced targeting to PML-NBs when introduced into the L19 construct or within a full-length ATRX–GFP fusion protein. Similarly, the ATPase dead mutant (Mu) showed
a reduced number of cells with speckle formation. This was not simply an effect of mutating ATRX since the PHD mutant had no effect on nuclear speckle formation. The Mu mutation lies outside
the TD2 domain within ATPase motif I of the SNF2 domain thereby suggesting that chromatin-remodeling activity may also be important for PML-NB localization/retention. Indeed, it has been
postulated that the association of satellite DNA with ATRX, Daxx and HP1 within PML-NBs at the G2 phase may facilitate re-establishment of condensed heterochromatin at satellite DNA.35
Certainly, one could speculate that proper targeting of ATRX to PML-NBs is essential for ATRX-directed chromatin remodeling of specific target genes tagged for silencing during neural
development. Mutations located within the SNF2 domain may attenuate (but not abolish) the chromatin-remodeling activity and/or the localization to the PML-NB such that the ATRX-specific
target genes are derepressed. Certainly, the inappropriate regulation of gene expression in the nervous system during development could result in a mental retardation phenotype and thus, the
identification of such targets remains a high priority of investigation. REFERENCES * Wilkie AO, Zeitlin HC, Lindenbaum RH _et al_: Clinical features and molecular analysis of the alpha
thalassemia/mental retardation syndromes. II. Cases without detectable abnormality of the alpha globin complex. _Am J Hum Genet_ 1990; 46: 1127–1140. CAS PubMed PubMed Central Google
Scholar * Gibbons RJ, Suthers GK, Wilkie AO, Buckle VJ, Higgs DR : X-linked alpha-thalassemia/mental retardation (ATR-X) syndrome: localization to Xq12-q21.31 by X inactivation and linkage
analysis. _Am J Hum Genet_ 1992; 51: 1136–1149. CAS PubMed PubMed Central Google Scholar * Gibbons RJ, Picketts DJ, Higgs DR : Syndromal mental retardation due to mutations in a
regulator of gene expression. _Hum Mol Genet_ 1995; 4: 1705–1709. Article CAS Google Scholar * Gibbons RJ, Higgs DR : Molecular-clinical spectrum of the ATR-X syndrome. _Am J Med Genet_
2000; 97: 204–212. Article CAS Google Scholar * Gibbons RJ, Pellagatti A, Garrick D _et al_: Identification of acquired somatic mutations in the gene encoding chromatin-remodeling factor
ATRX in the alpha-thalassemia myelodysplasia syndrome (ATMDS). _Nat Genet_ 2003; 34: 446–449. Article CAS Google Scholar * Steensma DP, Viprakasit V, Hendrick A _et al_: Deletion of the
{alpha}-globin gene cluster as a cause of acquired {alpha}-thalassemia in myelodysplastic syndrome. _Blood_ 2004; 103: 1518–1520. Article CAS Google Scholar * Steensma DP, Gibbons RJ,
Higgs DR : Acquired alpha-thalassemia in association with myelodysplastic syndrome and other hematologic malignancies. _Blood_ 2005; 105: 443–452. Article CAS Google Scholar * Steensma
DP, Higgs DR, Fisher CA, Gibbons RJ : Acquired somatic ATRX mutations in myelodysplastic syndrome associated with alpha thalassemia (ATMDS) convey a more severe hematologic phenotype than
germline ATRX mutations. _Blood_ 2004; 103: 2019–2026. Article CAS Google Scholar * Picketts DJ, Higgs DR, Bachoo S, Blake DJ, Quarrell OW, Gibbons RJ : ATRX encodes a novel member of the
SNF2 family of proteins: mutations point to a common mechanism underlying the ATR-X syndrome. _Hum Mol Genet_ 1996; 5: 1899–1907. Article CAS Google Scholar * Gibbons R : Alpha
