- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Mutations and deletions in the SPG4 gene are responsible for up to 40% of autosomal dominant hereditary spastic paraplegia (HSP). Patients have pyramidal signs in the lower limbs and some
present additional features including cognitive impairment such as executive dysfunction or subcortical dementia. We report 13 patients from three SPG4 families, who had spastic paraplegia
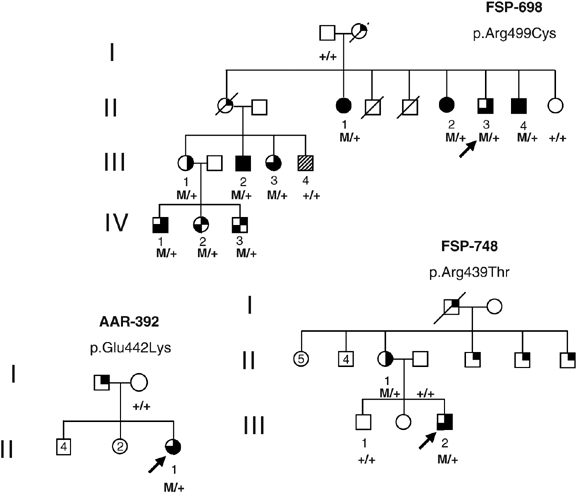
associated with mental retardation (n=1), extensive social dependence (n=10), or isolated psychomotor delay (n=2). In family FSP-698, 10 affected individuals had both HSP and mental
deficiency leading to social dependence in 9 and institutionalization in 5. The mean age at onset of spastic paraplegia was 11±20 years, ranging from 1 to 51 years. This phenotype segregated
either with a novel p.Glu442Lys mutation or the two previously described p.Arg459Thr and p.Arg499Cys substitutions in the SPG4 gene. Since two of these mutations were previously reported in
families with a pure form of the disease, another genetic factor linked to SPG4 could be responsible for this complex phenotype.
Hereditary spastic paraplegias (HSP) are clinically characterized by the presence of gait spasticity, muscle weakness and increased reflexes in the lower limbs (LL). Many patients may also
have sphincter disturbances and decreased vibration sense at ankles.1, 2, 3, 4 This phenotype defines clinically pure HSP, whereas complicated forms are associated with other neurological
signs such as peripheral neuropathy, mental retardation (MR), impairment of executive functions or dementia, cerebellar atrophy, optic atrophy or non-neurological signs such as
gastrooesophageal reflux.
HSP may be transmitted as autosomal dominant (AD), autosomal recessive (AR) or X-linked (XL) diseases. Autosomal dominant transmission accounts for 70–80% of all HSP.5, 6, 7 Among the nine
genes (SPG3A, SPG4, SPG6, SPG8, SPG10, SPG13, SPG17, SPG31, SPG33) and four loci (SPG9, SPG12, SPG19, SPG29) responsible for AD-HSP, SPG4 accounts for 15–40% of the families.1, 8, 9, 10 SPG4
encodes a 616-amino acid named spastin, which belongs to the AAA family (ATPases associated with diverse cellular activities). The AAA domain of spastin is located in the C terminus of the
protein between amino acids 342 and 599. Emerging evidence suggests that spastin plays a role in microtubule dynamics11 and that mutations in the spastin gene could lead to alteration of
intracellular organelles trafficking.12 All type of SPG4 mutations have been described, including missense, nonsense, splice-site mutations, small insertions and deletions9 as well as large
exonic deletions,13, 14 which strongly suggests that the pathogenic mechanism of SPG4 mutations is haploinsufficiency (ie, disease occurs once the level of functional spastin falls below a
critical level). So far, genotype–phenotype studies have not revealed a clear correlation between mutation types and age at onset or disease severity.9, 15, 16 The intragenic polymorphisms
S44L and P45Q are, however, associated with earlier age at onset.17, 18, 19
SPG4 mutations and deletions most often result in pure HSP with a mean age at onset of 29±17 years, ranging from infancy to 76 years.1, 9, 14 However, complicated forms associated to
variable degrees of cognitive decline, MR, epilepsy or cerebellar ataxia in addition to spasticity were described.20, 21, 22, 23, 24, 25, 26, 27, 28 We previously showed that carriers of
SPG4 mutation had subtle executive dysfunctions, which were absent in noncarriers relatives.28 Mental retardation was reported in SPG4 patients with cerebral malformations such as dysplasia
of the corpus callosum27 or congenital arachnoid cyst.22 We describe 13 patients from three SPG4 families, who have spastic paraplegia associated with mental deficiency without cerebral
malformation.
During diagnostic screening for SPG4 mutations in 543 families with available clinical information, we identified three families in whom MR, mental deficiency or psychomotor delay was
associated with spastic paraplegia and SPG4 mutations. The probands were seen at the outpatients clinic of the Department of Genetics and Cytogenetics, Salpêtrière Hospital in Paris. Their
relatives who could not come to the hospital were examined by one of us at their home or in their institutions. Families FSP-698 and FSP-748 originated from different regions of France,
whereas family AAR-392 originated from Morocco. Informed consent was obtained from each family member before blood sampling.
Age and sign at onset were noted for each patient, according to the patient and relatives. Gait spasticity was measured with a four-point scale, with 0=none, 1=mild, 2=moderate and 3=severe.
Functional impairment was assessed with a seven-points scale (0=none, 1=no functional impairment but signs at examination, 2=mild, 3=moderate, 4=walking with one cane, 5=walking with two
canes and 6=wheelchair-bounded). Psychomotor delay was assessed when patients acquired sitting position after the age of 9 months, gait after the age of 18 months or comprehensible speech
after the normal delay of 18–24 months. Social dependence was assessed when patients needed help for daily-life activities, such as finance management, self-direction, social skills, home
living, and so on. Mental retardation was defined by the DSM IV criteria, as having an IQ








