- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
_Dear Editor,_ TP73 shares significant structural homology with TP53.1 This predicts functional similarities for the regulation of cell cycle and apoptosis. Indeed, TAp73 becomes
transcriptionally activated upon DNA-damaging agents and oncogenes independent of p531,2 and ectopic TAp73 transactivates many p53 target genes involved in cell cycle arrest and apoptosis.1
Also, intact TAp73 function is an important determinant of cellular sensitivity to anticancer agents.3 However, the roles of TP53 and TP73 in mammalian tumorigenesis seem to be fundamentally
different. In contrast to TP53, TP73 is almost never targeted by inactivating mutations in human tumors.1 To the contrary, many different types of cancers overexpress TP73 (reviewed in Moll
_et al._4). Moreover, TP73 knockout mice lack a cancer phenotype but exhibit a developmental phenotype.5 These findings are inconsistent with a suppressor function of TP73. The different
roles of TP53 and TP73 in tumorigenesis might be based on the fact that TP73 gives rise to several different N- and C-terminal isoforms with opposing functions (reviewed in Moll _et al._4),
while p53 encodes a single suppressor protein. Aside from the transactivation-competent proapoptotic TAp73, four N-terminally truncated, dominant-negative isoforms are often concomitantly
overexpressed in human cancers. ΔN′p73, ΔNp73, Ex2p73 and Ex2/3p73 (collectively called ΔTAp73) lack the transactivation domain and are generated via alternate splicing of the P1 promoter
transcript6,7,8 or via the P2 promoter.9 ΔN and ΔN′p73 transcripts produce identical proteins. ΔTAp73 isoforms lack transactivation function and fail to induce cell cycle arrest and
apoptosis.9,10,11,12 They act as powerful inhibitors of p53 and TAp73 since they retain their DNA-binding and tetramerization competence.7,8,9,11,13,14 In cultured cells, ΔNp73 abrogates the
suppressive activity of p53 and TAp73 and inactivates their ability to induce apoptosis and cell cycle arrest.9 ΔNp73 overexpression results in malignant transformation of immortalized
NIH3T3 fibroblasts8 and also promotes immortalization in _primary_ cells and cooperates with oncogenic Ras in driving their transformation _in vivo_.15 Of note, human tumor data support the
notion that upregulation of ΔTAp73 alleviates the selection pressure to mutate p53.9 In gynecological cancers, significantly higher expression levels of ΔTAp73 are found in p53 wild-type
than in p53 mutant tumors. Moreover, the prevalence of upregulated ΔN and ΔN′p73 is higher in p53 wild-type than in p53 mutant tumors, while no such difference was seen for TAp73
upregulation.9,16 Also, ΔNp73 was an independent clinical prognostic marker for poor survival in neuroblastoma patients.17 Thus, there is mounting evidence that ΔTAp73 isoforms might act as
pathophysiologically relevant oncogenes, possibly outcompeting the opposing action of TAp73. This would readily explain the paucity of p73 mutations in cancers. Previously, we and others
demonstrated physical interaction between oncogenic and antioncogenic family members as a transdominant mechanism inhibiting the suppressor functions of wtp53 and TAp73.9,11,14 Mixed protein
complexes were found between endogenous ΔNp73_α_ or _β_ and either wtp53, TAp73_α_ or _β_ in human tumors and cultured tumor cells.9,11,14 The stoichiometric ratio of TA/ΔNp73 could be a
determinant in tumor formation. A small decrease in this ratio might be sufficient to convert TP73 from a tumor suppressor to an oncogene. Promoter competition by ΔNp73 at TAp73/p53 response
elements is another transdominant mechanism.11,12 Highly prevalent tumor-specific upregulation of ΔNp73 has already been found in several studies.8,9,16,17,18,19 For example, we reported