thalassaemia-mental retardation, X linked. _Orphanet J Rare Dis_ 2006; 1: 15. Article Google Scholar * Berube NG, Smeenk CA, Picketts DJ : Cell cycle-dependent phosphorylation of the ATRX
protein correlates with changes in nuclear matrix and chromatin association. _Hum Mol Genet_ 2000; 9: 539–547. Article CAS Google Scholar * Ishov AM, Vladimirova OV, Maul GG :
Heterochromatin and ND10 are cell-cycle regulated and phosphorylation-dependent alternate nuclear sites of the transcription repressor Daxx and SWI/SNF protein ATRX. _J Cell Sci_ 2004; 117:
3807–3820. Article CAS Google Scholar * McDowell TL, Gibbons RJ, Sutherland H _et al_: Localization of a putative transcriptional regulator (ATRX) at pericentromeric heterochromatin and
the short arms of acrocentric chromosomes. _Proc Natl Acad Sci USA_ 1999; 96: 13983–13988. Article CAS Google Scholar * Cardoso C, Timsit S, Villard L, Khrestchatisky M, Fontes M,
Colleaux L : Specific interaction between the XNP/ATR-X gene product and the SET domain of the human EZH2 protein. _Hum Mol Genet_ 1998; 7: 679–684. Article CAS Google Scholar * Tang J,
Wu S, Liu H _et al_: A novel transcription regulatory complex containing Daxx and the ATR-X syndrome protein. _J Biol Chem_ 2004; 279: 20369–20377. Article CAS Google Scholar * Xue Y,
Gibbons R, Yan Z _et al_: The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. _Proc Natl Acad Sci USA_ 2003; 100:
10635–10640. Article CAS Google Scholar * Gibbons RJ, McDowell TL, Raman S _et al_: Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA
methylation. _Nat Genet_ 2000; 24: 368–371. Article CAS Google Scholar * Cardoso C, Lutz Y, Mignon C _et al_: ATR-X mutations cause impaired nuclear location and altered DNA binding
properties of the XNP/ATR-X protein. _J Med Genet_ 2000; 37: 746–751. Article CAS Google Scholar * Berube NG, Jagla M, Smeenk C, De Repentigny Y, Kothary R, Picketts DJ :
Neurodevelopmental defects resulting from ATRX overexpression in transgenic mice. _Hum Mol Genet_ 2002; 11: 253–261. Article CAS Google Scholar * Sutherland HG, Mumford GK, Newton K _et
al_: Large-scale identification of mammalian proteins localized to nuclear sub-compartments. _Hum Mol Genet_ 2001; 10: 1995–2011. Article CAS Google Scholar * Ishov AM, Sotnikov AG,
Negorev D _et al_: PML is critical for ND10 formation and recruits the PML-interacting protein Daxx to this nuclear structure when modified by SUMO-1. _J Cell Biol_ 1999; 147: 221–234.
Article CAS Google Scholar * Torii S, Egan DA, Evans RA, Reed JC : Human Daxx regulates Fas-induced apoptosis from nuclear PML oncogenic domains (PODs). _EMBO J_ 1999; 18: 6037–6049.
Article CAS Google Scholar * Gibbons RJ, Picketts DJ, Villard L, Higgs DR : Mutations in a putative global transcriptional regulator cause X-linked mental retardation with
alpha-thalassemia (ATR-X syndrome). _Cell_ 1995; 80: 837–845. Article CAS Google Scholar * Ausio J, Levin DB, De Amorim GV, Bakker S, Macleod PM : Syndromes of disordered chromatin
remodeling. _Clin Genet_ 2003; 64: 83–95. Article CAS Google Scholar * Chelly J, Mandel JL : Monogenic causes of X-linked mental retardation. _Nat Rev Genet_ 2001; 2: 669–680. Article
CAS Google Scholar * Martinowich K, Hattori D, Wu H _et al_: DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. _Science_ 2003; 302: 890–893. Article
CAS Google Scholar * Chen WG, Chang Q, Lin Y _et al_: Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. _Science_ 2003; 302: 885–889. Article CAS
Google Scholar * Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T : Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. _Nat Genet_ 2005; 37: 31–40.
Article CAS Google Scholar * Alarcon JM, Malleret G, Touzani K _et al_: Chromatin acetylation, memory, and LTP are impaired in CBP+/- mice: a model for the cognitive deficit in
Rubinstein–Taybi syndrome and its amelioration. _Neuron_ 2004; 42: 947–959. Article CAS Google Scholar * Anderson J, Bhandari R, Kumar JP : A genetic screen identifies putative targets
and binding partners of CREB-binding protein in the developing _Drosophila_ eye. _Genetics_ 2005; 171: 1655–1672. Article CAS Google Scholar * Le Douarin B, Nielsen AL, Garnier JM _et
al_: A possible involvement of TIF1 alpha and TIF1 beta in the epigenetic control of transcription by nuclear receptors. _EMBO J_ 1996; 15: 6701–6715. Article CAS Google Scholar * Garrick
D, Sharpe JA, Arkell R _et al_: Loss of ATRX affects trophoblast development and the pattern of X-inactivation in extraembryonic tissues. _PLoS Genet_ 2006; 2: e58. Article Google Scholar
* Garrick D, Samara V, McDowell TL _et al_: A conserved truncated isoform of the ATR-X syndrome protein lacking the SWI/SNF-homology domain. _Gene_ 2004; 326: 23–34. Article CAS Google
Scholar * Nan X, Hou J, Maclean A _et al_: Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. _Proc Natl Acad Sci USA_
2007; 104: 2709–2714. Article CAS Google Scholar * Luciani JJ, Depetris D, Usson Y _et al_: PML nuclear bodies are highly organised DNA-protein structures with a function in
heterochromatin remodelling at the G2 phase. _J Cell Sci_ 2006. Download references ACKNOWLEDGEMENTS We thank Dr David Bazett-Jones (Hospital for Sick Children) for the DsRed-PML plasmid and
Dr Rashmi Kothary for critical reading of the manuscript. The work was supported by a grant to DJP from the Canadian Institutes of Health Research (MOP-66970). AUTHOR INFORMATION Author
notes * Nathalie G Bérubé Present address: 3Current address: Departments of Biochemistry and Paediatrics, University of Western Ontario; Child Health Research Institute and Lawson Health
Research Institute, London, Ontario, Canada., * Nathalie G Bérubé and Jasmine Healy: These authors have contributed equally to this work. AUTHORS AND AFFILIATIONS * Molecular Medicine
Program, Ottawa Health Research Institute, Ottawa, Ontario, Canada Nathalie G Bérubé, Jasmine Healy, Chantal F Medina, Shaobo Wu, Todd Hodgson & Magdalena Jagla * Departments of Medicine
and Biochemistry, Microbiology, and Immunology, University of Ottawa, Ottawa, Ontario, Canada David J Picketts Authors * Nathalie G Bérubé View author publications You can also search for
this author inPubMed Google Scholar * Jasmine Healy View author publications You can also search for this author inPubMed Google Scholar * Chantal F Medina View author publications You can
also search for this author inPubMed Google Scholar * Shaobo Wu View author publications You can also search for this author inPubMed Google Scholar * Todd Hodgson View author publications
You can also search for this author inPubMed Google Scholar * Magdalena Jagla View author publications You can also search for this author inPubMed Google Scholar * David J Picketts View
author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to David J Picketts. RIGHTS AND PERMISSIONS Reprints and permissions ABOUT
THIS ARTICLE CITE THIS ARTICLE Bérubé, N., Healy, J., Medina, C. _et al._ Patient mutations alter ATRX targeting to PML nuclear bodies. _Eur J Hum Genet_ 16, 192–201 (2008).
https://doi.org/10.1038/sj.ejhg.5201943 Download citation * Received: 07 June 2007 * Revised: 22 September 2007 * Accepted: 25 September 2007 * Published: 24 October 2007 * Issue Date:
February 2008 * DOI: https://doi.org/10.1038/sj.ejhg.5201943 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * ATR-X syndrome * ATRX * chromatin
remodeling * SWI/SNF * X-linked mental retardation * XLMR