that ΔNp73/ΔN′p73 transcripts are overexpressed in 73% of 37 gynecological cancers and ΔN′p73 transcripts are upregulated in 87% of 100 ovarian cancers.16 Since inhibitory interactions of
two proteins often lead to their stabilization, we asked whether ΔNp73 can affect TAp73 protein levels. First, we determined whether the variant N- or C-terminal region of a given p73
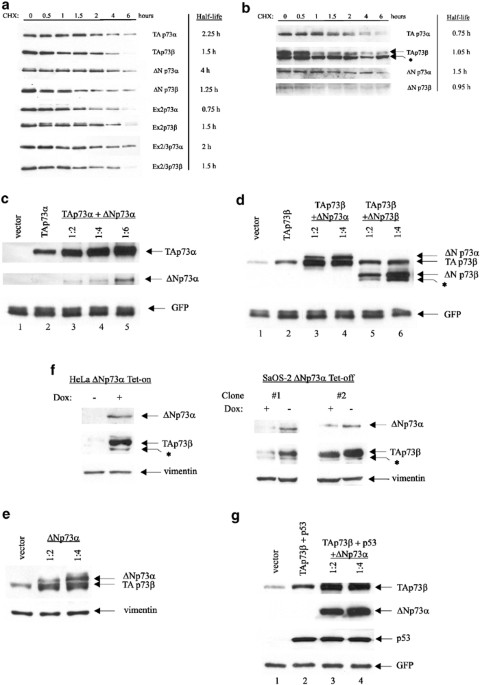
isoform impacts on its own steady-state level by determining the half-lives of human TAp73 and ΔTAp73_α_ and _β_ proteins in isolation (Figure 1a). To circumvent the problem of differential
sensitivities of different isoform-specific antibodies, we transfected Flag-tagged p73 isoforms into p53 null H1299, pulsed with protein synthesis inhibitor cycloheximide (CHX) and chased
protein levels over 6 h by Flag immunoblots. This analysis indicated that differences exist between C-terminal _α_- and _β_-isoforms, with _α_-forms generally being more stable than
_β_-forms (up to three-fold, see ΔNp73_α versus_ ΔNp73_β_) with the exception of Ex2p73, where the _β_-form is twice as stable as the _α_-form. Significantly, oncogenic ΔNp73_α_ is the most
stable of all eight proteins, with a half-life of 4 h. The N-terminus also modulates protein stability as seen among the _α_-subgroup, where differences range up to 5.3-fold (ΔNp73_α versus_
Ex2p73_α_ with 4 and 0.75 h, respectively). The shortest lived isoform is Ex2p73_α_, with a half-life of only 45 min. The N-terminus has only minimal influence among the _β_-subgroup (range
between 1 and 1.5 h). To compare stabilities of endogenous p73 isoforms, H1299 and RKO cells were treated with CHX and their degradation was followed (Figure 1b). The endogenous half-lives
were shorter but generally supported the trend seen with ectopic p73 isoforms. Endogenous ΔNp73_α_ again showed the longest half-life of ∼1.5 h. Taken together, the N- and C-terminal variant
regions moderately influence the stability of p73 proteins. Also, the relative longevity of ΔNp73 _versus_ TAp73 proteins, when in isolation, supports previous anecdotal observations that
ΔNp73 proteins are more stable than TAp73 in tumors.13,19 We next tested whether ΔNp73 proteins can induce stabilization of TAp73 proteins. H1299 cells were transfected with either TAp73_α_
or _β_ alone, or cotransfected with the same amount of TAp73_α_ or _β_ plus increasing amounts of ΔNp73α or ΔNp73_β_. Indeed, the steady-state levels of TAp73_α_ (Figure 1c) and TAp73_β_
(Figure 1d) increased proportionally with cotransfected ΔNp73_α_ or _β_. ΔNp73_α_ has a much stronger stabilizing effect towards TAp73 proteins than does ΔNp73_β_ (Figure 1d, compare lanes
2–4 _versus_ 2, 5 and 6 and data not shown). Identical data were obtained for endogenous TAp73 proteins in U2OS, H1299 and Phoenix cells (Figure 1e and data not shown). Marked TAp73_β_
stabilization was also observed upon induction of ΔNp73_α_ in Tet-inducible cell systems, such as HeLa Tet-on cells (Figure 1f, left) and in three independently derived SaOs-2 Tet-off clones
(Figure 1f, right and data not shown). Thus, TAp73 proteins are stabilized by coexpressed ΔNp73. Moreover, ΔNp73_α_ confers the main stabilization, possibly because the levels of ΔNp73_α_
are often higher than ΔNp73 which might in turn be related to the longer half life of ΔNp73_α_ compared to _β_. Given the homology between TP53 and TP73 and the existence of mixed protein
complexes between p53 and ΔNp73_α_,6,9,11,14 it is conceivable, at least in theory, that p53 protein levels could also be stabilized by ΔNp73_α_. However, p53 protein levels failed to
accumulate despite increasing amounts of co-expressed ΔNp73_α_ Figure 1g, compare lanes 2–4, while TAp73_β_ levels were induced to accumulate in the same cells. This differential is likely
related to different degradation pathways. While p53 is degradated by binding to its E3 ligase MDM2, MDM2 binding to p73 disrupts its interaction with p300/CBP, but does not mediate p73
degradation.20,21,22 Instead, p73 degradation is linked to SUMO-1 modification which renders it proteasome-degradable.23 It is possible that hetero-oligmerization prevents sumoylation of
p73, thereby stabilizing it. ΔNp73 strongly inhibits the transactivation function of p53 and TAp73 and this largely depends on forming mixed protein–protein complexes.9,11,14,15 For example,
we found that tetramerization-deficient mtΔNp73(L322P) almost completely abrogates the inhibitory function of ΔNp73 towards p53-mediated transactivation9 and abrogates the immortalization
function of ΔNp73 in primary fibroblasts.15 We therefore considered that the ability of ΔNp73 to form a heterocomplex with TAp73 could be critical to its ability to mediate accumulation of
TAp73 proteins. If this were the case, it would link ΔNp73-mediated TAp73 accumulation to its concomitant functional inhibition, that is, in such an inhibitory complex TAp73 accumulates but
is inactive. Indeed, after cotransfecting a constant amount of TAp73_β_ plasmid with increasing ratios of a p73 tetramerization domain plasmid (aa 313–404, called _p73tet_), TAp73_β_ levels
markedly accumulated and rapidly reached saturation at a 1 : 2 ratio (Figure 2a). Identical results were seen with endogenous TAp73_β_ protein (Figure 2b, lanes 1and 4). Also, cells
expressing the C-terminal fragment of human p73_α_ (aa 327–636), comprised of the tetramerization and the SAM domains (called p73DD2) accumulated endogenous TAp73_β_. In contrast, its mutant
version (mt p73DD), harboring a loss-of-function L371P mutation in the tetramerization domain, had lost much of this stabilizing activity (Figure 2b, lanes 5 and 6). Finally, while
coexpression of full-length ΔNp73_α_ induced the expected accumulation of endogenous TAp73_β_, its mutant counterpart mtΔNp73(L322P), which carries a mutation in the tetramerization domain
that inactivates its transdominant function,9 was able to do so only poorly (Figure 2b, lanes 1–3). Figure 2c shows that these polypeptides and their mutant counterparts were expressed at
equal levels. Taken together, these data show that the tetramerization domain is the minimal domain that is necessary and sufficient for ΔNp73 to induce accumulation of TAp73. However, other
region(s) of ΔNp73 may play an additional minor role, since tetramerization mutants do not completely revert this phenotype. Importantly, the ability of ΔNp73 to mediate TAp73 accumulation
clearly correlates with its functional activity as a dominant-negative inhibitor of TAp73. To confirm that ΔNp73_α_ is an inhibitor of TAp73_α_ and _β_ (and wtp53), we performed reporter
assays with TAp73, p53 and a TAp73/p53-responsive luciferase reporter in the absence or presence of an increasing ratio of ΔNp73_α_. As expected, ΔNp73_α_ completely suppressed the
transcriptional activity of TAp73_β_ and wt p53 in U2OS and AGS cells (both are wild type for p53) Figure 2d). Thus, these data show that the TAp73 protein, although stabilized by ΔNp73, is
functionally compromised or inactive. It has been proposed that ΔTAp73 inhibits the function of TAp73 and p53 in different ways: TAp73 is inhibited mainly at the oligomerization level,
forming an inactive hetero-oligomer.9,14 In the case of p53, in addition to forming inactive heterocomplexes, ΔTAp73 also exerts a dominant-negative effect by displacing p53 from its
promoters.11 In summary, we show here that ΔNp73 isoforms stabilize TAp73 proteins but, in doing so, inhibit their transactivation function. Individual p73 isoforms with variant N- and
C-termini differ only moderately in their half-lives. However, when coexpressed, TAp73_α_ and _β_ proteins become markedly stabilized by ΔNp73_α_. Similar results were seen with ΔNp73_β_,
albeit the effect is weaker. In contrast, p53 protein fails to accumulate via ΔNp73. Using tetramerization-deficient mutants, we show that the ability of ΔNp73 to mediate TAp73 accumulation
largely depends on its tetramerization domain and correlates with its ability to function as a dominant-negative inhibitor of TAp73. In the ongoing debate whether TAp73 is a relevant tumor
suppressor, we suggest that increased TAp73 protein levels should be interpreted with caution when levels are the only criteria that can be used to deduce TAp73 activity. This is
particularly the case in primary tumors where functional studies are not possible. REFERENCES * Melino G et al. (2002) _Nat. Rev. Cancer_ 2: 605–615 * Zaika A et al. (2001) _J. Biol. Chem._
276: 11310–11316 * Bergamaschi D et al. (2003) _Cancer Cell_ 3: 387–402 Article CAS Google Scholar * Moll UM et al. (2001) _Biochim. Biophys. Acta_ 1552: 47–59 * Yang A et al. (2000)
_Nature_ 404: 99–103 Article CAS Google Scholar * Pozniak CD et al. (2000) _Science_ 289: 304–306 Article CAS Google Scholar * Ishimoto O et al. (2002) _Cancer Res._ 62: 636–641 *
Stiewe T et al. (2002) _Cancer Res._ 62: 3598–3602 * Zaika AI et al. (2002) _J. Exp. Med._ 196: 765–780 * Fillippovich I et al. (2001) _Oncogene_ 20: 514–522 * Stiewe T et al. (2002) _J.
Biol. Chem._ 277: 14177–14185 Article CAS PubMed Google Scholar * Kartasheva NN et al. (2002) _Oncogene_ 21: 4715–4727 Article CAS Google Scholar * Grob TJ et al. (2001) _Cell Death
Differ._ 8: 1213–1223 Article CAS Google Scholar * Nakagawa T et al. (2002) _Mol. Cell. Biol._ 22: 2575–2585 * Petrenko O et al. (2003) _Mol. Cell. Biol._ 23: 5540–5555 * Concin N et al.
(2003) _Cancer Res._ submitted * Casciano I et al. (2002) _Cell Death Differ._ 9: 246–251 Article CAS Google Scholar * Putzer BM et al. (2003) _Cell Death Differ._ 10: 612–614 Article
Google Scholar * Douc-Rasy S et al. (2002) _Am. J. Pathol._ 160: 631–639 * Balint E et al. (1999) _Oncogene_ 18: 3923–3929 Article PubMed Google Scholar * Dobbelstein M et al. (1999)
_Oncogene_ 18: 2101–2106 Article CAS PubMed Google Scholar * Zeng X et al. (1999) _Mol. Cell. Biol._ 19: 3257–3266 Article CAS PubMed PubMed Central Google Scholar * Minty A et al.
(2000) _J. Biol. Chem._ 275: 36316–36323 Article CAS PubMed Google Scholar Download references AUTHOR INFORMATION Author notes * N Slade Present address: Department of Molecular
Medicine, Ruder Boskovic Insitute, Zagreb, 10 000, Croatia * AI Zaika Present address: Division of Gastroenterology and Hepatology, University of Virginia, Charlottesville, 22908, VA, USA
AUTHORS AND AFFILIATIONS * Department of Pathology Stony Brook University, Stony Brook, 11794, NY, USA N Slade, AI Zaika, S Erster & UM Moll Authors * N Slade View author publications
You can also search for this author inPubMed Google Scholar * AI Zaika View author publications You can also search for this author inPubMed Google Scholar * S Erster View author
publications You can also search for this author inPubMed Google Scholar * UM Moll View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR
Correspondence to UM Moll. RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Slade, N., Zaika, A., Erster, S. _et al._ ΔNp73 stabilises TAp73 proteins but
compromises their function due to inhibitory hetero-oligomer formation. _Cell Death Differ_ 11, 357–360 (2004). https://doi.org/10.1038/sj.cdd.4401335 Download citation * Published: 12
December 2003 * Issue Date: 01 March 2004 * DOI: https://doi.org/10.1038/sj.cdd.4401335 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative